
Abstract
Bone tumours are difficult to diagnose and treat, as they are rare and over 60 different subtypes are recognised. The emergence of next-generation sequencing has partly elucidated the molecular mechanisms behind these tumours, including the group of bone forming tumours (osteoma, osteoid osteoma, osteoblastoma and osteosarcoma). Increased knowledge on the molecular mechanism could help to identify novel diagnostic markers and/or treatment options. Osteoid osteoma and osteoblastoma are bone forming tumours without malignant potential that have overlapping morphology. They were recently shown to carry FOS and—to a lesser extent—FOSB rearrangements suggesting that these tumours are closely related. The presence of these rearrangements could help discriminate these entities from other lesions with woven bone deposition. Osteosarcoma is a malignant bone forming tumour for which different histological subtypes are recognised. High-grade osteosarcoma is the prototype of a complex karyotype tumour, and extensive research exploring its molecular background has identified phenomena like chromothripsis and kataegis and some recurrent alterations. Due to lack of specificity, this has not led to a valuable novel diagnostic marker so far. Nevertheless, these studies have also pointed towards potential targetable drivers of which the therapeutic merit remains to be further explored.
Similar content being viewed by others


Introduction
Bone tumours are rare and therefore considered difficult to diagnose and treat. They comprise a heterogeneous group of tumours, where most subtypes have a distinct clinical and histological presentation.
Histologically, over 60 different bone tumours are recognised. Some are difficult to separate as there can be extensive morphological and even immunohistochemical overlap. Distinction is important as these tumours differ in clinical behaviour and thus in required treatment. In recent years, many papers have been published unravelling the molecular background of several bone tumours, mostly using deep sequencing techniques. From the molecular point of view, these tumours can be roughly divided in two main groups, as a conceptual framework [1]: tumours can either have a simple or complex karyotype. The group of tumours with a simple karyotype are usually monomorphic and driven by a specific mutation or translocation. The tumours with complex karyotype are more often pleomorphic, show aneuploidy, with many copy number alterations and (random) translocations and mutations.
The group of skeletal tumours that are characterised by bone deposition contains osteoma, osteoid osteoma, osteoblastoma and osteosarcoma (Table 1). Osteoma is benign and composed of mature lamellar bone, has a simple karyotype and occurs more often in patients with Gardner’s syndrome, that harbour a germline mutation in the APC gene. Osteoid osteoma and osteoblastoma are histologically identical, have a simple karyotype and deep sequencing studies have recently unravelled a recurrent translocation [2]. This is in contrast with high-grade osteosarcoma, for which a complex karyotype showing aneuploidy, multiple copy number alterations, (random) translocations and mutations is the hallmark [3]. This review will focus on osteoid osteoma/osteoblastoma and high-grade osteosarcoma, as examples for simple karyotype, translocation driven versus complex karyotype tumours, respectively.
Osteoid osteoma and osteoblastoma
Novel FOS and FOSB rearrangements were recently found in osteoid osteoma and osteoblastoma [2]. These tumours account for 3% and 1% of all primary bone tumours, respectively [4]. These two entities are histologically similar and only slightly differ in their clinical presentation. At present, they are arbitrarily divided by tumour size below or above 2 cm in diameter, although the recent finding show that they share the same molecular alteration might suggest that they represent the same disease [4,5,6].
Clinical presentation
Osteoid osteoma and osteoblastoma typically present during the second decade of life, with men being overrepresented (male to female ratio 2:1) [4]. Osteoid osteoma is usually located at the long bones in the lower extremity, but other commonly described sites involve the spine, upper extremity, hands, feet and pelvis [4, 5, 7]. The most prominent clinical symptom of osteoid osteoma is frequent and severe night pain that responds adequately to nonsteroidal anti-inflammatory drugs (NSAIDs) [4, 5]. Osteoblastoma is larger in size, and the majority is localized in the posterior column of the spine [4, 5, 8], resulting in neurologic symptoms as a recurring sign [4]. Pain is frequently present, but in contrast to osteoid osteoma, it does not respond to administration of NSAIDs [4, 5]. Both osteoid osteoma and osteoblastomas have no malignant potential, although osteoblastoma can behave as a locally aggressive tumour [4]. For radiologists, the diagnosis of osteoid osteoma is usually straight forward, showing a characteristic oval radiolucency (nidus) with surrounding sclerosis, while osteoblastoma can be accompanied by a more broad differential diagnosis depending on its location, including aneurysmal bone cyst, giant cell tumour of bone and osteosarcoma [4, 9].
Histology
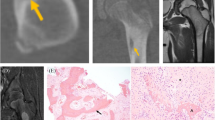
Osteoid osteoma and osteoblastoma are histologically indistinguishable [10] (Fig. 1a, b). Both tumours are composed of irregular trabeculae of woven bone, lined with active osteoblasts. In osteoid osteoma, the central area of the lesion (nidus) is sharply demarcated and surrounded by hyper-vascularized sclerotic bone. In between the trabeculae, there is loose vascularised stroma, and small osteoclast-like giant cells are frequently seen [7, 11]. Osteoblastoma can show slightly more haphazardly arranged trabeculae [6]. Additional aneurysmal bone cyst (ABC)-like changes can be present, especially in larger tumours [4]. The term epithelioid osteoblastoma is reserved for osteoblastomas with the presence of large osteoblasts with an epithelioid appearance. Surrounding cytoplasm is abundant, and nuclei are hyperchromatic or show prominent nucleoli [4]. The most important differential diagnosis includes osteoblastoma-like osteosarcoma, that is distinguished from osteoblastoma based on the presence of host-bone infiltration and lack of differentiation towards the periphery [12]. However, this can be difficult to appreciate in small biopsies or curettage specimens. Definitive diagnosis is always made based on radiological and clinicopathological correlation.

Osteoid osteoma and osteoblastoma. a Osteoid osteoma. b Osteoblastoma show identical morphology at haematoxylin and eosin staining, with deposition of woven bone by osteoblast-like tumour cells. c Fluorescence in situ hybridization (FISH) showing FOS rearrangement in osteoblastoma. d Immunohistochemical staining for FOS in osteoblastoma showing nuclear overexpression in the tumour cells. Scale bar is 50 μm
Molecular pathology
Before the elucidation of the genetic background of osteoid osteoma and osteoblastoma, clonal chromosome aberrations were reported in two osteoblastomas, with structural alterations involving 22q13.1 [13], and only non-recurrent rearrangements were found using cytogenetic studies [14]. In 2018, in a quiet genomic background with paucity of somatic alterations, recurrent FOS and—to a lesser extent—FOSB rearrangements were found in both osteoid osteoma and osteoblastoma using RNA sequencing, demonstrating that both tumours were similar at the molecular level. In 5 out of 6 cases, FOS rearrangements were present, while the remaining case showed rearrangements involving its paralogue, FOSB. All FOS breakpoints were exonic and involved exon 4. Rearrangement partners were both introns of others genes (ANKH, KIAA1199, MYO1B) or intergenic regions [2]. Equivalent to FOS rearranged epithelioid hemangioma [15, 16], stop codons were encountered at, or early after the break points, leading to truncation of the protein with retention of the leucine zipper, and therefore its function as a transcription factor. Functional studies in epithelioid hemangioma demonstrated that the truncated protein was more resistant to degradation [17]. In the FOSB rearranged osteoblastoma, rearrangement resulted in an in frame fusion connecting PPP1R10 to FOSB, leading to altered signalling, due to promotor swapping [2]. Strikingly, FOSB fusions were also involved in pseudomyogenic hemangioendothelioma and atypical epithelioid hemangioma, resulting in promoter swapping [18, 19]. As genetic alterations in these vascular tumours are identical to those found in osteoid osteoma and osteoblastoma, one can speculate that a comparable molecular mechanism of tumorigenesis is operable in osteoid osteoma and osteoblastoma.
These novel molecular findings have provided new tools to improve diagnostic accuracy, as both fluorescence in situ hybridization (FISH) and immunohistochemical staining can detect FOS rearrangements (Fig. 1c, d). FISH was performed in an independent cohort and showed in the majority of cases rearrangements involving FOS and to a lesser extent FOSB [2]. In a follow-up study, immunohistochemistry showed strong and diffuse nuclear staining in the majority (79%) of osteoid osteomas and osteoblastomas, using a FOS antibody against the N terminus [20]. However, a previously published small study cohort demonstrated that osteoid osteoma and osteoblastoma lacked strong nuclear expression of FOS, indicating variability in sensitivity between different antibodies [21]. In terms of specificity, strong nuclear expression of FOS has been detected in a subset of other bone forming tumours and was only rarely present in osteosarcoma [2, 21]. Notably, in mouse models, the c-fos oncogene caused osteosarcoma, when fused with a highly active promotor and the v-fos 3’ untranslated region [22]. This is intriguing as in human tumours FOS and FOSB rearrangements have so far only been identified in vascular and bone forming tumours lacking malignant potential [15, 16, 18, 19].
Osteosarcoma
Osteosarcoma is the most common primary malignant tumour of the bone [23]. The 5-year overall survival for osteosarcoma patients is 71% and has not improved in the last decades, clearly indicating that novel therapeutic strategies are needed [24]. Fortunately, many papers have been published gradually unravelling the pathogenesis of osteosarcoma, which might help develop new therapeutic targets.
Clinical presentation
Primary high-grade osteosarcoma occurs most often in young children and adolescents, but there is a second peak at a later age. In the latter group, osteosarcoma can occur secondary to radiation or Paget’s disease [25]. Osteosarcoma has a slight male predominance [26]. Patients with osteosarcoma often show signs of localised deep pain, especially manifest at night, developed over a longer period of a few weeks to months. This could also be in combination with limited mobility or localised warmth. A palpable mass can be present, which is tender during physical examination [27].
For diagnosis of conventional osteosarcoma, a radiograph is made in two planes, in which the lesion appears as lytic, sclerotic or mixed lytic and sclerotic. This lesion often expands into the surrounding soft tissue, with periosteal reaction and destruction of cortical bone [28]. MRI or CT imaging may provide additional information, guiding the subsequent biopsy of the lesion [28].
Histology
The presence of osteoid, the unmineralized extracellular matrix produced by the tumour cells, is the hallmark of osteosarcoma and visible as a pink dense structure in haematoxylin and eosin stained sections (Fig. 2a). Mineralization can occur. Osteosarcoma can arise in the medulla (central) or at the bone surface. Different osteosarcoma subtypes are recognised, based on their clinical presentation in combination with histological and molecular features (Table 2) [26]. High grade central osteosarcoma is the most common subtype, and most papers published over the last decade, as well as this review, focus on this subtype.

High-grade osteosarcoma. a Conventional osteoblastic osteosarcoma showing atypical cells with abundant deposition of osteoid (haematoxylin and eosin staining). Scale bar is 50 μm. b Combined binary ratio fluorescence in situ hybridization (COBRA-FISH) showing complex numerical and structural changes which is characteristic of high-grade osteosarcoma
Germline predisposition to osteosarcoma
Certain hereditary syndromes predispose to osteosarcoma, such as Li-Fraumeni syndrome (mutations in TP53 or, less frequently, CHEK2), retinoblastoma (mutations in RB1) and Rothmund-Thomson syndrome (mutations in RECQL4) [29,30,31]. Other hereditary syndromes with germline mutations in RecQ-like helicases, including RAPADILINO syndrome, Baller-Gerold syndrome, Werner syndrome and Bloom syndrome, also have an increased risk for osteosarcoma [32]. Another hereditary syndrome in which a helicase is mutated is ATR-X syndrome (alpha-thalassemia mental retardation syndrome). Patients with ATR-X syndrome show intellectual disability and skeletal abnormalities. Recently, two patients have been reported with ATR-X syndrome that developed osteosarcoma [33, 34].
Molecular alterations in osteosarcoma
High-grade osteosarcoma is characterised by a complex karyotype with many amplifications, deletions and (random) translocations (Fig. 2b). This complex genome hampers identification of the driver genes causing genome instability: very few recurrent alterations have been identified in osteosarcoma. One mechanism explaining the genomic instability in osteosarcoma is chromothripsis, the shattering of one or a few chromosomes into small fragments that are stitched together in a random order and orientation [35]. It was first discovered by Stephens et al. in chronic lymphocytic leukaemia, chordoma and osteosarcoma [35]: chromothripsis occurs in 3% of all cancers and in 30% of osteosarcomas. A more recent study confirmed chromothripsis in osteosarcoma, but showed a higher percentage—nearly 90%—where tumours with chromothripsis also frequently harbour amplifications [36]. The discrepancy may be attributed to the uncertain definition of chromothripsis. Exome sequencing shows a relatively low mutational burden in osteosarcoma ranging from 0.3–1.2 mutations per mega base; however, there is a pattern of localised hypermutation called kataegis in 50% of the tumours [3, 37]. These point mutations are non-recurrent, haphazard and cannot be considered as driver mutations. Further hampering the identification of driver genes is that no benign precursor of osteosarcoma is known. This is in contrast with for instance colorectal cancer, in which a benign precursor can be used to investigate multi-step progression behind tumorigenesis. Nevertheless, recent next-generation sequencing studies have revealed known and novel recurrent genetic alterations in osteosarcoma (Table 3). Most genes that were found to be altered are involved in maintaining genomic stability. Among the most commonly altered genes in osteosarcoma are the main players in maintaining genome stability: TP53 and RB1.
TP53 and RB1
Mutations in TP53 can be found in germline or can be sporadic. Previously, using immunohistochemistry or sequencing of the DNA binding domain of TP53, mutations were detected in only 20% of osteosarcomas [44]. Interestingly, the more sensitive whole genome sequencing studies can detect more sub clonal mutations and reveal a much higher percentage (47–90%) of osteosarcomas harbouring TP53 alterations [3, 36,37,38, 45]. Furthermore, many TP53 alterations involve structural alterations, most often consisting of translocations in the first intronic region of TP53, which is 10 kb in length. These alterations can only be detected with whole genome sequencing [46].
The second most frequently altered gene in osteosarcoma is RB1 (retinoblastoma 1), involved in blocking cells from entering S phase of the cell cycle [47]. Loss of Rb function in osteosarcoma therefore leads to a loss in Rb blockade of cell division. In addition to germline mutations, somatic mutations in RB1 were identified in 29–47% of osteosarcomas [3, 38].
The importance of TP53 and RB1 in osteosarcoma genesis is illustrated by the fact that patients with germline mutations in TP53 and RB1 are highly susceptible to cancer and frequently develop sarcomas. Different in vitro and in vivo studies confirm the important role of TP53 and RB1 mutations in sarcoma genesis [48, 49]. For example, homozygous deletion of TP53 and RB1 in osteogenic differentiated murine MSCs gives rise to osteosarcoma when injected into mice [49], while heterozygous deletion of TP53 is sufficient to induce osteosarcoma in a mouse model [48].
Regulators of p53 and Rb activity
MDM2 (mouse double minute 2 homologue) regulates p53 activity by ubiquitinating p53 protein leading to proteasomal degradation of p53 [50]. Up to 12% of high-grade osteosarcomas have amplification of the MDM2 gene at 12q13-15, but this is higher in low-grade central osteosarcoma and parosteal osteosarcoma, with around 29% and 67–79% MDM2 amplification, respectively [41, 51] (Table 2). The CDK4 gene (cyclin-dependent kinase 4) is located within the same region at 12q13-15 [52] and regulates Rb activity by phosphorylating Rb, resulting in deactivation of Rb. CDK4 and MDM2 are often co-amplified and overexpressed in osteosarcoma. CDK4 is amplified in 67% of parosteal osteosarcomas, but rarely in high-grade osteosarcoma (9%) [41, 53]. As the percentage of CDK4 and MDM2 amplifications in low-grade central osteosarcoma and parosteal osteosarcoma are much higher than in high-grade osteosarcoma, most likely the CDK4/MDM2 amplified high-grade tumours represent progression from low grade osteosarcoma [53].
Rb activity is also regulated by p16, which normally inhibits both CDK4 and CDK6. P16 is encoded by the CDKN2A gene at chromosome 9p21.3, that also encodes for p14. Homozygous deletion of the CDKN2A locus, which is associated with poor prognosis in osteosarcoma, eradicates both expression of p16Ink4A and p14ARF, of which the latter is a negative regulator of MDM2 [38, 54,55,56]. Therefore, deletion of p16 and p14, similar to co-amplification of CDK4 and MDM2, leads to inactivation of both the p53 and Rb pathway.
Other genome maintenance pathways
In addition to the p53 and Rb pathway, also other pathways involved in maintaining genome stability can be affected by mutations, both in sporadic as well as hereditary osteosarcoma. For instance, ATRX mutations can be found both as germline or somatic mutations [57], which is in contrast to mutations in RecQ-like helicases where only germline mutations have been identified. Around 29% of osteosarcomas harbour somatic mutations in ATRX [3]. The role of ATRX mutations in osteosarcoma genesis is largely unknown. ATRX is involved in chromatin remodelling and plays an important role in maintenance of chromosome stability [58]. Loss-of-function mutations in ATRX can lead to activation of the alternative lengthening of telomeres (ALT) pathway, maintaining the length of chromosome ends [59]. ALT is found in 59% of osteosarcomas, which is much higher as compared with other cancers such as carcinomas (5–15%) [60].
DNA repair is essential in maintaining genome stability. For instance, homologous recombination, the DNA repair pathway in which BRCA plays an important role, is crucial in maintaining genome stability. A recent whole exome sequencing (WES) study revealed a subset of osteosarcomas resemble features of BRCA mutant tumours [38]. These tumours show loss of heterozygosity, genomic instability and a mutation signature of substitutions and deletions that is also found in breast cancers with BRCA1/2 mutations. Around 80% of osteosarcomas show this BRCAness signature [38]. As this signature is linked to defects in homologous recombination, this vulnerability might be exploited with PARP inhibitors based on the principle of synthetic lethality. Indeed, different in vitro studies with osteosarcoma cell lines show that osteosarcoma cells are sensitive to PARP inhibitors [61, 62]. These results are promising, suggesting a possible new therapeutic strategy for osteosarcoma. However, further investigation on homologous recombination deficiency and PARP inhibitor sensitivity in osteosarcoma is needed.
Hormonal pathways
Although the genes that play a role in genome stability are among the most frequently mutated genes in osteosarcoma (RB1, TP53, CDK4, MDM2, ATRX), these genes function in essential cell survival pathways. Therefore, these genes are difficult to specifically target in the treatment of osteosarcoma. Fortunately, also mutations in other genes are frequently found that are easier to target as they are involved in hormonal pathways. For example, mutations in genes involved in IGF (insulin-like growth factor) signalling, including the IGF1 receptor (IGF1R), were identified in around 7–14% of osteosarcomas, with many of these genes having altered activity compared with normal human osteoblasts or mesenchymal stem cells [36, 63]. The IGF signalling pathway is known to be important in normal bone growth, bone development and bone metabolism, and it is therefore not surprising that it might also play a role in osteosarcoma pathogenesis [64, 65]. These findings provide a rationale to explore anti-IGFR therapy as a treatment strategy for a subset of osteosarcomas.
The oestrogen hormonal pathway is also altered in osteosarcoma. Healthy osteoblasts normally express oestrogen receptor alpha (ERα), but this is lacking in osteosarcoma [66]. Until recently, the mechanism behind the inactivation of oestrogen receptor in osteosarcoma was not known. In a recent study, it was found that ERα was hypermethylated in osteosarcoma, which can be ameliorated by the DNA methyltransferase inhibitor DAC [67]. DAC could re-express ERα and subsequently restored defective osteogenic differentiation and inhibited proliferation in osteosarcoma cells. This study illustrates that epigenetic alterations such as hypermethylation of genes are also important in osteosarcoma genesis.
What is driving osteosarcoma genesis?
Although recent sequencing efforts did not identify specific druggable osteosarcoma driver genes, they did reveal new and known recurrent genetic events involved in osteosarcoma that shed light on its initiation (Table 3). Most of the mutated genes function in genome maintenance pathways and the majority of osteosarcomas show genome instability in the form of chromoanagenesis [36]. Therefore, it is reasonable to assume a connection between these specific genetic mutations and chromoanagenesis, especially chromothripsis.
A main player in maintaining genome stability, TP53, was linked to chromothripsis in patients with Li- Fraumeni syndrome [68]. Furthermore, rats with a heterozygous deletion of TP53 developed osteosarcomas, with chromothripsis and other complex structural rearrangements [69]. As cells with aberrant TP53 have impaired cell cycle control [70], TP53 loss-of-function alterations can facilitate chromothripsis by allowing cell cycle progression despite DNA damage [68]. Thus, cells with inactive TP53 and DNA damage from chromothripsis will proliferate uncontrollably. Moreover, mutations in the DNA binding domain of TP53 can cause a neomorphic gain-of-function, that could very well contribute to the initiation of chromothripsis itself [71].
However, TP53 alone cannot explain all tumours with chromothripsis, as is evident from studies that illustrate there are tumours with functional TP53 with chromothripsis, and tumours with aberrant TP53 without chromothripsis [72, 73]. Genes functionally similar to TP53 might also be able to initiate and/or maintain chromothripsis. Whether this is the case for osteosarcomas needs to be further elucidated.
It is striking that for osteosarcoma, many different genes have been identified and with each new sequencing study, the list of potential driver genes is ever-growing. Whether a genetic alteration—in TP53 or other genes—is a cause or consequence of chromothripsis remains unknown. One could argue whether the identified altered genes in osteosarcoma should be called “driver events” if these genetic alterations are the consequence of a single catastrophic event, such as chromothripsis. Therefore, perhaps the answer to what causes osteosarcoma could be found by discovering what causes chromothripsis. Different mechanisms have been proposed to what causes chromothripsis, such as micronuclei formation with DNA damage, telomere attrition and chromosome pulverisation by DNA damaging agents [74, 75]. Which event is the ultimate cause of osteosarcoma, is not yet known.
Conclusion
There is an on-going shift from traditional cancer classification based solely on histopathology towards incorporation of molecular pathology in routine diagnostics, which ultimately can aid diagnostic decision making. Among the group of bone forming tumours of the skeleton, the use of deep sequencing has unravelled the molecular background of osteoid osteoma and osteoblastoma. The discovery of FOS and FOSB rearrangements found in osteoid osteoma and osteoblastoma have not only given insight in tumorigenesis, but have also provided the bone tumour pathologist with a novel diagnostic tool to improve diagnostic accuracy.
For high-grade osteosarcoma, due to its complex genomic background, no specific, recurrent genetic alteration has been found that can explain tumorigenesis, or can be used for diagnosis or treatment. Even though the number of publications on drugs that allegedly inhibit osteosarcoma growth has exponentially increased over the past few years, these claims are often based on in vitro studies including one single cell line [76]. Most of these publications are from Chinese institutes and often consist of investigations on the effect of traditional medicine on osteosarcoma. The remarkable increase of these studies is most probably the corollary of the convenient tissue culture properties of osteosarcoma cell lines and obscures findings of real significance.
Nevertheless, in the last years, several deep sequencing studies have been published that contribute towards the understanding of osteosarcoma pathogenesis. These next-generation sequencing studies have revealed underlying mechanisms, such as chromothripsis and kataegis, as well as a number of genes and pathways associated with osteosarcoma, especially those involved in genome maintenance (TP53, RB1, ATRX and homologous recombination) or hormonal signalling (IGF and ER signalling). The results from these studies could be the stepping stone towards the development of novel diagnostics/prognostic markers or treatment options. Since most of the alterations that were identified are not recurrent and involved in crucial processes in the cell such as genome stability, cell cycle and DNA repair, it will be a huge challenge for the coming decade to translate these findings into novel treatment options. In contrast to targeting genes involved in maintaining genome stability, such as TP53 and RB1, targeting the hormonal pathways, especially IGF and oestrogen or targeting DNA repair, for example by PARP inhibition, seem more promising.
References
Lam SW, van Ijzendoorn DGP, Cleton-Jansen A-M, Szuhai K, Bovée JVMG (2019) Molecular pathology of bone tumors. J Mol Diagn 21(2):171–182. https://doi.org/10.1016/j.jmoldx.2018.11.002
Fittall MW, Mifsud W, Pillay N, Ye H, Strobl AC, Verfaillie A, Demeulemeester J, Zhang L, Berisha F, Tarabichi M, Young MD, Miranda E, Tarpey PS, Tirabosco R, Amary F, Grigoriadis AE, Stratton MR, Van Loo P, Antonescu CR, Campbell PJ, Flanagan AM, Behjati S (2018) Recurrent rearrangements of FOS and FOSB define osteoblastoma. Nat Commun 9(1):2150. https://doi.org/10.1038/s41467-018-04530-z
Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, Parker M, Rusch M, Nagahawatte P, Wu J, Mao S, Boggs K, Mulder H, Yergeau D, Lu C, Ding L, Edmonson M, Qu C, Wang J, Li Y, Navid F, Daw NC, Mardis ER, Wilson RK, Downing JR, Zhang J, Dyer MA, St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome P (2014) Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7(1):104–112. https://doi.org/10.1016/j.celrep.2014.03.003
Atesok KI, Alman BA, Schemitsch EH, Peyser A, Mankin H (2011) Osteoid osteoma and osteoblastoma. J Am Acad Orthop Surg 19(11):678–689
Greenspan A (1993) Benign bone-forming lesions: osteoma, osteoid osteoma, and osteoblastoma. Clinical, imaging, pathologic, and differential considerations. Skelet Radiol 22(7):485–500
Schajowicz F, Lemos C (1970) Osteoid osteoma and osteoblastoma. Closely related entities of osteoblastic derivation. Acta Orthop Scand 41(3):272–291
Jaffe HL (1953) Osteoid-osteoma. Proc R Soc Med 46(12):1007–1012
Cerase A, Priolo F (1998) Skeletal benign bone-forming lesions. Eur J Radiol 27(Suppl 1):S91–S97
Kroon HM, Schurmans J (1990) Osteoblastoma: clinical and radiologic findings in 98 new cases. Radiology 175(3):783–790. https://doi.org/10.1148/radiology.175.3.2343130
Frassica FJ, Waltrip RL, Sponseller PD, Ma LD, McCarthy EF Jr (1996) Clinicopathologic features and treatment of osteoid osteoma and osteoblastoma in children and adolescents. Orthop Clin North Am 27(3):559–574
Jaffe HL (1935) “Osteoid-osteoma”: a bening osteoblastic tumor composed of osteoid and atypical bone. Arch Surg 31(5):709–728. https://doi.org/10.1001/archsurg.1935.01180170034003
Bertoni F, Unni KK, McLeod RA, Dahlin DC (1985) Osteosarcoma resembling osteoblastoma. Cancer 15(55):416–426
Nord KH, Nilsson J, Arbajian E, Vult von Steyern F, Brosjo O, Cleton-Jansen AM, Szuhai K, Hogendoorn PCW (2013) Recurrent chromosome 22 deletions in osteoblastoma affect inhibitors of the Wnt/beta-catenin signaling pathway. PLoS One 8(11):e80725. https://doi.org/10.1371/journal.pone.0080725
Giannico G, Holt GE, Homlar KC, Johnson J, Pinnt J, Bridge JA (2009) Osteoblastoma characterized by a three-way translocation: report of a case and review of the literature. Cancer Genet Cytogenet 195(2):168–171. https://doi.org/10.1016/j.cancergencyto.2009.06.024
van IJzendoorn DGP, de Jong D, Romagosa C, Picci P, Benassi MS, Gambarotti M, Daugaard S, van de Sande M, Szuhai K, Bovée JVMG (2015) Fusion events lead to truncation of FOS in epithelioid hemangioma of bone. Genes Chromosom Cancer 54(9):565–574. https://doi.org/10.1002/gcc.22269
Huang SC, Zhang L, Sung YS, Chen CL, Krausz T, Dickson BC, Kao YC, Agaram NP, Fletcher CD, Antonescu CR (2015) Frequent FOS gene rearrangements in epithelioid hemangioma: a molecular study of 58 cases with morphologic reappraisal. Am J Surg Pathol 39(10):1313–1321. https://doi.org/10.1097/PAS.0000000000000469
van IJzendoorn DGP, Forghany Z, Liebelt F, Vertegaal AC, Jochemsen AG, Bovée JVMG, Szuhai K, Baker DA (2017) Functional analyses of a human vascular tumor FOS variant identify a novel degradation mechanism and a link to tumorigenesis. J Biol Chem 292(52):21282–21290. https://doi.org/10.1074/jbc.C117.815845
Walther C, Tayebwa J, Lilljebjorn H, Magnusson L, Nilsson J, von Steyern FV, Ora I, Domanski HA, Fioretos T, Nord KH, Fletcher CD, Mertens F (2014) A novel SERPINE1-FOSB fusion gene results in transcriptional up-regulation of FOSB in pseudomyogenic haemangioendothelioma. J Pathol 232(5):534–540. https://doi.org/10.1002/path.4322
Antonescu CR, Chen HW, Zhang L, Sung YS, Panicek D, Agaram NP, Dickson BC, Krausz T, Fletcher CD (2014) ZFP36-FOSB fusion defines a subset of epithelioid hemangioma with atypical features. Genes Chromosom Cancer 53(11):951–959. https://doi.org/10.1002/gcc.22206
Lam SW, Cleven AHG, Kroon HM, Briaire-de Bruijn IH, Szuhai K, Bovée JVMG (2019) Utility of FOS as diagnostic marker for osteoid osteoma and osteoblastoma. Virchows Archiv, in press
Franchi A, Calzolari A, Zampi G (1998) Immunohistochemical detection of c-fos and c-jun expression in osseous and cartilaginous tumours of the skeleton. Virchows Arch 432(6):515–519
Grigoriadis AE, Schellander K, Wang Z, Wagner EF (1993) Osteoblasts are target cells for transformation in c-fos transgenic mice. J Cell Biol 122(3):685–701
Mirabello L, Troisi RJ, Savage SA (2009) Osteosarcoma incidence and survival rates from 1973 to 2004: data from the surveillance, epidemiology, and end results program. Cancer 115(7):1531–1543. https://doi.org/10.1002/cncr.24121
Smeland S, Bielack SS, Whelan J, Bernstein M, Hogendoorn PCW, Krailo MD, Gorlick R, Janeway KA, Ingleby FC, Anninga J, Antal I, Arndt C, Brown KLB, Butterfass-Bahloul T, Calaminus G, Capra M, Dhooge C, Eriksson M, Flanagan AM, Friedel G, Gebhardt MC, Gelderblom H, Goldsby R, Grier HE, Grimer R, Hawkins DS, Hecker-Nolting S, Sundby Hall K, Isakoff MS, Jovic G, Kuhne T, Kager L, von Kalle T, Kabickova E, Lang S, Lau CC, Leavey PJ, Lessnick SL, Mascarenhas L, Mayer-Steinacker R, Meyers PA, Nagarajan R, Randall RL, Reichardt P, Renard M, Rechnitzer C, Schwartz CL, Strauss S, Teot L, Timmermann B, Sydes MR, Marina N (2019) Survival and prognosis with osteosarcoma: outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer 109:36–50. https://doi.org/10.1016/j.ejca.2018.11.027
Ottaviani G, Jaffe N (2009) The epidemiology of osteosarcoma. Cancer Treat Res 1552(3)
Inwards C, Squire J, Rosenberg AE, Cleton-Jansen AM, de Pinieux G, Deyrup AT, Hauben E, Oliveira AM, Okada K, Kalil RK, Lazar A, Mertens F, Montag AG, Wold LE, McCarthy EF, Osteogenic tumors (2013). In: Fletcher C, Bridge JA, Hogendoorn PCW, Mertens F (eds) WHO classification of tumours of soft tissue and bone, 4th. IARC, Lyon, pp 281–296
Widhe B, Widhe T (2000) Initial symptoms and clinal features in osteosarcoma and Ewing sarcoma. J Bone Joint Surg 82(5):667–674
Casali PG, Bielack S, Abecassis N, Aro HT, Bauer S, Biagini R, Bonvalot S, Boukovinas I, Bovée JVMG, Brennan B, Brodowicz T, Broto JM, Brugières L, Buonadonna A, De Álava E, Dei Tos AP, Del Muro XG, Dileo P, Dhooge C, Eriksson M, Fagioli F, Fedenko A, Ferraresi V, Ferrari A, Ferrari S, Frezza AM, Gaspar N, Gasperoni S, Gelderblom H, Gil T, Grignani G, Gronchi A, Haas RL, Hassan B, Hecker-Nolting S, Hohenberger P, Issels R, Joensuu H, Jones RL, Judson I, Jutte P, Kaal S, Kager L, Kasper B, Kopeckova K, Krákorová DA, Ladenstein R, Le Cesne A, Lugowska I, Merimsky O, Montemurro M, Morland B, Pantaleo MA, Piana R, Picci P, Piperno-Neumann S, Pousa AL, Reichardt P, Robinson MH, Rutkowski P, Safwat AA, Schöffski P, Sleijfer S, Stacchiotti S, Strauss SJ, Sundby Hall K, Unk M, Van Coevorden F, van der Graaf WTA, Whelan J, Wardelmann E, Zaikova O, Blay JY (2018) Bone sarcomas: ESMO–PaedCan–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol 29(Supplement_4):iv79–iv95. https://doi.org/10.1093/annonc/mdy310
Ruijs MWG, Broeks A, Menko FH, Ausems MGEM, Wagner A, Oldenburg R, Meijers-Heijboer H, van’t Veer LJ, Verhoef S (2009) The contribution of CHEK2 to the TP53-negative Li-Fraumeni phenotype. Heredit Cancer Clin Pract 7(1):4. https://doi.org/10.1186/1897-4287-7-4
Ruijs MWG, Verhoef S, Rookus MA, Pruntel R, van der Hout AH, Hogervorst FBL, Kluijt I, Sijmons RH, Aalfs CM, Wagner A, Ausems MGEM, Hoogerbrugge N, van Asperen CJ, Gomez Garcia EB, Meijers-Heijboer H, ten Kate LP, Menko FH, van’t Veer LJ (2010) TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet 47(6):421–428. https://doi.org/10.1136/jmg.2009.073429
Marees T, Moll AC, Imhof SM, de Boer MR, Ringens PJ, van Leeuwen FE (2008) Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst 100(24):1771–1779. https://doi.org/10.1093/jnci/djn394
Hameed M, Mandelker D (2018) Tumor syndromes predisposing to osteosarcoma. Adv Anat Pathol 25:217–222
Ji J, Quindipan C, Parham D, Shen L, Ruble D, Bootwalla M, Maglinte DT, Gai X, Saitta SC, Biegel JA, Mascarenhas L (2017) Inherited germline ATRX mutation in two brothers with ATR-X syndrome and osteosarcoma. Am J Med Genet A 173(5):1390–1395. https://doi.org/10.1002/ajmg.a.38184
Smolle MA, Heitzer E, Geigl JB, Al Kaissi A, Liegl-Atzwanger B, Seidel MG, Holzer LA, Leithner A (2017) A novel mutation in ATRX associated with intellectual disability, syndromic features, and osteosarcoma. Pediatr Blood Cancer 64(10). https://doi.org/10.1002/pbc.26522
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144(1):27–40. https://doi.org/10.1016/j.cell.2010.11.055
Behjati S, Tarpey PS, Haase K, Ye H, Young MD, Alexandrov LB, Farndon SJ, Collord G, Wedge DC, Martincorena I, Cooke SL, Davies H, Mifsud W, Lidgren M, Martin S, Latimer C, Maddison M, Butler AP, Teague JW, Pillay N, Shlien A, McDermott U, Futreal PA, Baumhoer D, Zaikova O, Bjerkehagen B, Myklebost O, Amary MF, Tirabosco R, Van Loo P, Stratton MR, Flanagan AM, Campbell PJ (2017) Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat Commun 8:15936. https://doi.org/10.1038/ncomms15936
Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, Cowley GS, Bhatt AS, Rheinbay E, Pedamallu CS, Helman E, Taylor-Weiner A, McKenna A, DeLuca DS, Lawrence MS, Ambrogio L, Sougnez C, Sivachenko A, Walensky LD, Wagle N, Mora J, de Torres C, Lavarino C, Dos Santos AS, Yunes JA, Brandalise SR, Mercado-Celis GE, Melendez-Zajgla J, Cardenas-Cardos R, Velasco-Hidalgo L, Roberts CW, Garraway LA, Rodriguez-Galindo C, Gabriel SB, Lander ES, Golub TR, Orkin SH, Getz G, Janeway KA (2014) Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A 111(51):E5564–E5573. https://doi.org/10.1073/pnas.1419260111
Kovac M, Blattmann C, Ribi S, Smida J, Mueller NS, Engert F, Castro-Giner F, Weischenfeldt J, Kovacova M, Krieg A, Andreou D, Tunn PU, Durr HR, Rechl H, Schaser KD, Melcher I, Burdach S, Kulozik A, Specht K, Heinimann K, Fulda S, Bielack S, Jundt G, Tomlinson I, Korbel JO, Nathrath M, Baumhoer D (2015) Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun 6:8940. https://doi.org/10.1038/ncomms9940
Sayles LC, Breese MR, Koehne AL, Leung SG, Lee AG, Liu HY, Spillinger A, Shah AT, Tanasa B, Straessler K, Hazard FK, Spunt SL, Marina N, Kim GE, Cho SJ, Avedian RS, Mohler DG, Kim MO, DuBois SG, Hawkins DS, Sweet-Cordero EA (2019) Genome-informed targeted therapy for osteosarcoma. Cancer Discov 9(1):46–63. https://doi.org/10.1158/2159-8290.CD-17-1152
Shao YW, Wood GA, Lu J, Tang QL, Liu J, Molyneux S, Chen Y, Fang H, Adissu H, McKee T, Waterhouse P, Khokha R (2019) Cross-species genomics identifies DLG2 as a tumor suppressor in osteosarcoma. Oncogene 38(2):291–298. https://doi.org/10.1038/s41388-018-0444-4
Mejia-Guerrero S, Quejada M, Gokgoz N, Gill M, Parkes RK, Wunder JS, Andrulis IL (2010) Characterization of the 12q15MDM2and 12q13-14CDK4amplicons and clinical correlations in osteosarcoma. Genes, Chromosomes Cancer. https://doi.org/10.1002/gcc.20761
Kuijjer ML, van den Akker BEWM, Hilhorst R, Mommersteeg M, Buddingh EP, Serra M, Burger H, Hogendoorn PCW, Cleton-Jansen AM (2014) Kinome and mRNA expression profiling of high-grade osteosarcoma cell lines implies Akt signaling as possible target for therapy. BMC Med Genet 7(4)
Baranski Z, Booij TH, Kuijjer ML, de Jong Y, Cleton-Jansen AM, Price LS, van de Water B, Bovée JVMG, Hogendoorn PCW, Danen EHJ (2015) MEK inhibition induces apoptosis in osteosarcoma cells with constitutive ERK1/2 phosphorylation. Genes Cancer 6(11):503–512
Radig K, Schneider-Stock R, Oda Y, Neumann W, Mittler U, Roessner A (1996) Mutation spectrum of p53 gene in highly malignant human osteosarcomas. Gen Diagn Pathol 142(1):25–32
Salk JJ, Schmitt MW, Loeb LA (2018) Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet 19(5):269–285. https://doi.org/10.1038/nrg.2017.117
Macintyre G, Ylstra B, Brenton JD (2016) Sequencing structural variants in cancer for precision therapeutics. Trends Genet 32(9):530–542. https://doi.org/10.1016/j.tig.2016.07.002
Weinberg R (1995) The retinoblastoma protein and cell cycle control. Cell 81:323–330
Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, Rodda SJ, Snay E, Dunning P, Fahey FH, Alt FW, McMahon AP, Orkin SH (2008) Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev 22(12):1662–1676. https://doi.org/10.1101/gad.1656808
Rubio R, Gutierrez-Aranda I, Saez-Castillo AI, Labarga A, Rosu-Myles M, Gonzalez-Garcia S, Toribio ML, Menendez P, Rodriguez R (2013) The differentiation stage of p53-Rb-deficient bone marrow mesenchymal stem cells imposes the phenotype of in vivo sarcoma development. Oncogene 32(41):4970–4980. https://doi.org/10.1038/onc.2012.507
Freedman DA, Levine AJ (1999) Functions of the MDM2 oncoprotein. Cell Mol Life Sci 55:96–107
Salinas-Souza C, De Andrea C, Bihl M, Kovac M, Pillay N, Forshew T, Gutteridge A, Ye H, Amary MF, Tirabosco R, Toledo SRC, Baumhoer D, Flanagan AM (2015) GNAS mutations are not detected in parosteal and low-grade central osteosarcomas. Mod Pathol 28(10):1336–1342. https://doi.org/10.1038/modpathol.2015.91
Wunder JS, Eppert K, Burrow SR, Gogkoz N, Bell RS, Andrulis IL (1999) Co-amplification and overexpression of CDK4, SAS and MDM2 occurs frequently in human parosteal osteosarcomas. Oncogene 18:783–788
Dujardin F, Binh MB, Bouvier C, Gomez-Brouchet A, Larousserie F, Muret A, Louis-Brennetot C, Aurias A, Coindre JM, Guillou L, Pedeutour F, Duval H, Collin C, de Pinieux G (2011) MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Mod Pathol 24(5):624–637. https://doi.org/10.1038/modpathol.2010.229
Mohseny AB, Szuhai K, Romeo S, Buddingh EP, Briaire-de Bruijn I, de Jong D, van Pel M, Cleton-Jansen AM, Hogendoorn PCW (2009) Osteosarcoma originates from mesenchymal stem cells in consequence of aneuploidization and genomic loss of Cdkn2. J Pathol 219(3):294–305. https://doi.org/10.1002/path.2603
Mohseny AB, Tieken C, van der Velden PA, Szuhai K, de Andrea C, Hogendoorn PCW, Cleton-Jansen AM (2010) Small deletions but not methylation underlie CDKN2A/p16 loss of expression in conventional osteosarcoma. Genes Chromosom Cancer 49(12):1095–1103. https://doi.org/10.1002/gcc.20817
Park Y-B, Park MJ, Kimura K, Shimizu K, Lee SH, Yokota J (2002) Alterations in the Ink4a/ARF locus and their effects on the growth of human osteosarcoma cell lines. Cancer Genet Cytogenet 133:105–111
Helman LJ, Meltzer P (2003) Mechanisms of sarcoma development. Nat Rev Cancer 3(9):685–694. https://doi.org/10.1038/nrc1168
De La Fuente R, Baumann C, Viveiros MM (2011) Role of ATRX in chromatin structure and function: implications for chromosome instability and human disease. Reproduction 142(2):221–234. https://doi.org/10.1530/REP-10-0380
Yost KE, Clatterbuck Soper SF, Walker RL, Pineda MA, Zhu YJ, Ester CD, Showman S, Roschke AV, Waterfall JJ, Meltzer PS (2019) Rapid and reversible suppression of ALT by DAXX in osteosarcoma cells. Sci Rep 9(1):4544. https://doi.org/10.1038/s41598-019-41058-8
Henson JD, Reddel RR (2010) Assaying and investigating alternative lengthening of telomeres activity in human cells and cancers. FEBS Lett 584(17):3800–3811. https://doi.org/10.1016/j.febslet.2010.06.009
Engert F, Kovac M, Baumhoer D (2017) Osteosarcoma cells with genetic signatures of BRCAness are susceptible to the PARP inhibitor talazoparib alone or in combination with chemotherapeutics. Oncotarget 8(30):48794–48806
Holme H, Gulati A, Brough R, Fleuren EDG, Bajrami I, Campbell J, Chong IY, Costa-Cabral S, Elliott R, Fenton T, Frankum J, Jones SE, Menon M, Miller R, Pemberton HN, Postel-Vinay S, Rafiq R, Selfe JL, von Kriegsheim A, Munoz AG, Rodriguez J, Shipley J, van der Graaf WTA, Williamson CT, Ryan CJ, Pettitt S, Ashworth A, Strauss SJ, Lord CJ (2018) Chemosensitivity profiling of osteosarcoma tumour cell lines identifies a model of BRCAness. Sci Rep 8(1). https://doi.org/10.1038/s41598-018-29043-z
Kuijjer M, Peterse EFP, Van den Akker BEWM, Briare-de Brujin IH, Serra M, Meza-Zepeda LA, Myklebost O, Bassim Hassan A, Hogendoorn PCW, Cleton-Jansen AM (2013) IRIGF1R signaling as potential target for treatment of high-grade osteosarcoma. BMC Cancer 13(245)
Conover CA (2008) Insulin-like growth factor-binding proteins and bone metabolism. Am J Physiol Endocrinol Metab 294(1):E10–E14. https://doi.org/10.1152/ajpendo.00648.2007
McCarthy TL, Centrella M (2001) Local IGF-I expression and bone formation. Growth Hormon IGF Res 11(4):213–219. https://doi.org/10.1054/ghir.2001.0236
Dohi O, Hatori M, Suzuki T, Ono K, Hosaka M, Akahira J, Miki Y, Nagasaki S, Itoi E, Sasano H (2008) Sex steroid receptors expression and hormone-induced cell proliferation in human osteosarcoma. Cancer Sci 99(3):518–523. https://doi.org/10.1111/j.1349-7006.2007.00673.x
Lillo Osuna MA, Garcia-Lopez J, El Ayachi I, Fatima I, Khalid AB, Kumpati J, Slayden AV, Seagroves TN, Miranda-Carboni GA, Krum SA (2019) Activation of estrogen receptor alpha by decitabine inhibits osteosarcoma growth and metastasis. Cancer Res 79(6):1054–1068. https://doi.org/10.1158/0008-5472.CAN-18-1255
Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, Pfaff E, Tica J, Wang Q, Massimi L, Witt H, Bender S, Pleier S, Cin H, Hawkins C, Beck C, von Deimling A, Hans V, Brors B, Eils R, Scheurlen W, Blake J, Benes V, Kulozik AE, Witt O, Martin D, Zhang C, Porat R, Merino DM, Wasserman J, Jabado N, Fontebasso A, Bullinger L, Rucker FG, Dohner K, Dohner H, Koster J, Molenaar JJ, Versteeg R, Kool M, Tabori U, Malkin D, Korshunov A, Taylor MD, Lichter P, Pfister SM, Korbel JO (2012) Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148(1-2):59–71. https://doi.org/10.1016/j.cell.2011.12.013
Hermsen R, Toonen P, Kuijk E, Youssef SA, Kuiper R, van Heesch S, de Bruin A, Cuppen E, Simonis M (2015) Lack of major genome instability in tumors of p53 null rats. PLoS One 10(3):e0122066. https://doi.org/10.1371/journal.pone.0122066
Shaw PH (1996) The role of p53 in cell cycle regulation. Pathol Res Pract 192(7):669–675. https://doi.org/10.1016/s0344-0338(96)80088-4
Muller PA, Vousden KH (2014) Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25(3):304–317. https://doi.org/10.1016/j.ccr.2014.01.021
Mehine M, Kaasinen E, Makinen N, Katainen R, Kampjarvi K, Pitkanen E, Heinonen HR, Butzow R, Kilpivaara O, Kuosmanen A, Ristolainen H, Gentile M, Sjoberg J, Vahteristo P, Aaltonen LA (2013) Characterization of uterine leiomyomas by whole-genome sequencing. N Engl J Med 369(1):43–53. https://doi.org/10.1056/NEJMoa1302736
Cohen A, Sato M, Aldape K, Mason CC, Alfaro-Munoz K, Heathcock L, South ST, Abegglen LM, Schiffman JD, Colman H (2015) DNA copy number analysis of grade II-III and grade IV gliomas reveals differences in molecular ontogeny including chromothripsis associated with IDH mutation status. Acta Neuropathol Commun 3:34. https://doi.org/10.1186/s40478-015-0213-3
Ly P, Cleveland DW (2017) Rebuilding chromosomes after catastrophe: emerging mechanisms of chromothripsis. Trends Cell Biol 27(12):917–930. https://doi.org/10.1016/j.tcb.2017.08.005
Koltsova AS, Pendina AA, Efimova OA, Chiryaeva OG, Kuznetzova TV, Baranov VS (2019) On the complexity of mechanisms and consequences of chromothripsis: An Update. Front Genet 10:393. https://doi.org/10.3389/fgene.2019.00393
Peterse EFP, van Leeuwen TN, Cleton-Jansen AM (2017) A researcher’s perspective on the quantity of osteosarcoma in vitro studies. J Bone Oncol 7:29–31. https://doi.org/10.1016/j.jbo.2017.04.004
Acknowledgements
The authors would like to thank Dr. K. Szuhai, Department of Cell and Chemical Biology, Leiden University Medical Centre, for providing Fig. 2b.
Funding
Our laboratory is financially supported by The Netherlands Organisation for Scientific Research Grant ZON-MW VICI 016.VICI.170.055 (to J. V. M. G. B.).
Author information
Authors and Affiliations
Contributions
All authors contributed to and approved the final manuscript.
Corresponding author
Ethics declarations
Not applicable.
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Franceschini, N., Lam, S.W., Cleton-Jansen, AM. et al. What’s new in bone forming tumours of the skeleton?. Virchows Arch 476, 147–157 (2020). https://doi.org/10.1007/s00428-019-02683-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-019-02683-w