
Abstract
Purpose
Niraparib is a highly selective inhibitor of PARP-1 and PARP-2 approved in the United States for maintenance treatment of adult patients with recurrent ovarian cancer in complete or partial response to platinum-based chemotherapy. In this open-label crossover study, we evaluated the effects of food on niraparib pharmacokinetics (PK) and safety.
Methods
Patients received a single 300-mg dose of niraparib either after a high-fat meal or under fasting conditions. After a 7-day PK assessment, all patients received a second 300-mg dose of niraparib under the opposite condition, followed by 7-day PK assessment. Blood samples for PK analyses were collected at baseline (on days 1 and 8) and up to 168 h post-dose. Bioequivalence between conditions was defined by the 90% confidence intervals (CIs) for area under the plasma concentration–time curve (AUC) from 0 to last measurable concentration (AUC0–last) and from 0 to infinity (AUC0–∞) being within the 80–125% range.
Results
The high-fat meal/fasting ratios of geometric least-squares means for AUC0–last and AUC0–∞ were 106.8 (90% CI 97.8–116.6) and 110.1 (90% CI 99.7–121.6), respectively, indicating bioequivalence between conditions. Mean half-life, maximum plasma concentration (Cmax), and time to Cmax after the high-fat meal were similar to, 27% smaller than, and 128% greater than after fasting, respectively. Adverse events were similar between conditions.
Conclusions
A high-fat meal did not impact the PK profile of niraparib, indicating that niraparib can be taken with or without food. Niraparib was safe and well-tolerated.
Similar content being viewed by others

Introduction
Ovarian cancer is the second-leading cause of gynecologic cancer deaths worldwide [1, 2]. Platinum-based chemotherapy, usually given in 4–6 cycles, is used in the setting of disease recurrence after a response lasting at least 6 months to a prior platinum-based chemotherapy, and is typically not intended to be used until progression of disease. Despite high response rates to second-line platinum-based treatment in patients with recurrent ovarian cancer, the effectiveness of platinum-based chemotherapy diminishes over time; progression-free survival (PFS), response rates, and platinum-free intervals generally decrease after each subsequent treatment with a platinum regimen [3].
Poly(ADP–ribose) polymerase (PARP)–1 and PARP–2 are proteins that play a crucial role in DNA repair by detecting DNA damage, binding DNA sites with single-strand breaks, and prompting catalytic polymerization [4, 5]. PARP inhibition in sensitive tumor cells produces PARP–DNA complexes, irreparable double-strand DNA breaks, and genomic instability that collectively result in cell death [6, 7]. Clinical studies have shown that PARP inhibitors are effective in patients with recurrent ovarian cancer, particularly those with breast cancer gene (BRCA) mutations and platinum-sensitive disease [8,9,10,11,12].
Niraparib is a highly selective inhibitor of PARP-1 and PARP-2 [13] approved in the United States for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy. In a phase 1 dose-escalation study in patients with advanced solid tumors, including those harboring BRCA1/2 mutations, niraparib was well tolerated and had antitumor activity in patients who had platinum-sensitive or platinum-resistant ovarian cancer with or without germline BRCA (gBRCA) mutations [14]. A recent, randomized, placebo-controlled phase 3 study was conducted in patients with recurrent ovarian cancer following complete or partial response to chemotherapy (ENGOT-OV16/NOVA; NCT01847274) [15]. This study demonstrated that PFS was significantly longer in patients receiving 300 mg niraparib once daily than in those receiving placebo in both the gBRCA cohort (21.0 months vs. 5.5 months; hazard ratio 0.27; 95% confidence interval [CI] 0.17–0.41; p < 0.001) and the overall non-gBRCA cohort 9.3 vs. 3.9 months; (hazard ratio 0.45; 95% CI 0.34–0.61; p < 0.001). In addition, the side-effect profile of niraparib was consistent with that observed in previous studies, and no new safety signals were identified.
Food may change the bioavailability of oral oncology drugs by altering absorption processes or interfering with metabolism (e.g., by inhibiting cytochrome P450 enzymes) [16, 17], ultimately affecting these drugs’ clinical efficacy and safety profile. Thus, oral oncology drugs are commonly indicated for administration to fasting patients.
We present here the findings of a 14-day, open-label, crossover ENGOT-OV16/NOVA substudy conducted to evaluate the effects of food in patients with ovarian cancer. Specifically, the goal was to assess the effect of a high-fat meal on the pharmacokinetics (PK) of a single 300-mg dose of niraparib. The safety of niraparib was also evaluated in the same subset of patients. Efficacy was not evaluated in this substudy.
Materials and methods
Subjects
Patients of at least 18 years old were deemed eligible for this food effect substudy if they had a histological diagnosis of ovarian cancer, fallopian tube cancer, or primary peritoneal cancer, regardless of platinum sensitivity and burden of disease, and if no standard therapy existed or the patient had refused standard therapy. To be eligible, patients also had to have an Eastern Cooperative Oncology Group (ECOG) performance status between 0 and 2, be able to eat a high-fat meal, and be able to fast for 12 h.
Patients were excluded if they had received chemotherapy within 3 weeks of study start or palliative radiotherapy within 1 week of enrollment (encompassing > 20% of the bone marrow); had symptomatic, uncontrolled brain or leptomeningeal metastases; had persistent grade > 2 toxicity from prior cancer therapy or a known hypersensitivity to the components of niraparib; were pregnant or breastfeeding or expecting to conceive children within the projected duration of the study treatment; were immunocompromised (with the exception of patients with splenectomy, who were allowed in the study); had a baseline QT prolongation > 470 milliseconds; or had taken a proton pump inhibitor, antacids, or an H2 blocker within 48 h of dosing.
Food effect substudy design
Patients were randomized 72 h prior to a 300-mg dose of niraparib in a crossover design (Fig. 1) to either sequence AB or sequence BA. In sequence AB, patients fasted (had nothing to eat or drink, except water) for at least 10 h before receiving a single dose of 300 mg niraparib, and continued to fast for at least 2 h after the dose. In sequence BA, patients fasted for at least 10 h before consuming a high-fat meal; within 5 min of finishing the meal, a single dose of 300 mg niraparib was administered orally, and then patients resumed fasting for at least 4 h. The high-fat meal (with fat representing approximately 50% of total caloric content), which was also high in calories (approximately 800–1000 calories), is recommended for food effect bioavailability and fed bioequivalence studies [18].

Design of food effect substudy. PK pharmacokinetic
After a 7-day PK assessment and washout period, all patients received a second 300-mg dose of niraparib under the opposite condition, followed by a second 7-day PK assessment (Fig. 1). Patients in sequence AB received niraparib after a high-fat meal, and patients in sequence BA received niraparib under fasting conditions. Approximately 2 weeks after completion of this substudy, patients continued with open-label niraparib at 300 mg once daily until disease progression, unacceptable toxicity, death, withdrawal of consent, or loss to follow-up, whichever occurred first. (This is referred to as the once-daily dosing phase.)
Assessments
Patients were included in the PK population if they received niraparib in each condition; a sufficient number of blood samples were collected from each patient in each condition. Blood samples for PK analyses were collected at baseline (on days 1 and 8 within 30 min prior to dosing) and 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, 72, 96, 120, and 168 h post dose.
To assess differences in PK parameters between the fasting and the high-fat condition, 90% CIs for the ratios of geometric means for maximum observed concentration (Cmax), area under the plasma concentration–time curve (AUC) from 0 to last measurable concentration (AUC0–last), and AUC from 0 to infinity (AUC0–∞) were calculated.
Safety
The safety population consisted of all patients who received at least one dose of study treatment. In the 14-day substudy, safety data were assessed on days 1 and 8 when patients received niraparib (crossover phase); after completion of the substudy, safety data were assessed once daily while patients received niraparib. Safety evaluations included assessment of treatment-emergent adverse events (TEAEs), changes in clinical laboratory parameters (hematology and chemistry), vital signs, electrocardiogram (ECG) parameters, physical examinations, and use of concomitant medications. Adverse events were graded according to Common Terminology Criteria for Adverse Events v.4.02 and were monitored throughout the study. Triplicate ECGs were performed on day 1 at baseline (predose) and at 1, 1.5, 2, 3, 4, 6, and 8 h post dose to coincide with PK assessments. ECGs were performed 2–5 min apart and prior to blood sampling for PK analysis.
Statistical analysis
An analysis of variance within WinNonlin™ Phoenix (Version 6.3) was used to analyze maximum observed concentration (Cmax), AUC0–last, and AUC0–∞. The model included terms for treatment (high-fat meal or fasted), treatment sequence (high-fat meal then fasted or fasted then high-fat meal), and period (day 1 or day 8). The two treatment groups were deemed bioequivalent if the CIs for AUC0–last and AUC0–∞ were within the 80–125% range.
This study was approved by the appropriate institutional or national research ethics committee or both, and was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Results
Subjects
A total of 17 patients were enrolled in the food effect substudy (Table 1). Patient demographics and baseline characteristics were generally well-balanced between groups. Across the two groups, patients were between 47 and 69 years of age, most (88.2%) were white, and all had an ECOG performance status of 0 or 1 at study entry. The median age overall was 64 years, which is consistent with ovarian cancer primarily being reported in women over 50 years of age. Patient demographics in this study were consistent with those reported in the ENGOT-OV16/NOVA study [15]. All patients had ECOG performance status values of 0 (70.6%) or 1 (29.4%), indicating that all were fully active or ambulatory and capable of self-care at the time of screening.
Pharmacokinetics
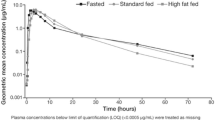
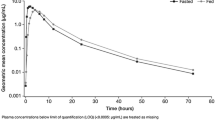
Of the 17 patients enrolled in the food effect substudy, the PK population comprised 16 patients for analysis during the fasting condition and 15 patients for analysis during the high-fat meal condition (Table 2). One patient vomited after the first dose and was excluded from both the fasting and high-fat conditions; a second patient who received a high-fat meal had only two PK concentrations in this condition and was, therefore, excluded from the PK analysis. The mean plasma concentration–time profiles of niraparib in the linear and semilogarithmic scales are shown in Fig. 2a, b, respectively. The high-fat meal and fasting conditions were associated with comparable niraparib plasma concentrations; higher maximum concentrations were apparent following the fasting condition. Predose concentrations were measurable in all patients that received niraparib treatment on day 8 and were included in the analysis. These measurable concentrations ranged from 8.26 to 95.7 ng/mL and were consistent with what was expected, based on the half-life (t½) of niraparib. The mean and standard deviation (SD) of the mean for niraparib PK parameters are presented in Table 2. Both treatment conditions had similar t½, AUC0–last, and AUC0–∞. For example, the t½ for niraparib was 47.9 ± 17.5 h for the high-fat meal condition and 50.5 ± 17.9 h for the fasting condition. The mean (SD) Cmax for the high-fat condition was 27% smaller than for the fasting condition [582.1 (228.6) vs. 803.7 (403.3) ng/mL]. The mean (SD) time to Cmax (Tmax) for the high-fat condition was 128% greater than for the fasting condition [8.0 (4.9) vs. 3.5 (1.2) h]. Statistical evaluation of the PK parameters AUC0–last, AUC0–∞, and Cmax is presented in Table 3. The high-fat meal/fasting ratios of geometric least-squares (LS) means for AUC0–last and AUC0–∞ were 106.8 and 110.1, respectively. The corresponding 90% CIs were within the 80–125% bioequivalence statistical limits. The geometric LS mean ratio for Cmax was 78.5, with the lower limit of the 90% CI (69.5) below the bioequivalence acceptance range.

Niraparib mean plasma concentration–time plots on a linear and b semilogarithmic scales
Safety
The safety population included 17 patients. In the crossover phase, grade ≥ 3 TEAEs included anemia (deemed treatment-related and occurring in one patient) and urinary tract infection (occurring in one patient) in the fasted condition (Table 4). Five of 17 patients (29.4%) experienced at least one treatment-related TEAE. A treatment-emergent serious adverse event (urinary tract infection) occurred in one patient in the fasted condition. Hypokalemia was the only TEAE reported in ≥ 10% of patients. During the once-daily niraparib dosing phase (i.e., after the substudy), the grade ≥ 3 TEAE profile (Table 5) was consistent with the safety profile seen in other niraparib studies [15] and other PARP inhibitors [19,20,21]. There were no reports of clinically significant abnormal ECG changes, including QTc interval prolongation, attributed to niraparib.
Discussion
The antitumor activity of niraparib and the favorable safety profile of niraparib were initially demonstrated in a phase 1 dose escalating study in patients with advanced solid tumors, including those with gBRCA1/2 mutations [14]. More recently, the randomized, double-blind, placebo-controlled phase 3 ENGOT-OV16/NOVA trial [15], conducted in patients with recurrent ovarian cancer who had a complete or partial response to platinum-based chemotherapy, showed that niraparib significantly improved progression-free survival compared with placebo. The results of this ENGOT-OV16/NOVA substudy demonstrate that food did not impact the PK profile of niraparib compared to fasting in recurrent ovarian cancer patients. The t½ of niraparib was similar in the two conditions. Food increased the Tmax approximately by 128% compared with fasting, indicating a delay in the rate of absorption. Food also decreased the Cmax by approximately 27% compared with fasting. The high-fat meal/fasting condition ratios for AUC0–last and AUC0–∞ were 106.8 and 110.1, respectively, and the corresponding 90% CIs were within the bioequivalence acceptance range, indicating that the extent of absorption did not change. The potential for the modulation of absorption by food is typically limited with highly (e.g., > 50%) bioavailable drugs such as niraparib. In a similarly-sized phase 1, open-label, randomized study conducted in 32 patients with refractory/resistant advanced solid tumors [20], a high-fat or standard meal delayed the rate of absorption (Tmax) of olaparib by approximately 2 h and increased the extent of absorption (AUC) by approximately 20%.
Oral oncology drugs are typically administered on an empty stomach, as studies have demonstrated that food may increase the bioavailability of drugs by 4- to 10-fold [16, 17] and thus increase their toxicity. The results of the current substudy suggest that patients should be made aware that they can take niraparib with or without food, since eating does not change the expected efficacy and safety profile of this PARP inhibitor.
The safety results were in line with those of the overall ENGOT-OV16/NOVA study [15] and with the known safety profile of other PARP inhibitors [20,21,22], and no new safety signals were observed. The most frequently reported treatment-related TEAEs post substudy (in the once-daily dosing phase) were decreased appetite, anemia, and thrombocytopenia.
In this substudy, PK parameters for oral niraparib were similar following a 10-hour fast or high-fat meal, and the safety profile was similar to data shown previously [14]. In conclusion, the lack of food effect on PK parameters indicates that niraparib can be taken with or without food according to patient preference or individual tolerance.
References
Sankaranarayanan R, Ferlay J (2010) Worldwide burden of gynecological cancer. In: Preedy VR, Watson RR (eds) Handbook of disease burdens and quality of life measures. Springer, New York, pp 803–823
Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64(1):9–29. https://doi.org/10.3322/caac.21208
Hanker LC, Loibl S, Burchardi N, Pfisterer J, Meier W, Pujade-Lauraine E, Ray-Coquard I, Sehouli J, Harter P, du Bois A, Ago, group Gs (2012) The impact of second to sixth line therapy on survival of relapsed ovarian cancer after primary taxane/platinum-based therapy. Ann Oncol 23(10):2605–2612. https://doi.org/10.1093/annonc/mds203
De Lorenzo SB, Patel AG, Hurley RM, Kaufmann SH (2013) The elephant and the blind men: making sense of PARP inhibitors in homologous recombination deficient tumor cells. Front Oncol 3:228. https://doi.org/10.3389/fonc.2013.00228
Jones P, Wilcoxen K, Rowley M, Toniatti C (2015) Niraparib: a poly(ADP-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. J Med Chem 58(8):3302–3314. https://doi.org/10.1021/jm5018237
Kaelin WGJ (2005) The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 5(9):689–698. https://doi.org/10.1038/nrc1691
Underhill C, Toulmonde M, Bonnefoi H (2011) A review of PARP inhibitors: from bench to bedside. Ann Oncol 22(2):268–279. https://doi.org/10.1093/annonc/mdq322
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, Wickens M, Tutt A (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376(9737):245–251. https://doi.org/10.1016/S0140-6736(10)60893-8
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361(2):123–134. https://doi.org/10.1056/NEJMoa0900212
Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, Gandara DR, Allen D, Kiesel B, Beumer JH, Newman EM, Rubinstein L, Chen A, Zhang Y, Wang L, Kinders RJ, Parchment RE, Tomaszewski JE, Doroshow JH (2012) A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res 18(6):1726–1734. https://doi.org/10.1158/1078-0432.CCR-11-2821
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C, Carmichael J, Matulonis U (2012) Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 366(15):1382–1392. https://doi.org/10.1056/NEJMoa1105535
Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, Oza A (2011) Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 12(9):852–861. https://doi.org/10.1016/S1470-2045(11)70214-5
Jones P, Altamura S, Boueres J, Ferrigno F, Fonsi M, Giomini C, Lamartina S, Monteagudo E, Ontoria JM, Orsale MV, Palumbi MC, Pesci S, Roscilli G, Scarpelli R, Schultz-Fademrecht C, Toniatti C, Rowley M (2009) Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J Med Chem 52(22):7170–7185. https://doi.org/10.1021/jm901188v
Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, Kreischer N, Thway K, Gevensleben H, Sun L, Loughney J, Chatterjee M, Toniatti C, Carpenter CL, Iannone R, Kaye SB, de Bono JS, Wenham RM (2013) The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol 14(9):882–892. https://doi.org/10.1016/S1470-2045(13)70240-7
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, Ben-Baruch NE, Marth C, Madry R, Christensen RD, Berek JS, Dorum A, Tinker AV, du Bois A, Gonzalez-Martin A, Follana P, Benigno B, Rosenberg P, Gilbert L, Rimel BJ, Buscema J, Balser JP, Agarwal S, Matulonis UA, Investigators E-ON (2016) Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. https://doi.org/10.1056/NEJMoa1611310
Ratain MJ (2011) Flushing oral oncology drugs down the toilet. J Clin Oncol 29(30):3958–3959. https://doi.org/10.1200/JCO.2011.37.1617
Kaitin KI, DiMasi JA (2011) Pharmaceutical innovation in the 21st century: new drug approvals in the first decade, 2000–2009. Clin Pharmacol Ther 89(2):183–188. https://doi.org/10.1038/clpt.2010.286
US Food and Drug Administration, Center for Drug Evaluation and Research (2002) Guidance for industry: food-effect bioavailability and fed bioequivalence studies. http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126833.pdf
Matulonis UA, Penson RT, Domchek SM, Kaufman B, Shapira-Frommer R, Audeh MW, Kaye S, Molife LR, Gelmon KA, Robertson JD, Mann H, Ho TW, Coleman RL (2016) Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Ann Oncol 27(6):1013–1019. https://doi.org/10.1093/annonc/mdw133
Rolfo C, Swaisland H, Leunen K, Rutten A, Soetekouw P, Slater S, Verheul HM, Fielding A, So K, Bannister W, Dean E (2015) Effect of food on the pharmacokinetics of olaparib after oral dosing of the capsule formulation in patients with advanced solid tumors. Adv Ther 32(6):510–522. https://doi.org/10.1007/s12325-015-0214-4
van der Noll R, Marchetti S, Steeghs N, Beijnen JH, Mergui-Roelvink MW, Harms E, Rehorst H, Sonke GS, Schellens JH (2015) Long-term safety and anti-tumour activity of olaparib monotherapy after combination with carboplatin and paclitaxel in patients with advanced breast, ovarian or fallopian tube cancer. Br J Cancer 113(3):396–402. https://doi.org/10.1038/bjc.2015.256
Matulonis UA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Spencer S, Parry D, Grinsted L, Ledermann JA (2016) Olaparib maintenance therapy in patients with platinum-sensitive, relapsed serous ovarian cancer and a BRCA mutation: overall survival adjusted for postprogression poly(adenosine diphosphate ribose) polymerase inhibitor therapy. Cancer 122(12):1844–1852. https://doi.org/10.1002/cncr.29995
Acknowledgements
This study was funded by Tesaro, Inc. Writing and editorial support was funded by Tesaro, Inc. Ruggero Galici, PhD (Ashfield Healthcare Communications, Middletown, CT, USA), drafted and revised the manuscript based on a content outline and comments provided by the authors. Joshua Safran (Ashfield Healthcare Communications) copyedited and styled the manuscript per journal requirements.
Funding
This study was funded by Tesaro, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Kathleen Moore has served on advisory boards for Advaxis, AstraZeneca, Clovis Oncology, Genentech, Inc., ImmunoGen, Tesaro, Inc., and VBL Therapeutics. Zhi-Yi Zhang, Shefali Agarwal, and Vikram Kansra are employees and stockholders of Tesaro, Inc. Howard Burris has nothing to disclose. Mahish R. Patel has served on speakers’ bureaus for Celgene, Exelixis, Genentech, Inc., and Taiho.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional or national research committee or both and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Moore, K., Zhang, ZY., Agarwal, S. et al. The effect of food on the pharmacokinetics of niraparib, a poly(ADP-ribose) polymerase (PARP) inhibitor, in patients with recurrent ovarian cancer. Cancer Chemother Pharmacol 81, 497–503 (2018). https://doi.org/10.1007/s00280-017-3512-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-017-3512-5