
Abstract
Objective
This study investigated the efficacy, safety, tolerability, and pharmacokinetics of a novel cholesterol absorption inhibitor, FM-VP4, comprising disodium ascorbyl sitostanol phosphate (DASP) and disodium ascorbyl campestanol phosphate (DACP).
Methods
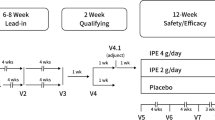
In phase 1, 30 men received a single dose of 100, 200, 400, 800, 1,600, or 2,000 mg FM-VP4 or placebo. In phase 2, 100 men were treated with 100, 200, 400, or 800 mg/day of FM-VP4 or placebo for 4 weeks.
Results
The drug was well tolerated at each single or multiple dose level. After 4 weeks of treatment, low-density lipoprotein cholesterol (LDL-C) levels changed by 2.7% in the placebo group and by 2.9%, −4.2%, and −4.6% in the 100, 200, and 800 mg/day groups, respectively, which was not statistically significant. However, 400 mg/day of FM-VP4 significantly decreased LDL-C by 6.5% (p=0.02). Phase 1 showed that DACP and DASP were absorbed into plasma with a median tmax of 12 h for both components, and clearance was slow with a mean t1/2λ of 57 h. During 4 weeks of treatment, steady state was reached by approximately 8 days.
Conclusion
This study demonstrated that up to 800 mg/day of FM-VP4 is safe and well tolerated for at least 4 weeks. Furthermore, the higher doses significantly reduced LDL-C by 7% compared with baseline or by 10% compared with placebo, with the maximum effect reached at 400 mg/day.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Worldwide, cardiovascular disease (CVD) is the most common cause of death, with atherosclerotic vascular disease as the underlying cause. It has been well established that elevated plasma concentrations of low-density lipoprotein cholesterol (LDL-C) lead to an increased risk of atherosclerosis and coronary heart disease [1]. Dietary modification can improve the lipid profile and the potential risk of CVD, but for a significant portion of the population, treatment with lipid-lowering agents is necessary to reduce blood cholesterol effectively. Statins are the primary class of drugs used for managing LDL-C and are the most potent agents in lowering LDL-C. In fact, the lowering of LDL-C by statin therapy has been shown to decrease the number of fatal and nonfatal myocardial events in both primary and secondary prevention studies [2–6]. However, statins have not provided the final answer in terms of CVD prevention, and their use is associated with side effects, albeit rare, at higher doses. Therefore, cholesterol-lowering agents that act via other mechanisms are of great interest.
Plant sterols, or phytosterols, are naturally occurring compounds in vegetable oil, seeds, and nuts that are structurally related to cholesterol. Daily intake of 2,000 mg of plant sterols or their saturated counterparts, plant stanols, or phytostanols, has been shown to decrease LDL-C by 9–14% without affecting high-density lipoprotein cholesterol (HDL-C) concentrations [7, 8]. Plant sterols and stanols are thought to decrease plasma cholesterol concentrations by inhibiting cholesterol absorption in the intestine [9]. When cholesterol absorption is decreased, the hepatic cholesterol pool is reduced, resulting in enhanced cholesterol synthesis by the liver. At the same time, LDL receptors are upregulated, with ensuing lower LDL-C concentrations in plasma [10].
A new water soluble plant stanol derivative, disodium ascorbyl phytostanol phosphate (FM-VP4, Fig. 1) has been developed as a cholesterol-absorption inhibitor. FM-VP4 consists of a mixture of sitostanol and campestanol to which ascorbate is covalently bound via a phosphodiester linkage. It has been shown to inhibit the in vitro uptake of micellar [3H]-cholesterol by approximately 50% in human and rat enterocyte cell lines [11, 12]. In vivo, FM-VP4 led to a dose-related inhibition of [3H]-cholesterol absorption in rats, as shown by a maximally 80% reduced area under the concentration time curve (AUC) of orally administered micellar [3H]-cholesterol [13]. Furthermore, the LDL-lowering activity of FM-VP4 was observed in a broad range of LDL-sensitive species, including gerbils [14, 15], hamsters [16], and apolipoprotein E (ApoE)-deficient transgenic mice [17]. In hamsters, FM-VP4 was a more potent LDL-lowering agent than unesterified plant stanols [16], and in ApoE deficient mice, the LDL-lowering effect of FM-VP4 was correlated with retardation of atherosclerotic plaque development [17]. Other effects of FM-VP4 included the decrease of plasma triglyceride levels and the reduction of abdominal fat or body-weight gain in gerbils [14, 15] and mice [18]. In all of these cases, FM-VP4 showed no side effects to the animals tested.

Chemical structure of FM-VP4, composed of disodium ascorbyl campestanol phosphate (R=CH3) and disodium ascorbyl sitostanol phosphate (R=C2H5)
Preclinical data suggested that FM-VP4 is a potent cholesterol-lowering agent with no significant toxic effects. Here, we report the first human study designed to assess the efficacy, safety, tolerability, and pharmacokinetics of single and multiple doses of this water-soluble plant stanol analogue.
Subjects and methods
Subjects
Subjects were recruited via advertisements in local newspapers. The study protocol was carefully explained before subjects were asked to give their written informed consent. The study protocol was approved by the Institutional Review Board of the Academic Medical Centre. Subjects were eligible if they were male, 18–75 years old, healthy as reviewed by medical history and physical examination, had LDL-C concentrations ≥3.0 mmol/L during one of the screening visits or at baseline and triglyceride (TG) concentration ≤4.5 mmol/L at the first visit, had a body mass index (BMI) <35 kg/m2, and did not use any steroids, β-blockers, corticosteroids, thiazide diuretics, or antiepileptics. Subjects with a history of hypertension, arterial diseases, diabetes mellitus type I or II, hypothyroidism, obstructive biliary disorders, pancreatitis, collagen disorders, or autoimmune disease were excluded, as were subjects with history of malignancy during the previous 3 years, significant hepatic, renal, cardiac, or cerebral disease, or plasma levels of hepatic transaminases higher than two times the upper limit of normal (ULN). Subjects of phase 1 were not allowed to use any lipid-lowering drugs at inclusion, and the use of plant sterol- or stanol-containing products had to be discontinued at the inclusion visit. Subjects of phase 2 had to discontinue the use of plant sterol or stanol products or fish oils at the inclusion visit and statin treatment 6 weeks (40 days) before the start of study treatment. Thirty men participated in the phase 1 trial and 100 men in phase 2.
Drugs
Two types of tablets were used: 100 mg oval FM-VP4 tablets or a matching placebo. Both investigational products were supplied through Forbes Medi-Tech Inc. (Vancouver, Canada). FM-VP4 is a semisynthetic esterified plant stanol derivative produced as a disodium salt. The two major components of FM-VP4 are disodium ascorbyl sitostanol phosphate (DASP) and disodium ascorbyl campestanol phosphate (DACP) (Fig. 1), which are present in the proportion of approximately 2:1, respectively.
Design
This single-center, double-blind, placebo-controlled, dose-escalation trial comprised two parts: In phase 1, 30 men received a single dose of FM-VP4. Five subjects were assigned to each dose group (100, 200, 400, 800, 1,600, or 2,000 mg), and within each group, one subject was randomly assigned to placebo. Once a complete cohort of five subjects was treated and the safety parameters had been reviewed, the following dose was initiated. In the subsequent phase 2 trial, 100 men were treated for 28 days (4 weeks). Twenty-five subjects were assigned to each dose group (100, 200, 400, or 800 mg/day), and within each group, five subjects were randomly assigned to placebo. The first five subjects in each cohort were hospitalized for 5 days. Escalation to the next dosing level was only allowed once these five subjects completed treatment and all results and safety data were evaluated.
Phase 1
A week before treatment, subjects visited the hospital for screening. Informed consent was obtained, and subjects underwent a physical examination. A fasting blood sample was taken to measure lipids, biochemistry, and hematology. Within 3 days before treatment, subjects visited the hospital for another blood sample, and urinalysis and electrocardiogram (EKG) were performed. If subjects met all inclusion criteria, they were hospitalized for 24 h on the day of treatment. In the morning, subjects’ weight, supine blood pressure (BP), and heart rate were measured, and a predose fasting blood sample was taken for baseline safety parameters and pharmacokinetics. Subsequently, subjects were administered one to 20 tablets containing 100 mg FM-VP4 each or placebo. Tablets were swallowed with 250 mL of water. Breakfast followed 30 min later. Weight, supine BP, and heart rate were recorded at 3, 6, 9, and 12 h after dosing. A blood sample for pharmacokinetics was taken 6 and 12 h after dosing. Any spontaneous complaints were recorded as adverse events and closely monitored. Subjects were detained overnight under observation, and 24 h after dosing, another blood sample was taken for safety parameters and pharmacokinetics. Subjects’ weight, supine BP, and heart rate were measured, and urinalysis and an EKG were performed. Once the EKG, biochemistry, and hematology of the 24-h postdosing sample had been reviewed, subjects were discharged from the hospital. They returned to hospital 48, 96, and 144 or 168 h (6 or 7 days) after treatment for weight, supine BP, and heart rate measurements and for a blood sample to measure safety parameters and pharmacokinetics. At the last visit, a final physical examination was performed.
Phase 2
Four to 8 weeks before treatment, subjects visited the hospital for screening. Informed consent was obtained, and subjects underwent a physical examination. A fasting blood sample was taken to measure lipids, biochemistry, and hematology. Subjects were instructed to follow a diet adapted from the National Cholesterol Education Program (NCEP) Step 1 diet during the entire study, including the 4- to 8-week run-in period. Consumption of plant sterol- or stanol-containing products and the use of fish oils had to be discontinued at the screening visit, and statin treatment had to be discontinued 6 weeks before treatment with FM-VP4. If they met all inclusion criteria, subjects visited the hospital halfway through the run-in period and within 3 days before study treatment for baseline blood lipids, biochemistry, and hematology measurements. At the latter visit, urinalysis was performed and an EKG recorded.
The first five subjects per dose cohort, of which four were on active treatment and one on placebo, were hospitalized for 5 days. Each morning, subjects’ weight, supine BP, and heart rate were measured, and a predose fasting blood sample was taken for safety parameters and pharmacokinetics. Subsequently, subjects were administered one to four tablets containing 100 mg FM-VP4 each, depending on the dose cohort, or placebo. Tablets were swallowed entirely with up to 100 mL of water. Breakfast followed 30 min later. All doses were divided and given twice per day; another one to four tablets were administered 30 min before dinner. The morning and evening doses were packaged in separate bottles. Subjects in the 100-mg group received one FM-VP4 tablet and one placebo tablet. Supine BP and heart rate were also recorded daily after lunch and dinner. Any spontaneous complaints were recorded as adverse events and closely monitored. Subjects were observed overnight and discharged 5 days after the first dosing. All subjects visited the hospital for efficacy and safety measurements once per week during and 14 days after treatment. At the last visit, a final physical examination and an EKG were performed. Compliance was calculated based on the number of tablets supplied to the patient minus the number returned.
Plasma analyses
Complete blood count, fibrinogen, and the biochemical profile [alanine aminotransferase (ALT) and aspartate aminotransferase (AST), bilirubin, creatine kinase (CK), creatinine, glucose, and C-reactive protein (CRP)] were assessed using the local hospital laboratory. Changes in laboratory parameters were considered abnormal in case of ALT or AST levels greater than three times ULN, a CK level greater than five times ULN, a creatinine increase of ≥40 μmol/L compared with baseline, a creatinine level >177 μmol/L, white blood cell count <3 × 109/L, and a decrease in hemoglobin of at least 1.5 g/dL compared with baseline. Thyroid-stimulating hormone (TSH) was measured at the first screening visit. Urinalysis for blood, glucose, protein, pH, and specific gravity was performed by dipstick within 3 days before and 1 day after dosing in phase 1 and within 3 days before, after 4 weeks of treatment, and at the last visit of phase 2. Urinalysis by wet microscopic slide was performed if the dipstick analysis was abnormal. Lipid analyses were performed by an external laboratory (MRL International, Zaventem, Belgium) by using standardized procedures, and LDL-C was calculated using the Friedewald equation [19]. The results were kept blinded to the investigators.
Plasma was stored at −80°C for further analyses. ApoB, ApoAI, and lipoprotein a [Lp(a)] as well as vitamin E and vitamin A were analyzed in one run after the study had been completed. ApoAI, ApoB, and Lp(a) levels were determined by nephelometry with a Beckman Array (Mijdrecht, the Netherlands) according to the manufacturer’s instructions, and vitamin E and vitamin A were measured by high-performance liquid chromatography (HPLC) with fluorescence detection using a Chromsep Glass, 100*3 mm, inertsil 5, ODS-3 column (Varian-Chrompack, Middelburg, the Netherlands).
All DASP and DACP plasma concentrations were assayed by a validated liquid chromatography/mass spectrometry/mass spectrometry method (LC/MS/MS) based on a previously described method [20]. The lower limit of quantification (LLOQ) was 30.8 ng/mL. Data analysis was carried out using Microsoft Excel 97, and pharmacokinetic data was analyzed using Pharsight WinNonlin software, version 3.1 build 168 (Pharsight WinNonlin, Mountain View, CA, USA) based on noncompartmental kinetics analysis. For phase 1, concentrations of DASP and DACP in plasma were measured at baseline and 6, 12, 24, 48, 96, and 144 or 168 h after treatment. DASP and DACP concentrations were plotted against time, and the half-lives of the compounds were estimated by the method of residuals. The area under the DACP and DASP concentration time curve (AUC 0-t) was estimated by the trapezoidal rule [21]. In phase 2, trough concentrations of DASP and DACP were measured at baseline, after 8 and 28 days of treatment, and 14 days after treatment (day 42). In subjects who were hospitalized for the first 5 days of treatment, trough DASP and DACP concentrations were also measured each morning of hospitalization (day 1 through day 5).
Plant sterols and stanols β-sitosterol, campesterol, sitostanol, and campestanol were assessed by selective ion monitoring gas chromatography mass spectrometry (SIM-GC-MS). High-purity solvents were purchased from Merck, Germany. Bis-(Trimethylsilyl)trifluoroacetamide (BSTFA) was obtained from Sigma (Steinneim, Germany) and pyridine from Pierce (Rockford, IL, USA). Beta-sitosterol (24β-ethylcholesterol), β-sitostanol (24α-ethyl-5α-cholestan-3β-ol), and campesterol (24α-methyl-5-cholesten-3β-ol) were purchased from Sigma, and stigmasterol from Lacoclau AB, Sweden. For sterol extraction, 500 μl of plasma was mixed with 100 μl 0.01 mg/ml stigmasterol and saponified for 60 min at 60°C in 1 ml of 4% (w/v) KOH in 90% ethanol. After saponification, the samples were mixed with 1 ml of water and extracted two times with 2 ml of hexane. The pooled hexane extracts were dried under nitrogen and derivatized with 50 μl BSTFA/pyridine (v/v 5:1) at 60°C for 60 min. For SIM-GC-MS, 2 μl of derivative mixture were delivered by automatic injection to an HP-5890 gas chromatograph split-injection port (1:20) leading to a 0.2 mm × 25 m Chrompack CP-sil 19 CB (WCOT Fused Silica) capillary column. The injection port contained a glass wool liner. The carrier gas was helium at a linear rate of 1 ml/min. The oven temperature started at 120°C and was raised to 260°C at 20°C/min, then to 280°C at 2°C/min, and finally to 300°C at 40°C/min and held for 5 min. An HP-5989B mass spectrometer was used as detector. Measurements were done in the electron impact mode at 70 eV with an ion source temperature of 250°C. The quadropole temperature was 150°C. Mass spectrometric data were collected in the elected ion mode at m/z=396 and 357 for β-sitosterol, m/z=488 and 373 for β-sitostanol, m/z=382 and 343 for campesterol, m/z=369 and 384 for campestanol, and m/z=255 and 394 for stigmasterol. Calibration curves were constructed by mixing 100 μl of 0.01 mg/ml stigmasterol with a series of 0- to 500-μl samples of a standard solution containing 10 μmol β-sitosterol, 2 μmol/l sitostanol, and 20 μmol/l campesterol.
Statistical analyses
The primary efficacy variable was the percent change in LDL after 4 weeks of treatment compared with baseline levels in phase 2. Differences in percentage LDL-C changes between the five treatment groups were calculated using analyses of variance (ANOVA) in SAS. P < 0.05 was considered statistically significant. If there was a significant difference between dose groups by ANOVA, each active dose was compared with placebo by a one-sided t test, with p < 0.025 being statistically significant. This procedure was also performed for percentage changes in total cholesterol (TC), HDL-C, and TG. For ApoB, ApoAI, Lp(a), vitamin A, vitamin E, and plant sterols, only ANOVA was performed to test differences in percentage changes between the five treatment groups. Safety data are presented descriptively.
Results
In phase 1, 30 male volunteers completed the study in accordance with the protocol. There were no withdrawals. The majority of subjects (93%) were of Caucasian descent. Mean subject age was 50.4 (range 24–63) years. In phase 2, 101 male subjects were enrolled, but one subject withdrew consent prior to receiving study medication due to personal reasons and therefore contributed no safety or efficacy data. The remaining 100 subjects completed the study. The majority of subjects (89%) were Caucasian, with the remaining being Asian or another race. Mean subject age was 53.7 (range 23–75) years. As calculated from returned tablets, the mean percentage of received tablets was 97%. One subject in the 800-mg group had a compliance <80%. All other patients received >80% of the planned number of tablets during the treatment period. Five subjects received statin treatment within 40 days before the first dose of study treatment (29–35 days). Mean weight ranged between 83.9 ± 13.1 and 84.6 ± 13.0 kg during the entire study.
Adverse events
In phase 1, 23 treatment-emergent adverse events were reported by 16 subjects. Of these events, five (21.7%) occurred in the 100-mg group, three (13.0%) in both the 200- and 400-mg groups, four (17.4%) in the 800-mg group, two (8.7%) in the 1,600-mg group, and three (13.0%) in both the 2,000 mg and placebo groups. All reported events were mild. The most common events were dizziness, headache, and fatigue. Other adverse events were loose stools, vasovagal attack, influenza, upper respiratory tract infection, elevated bilirubin, elevated BP, arthralgia, difficult micturition, polyuria, and pharyngolaryngeal pain. Three events were considered to be possibly related to the study drug. One was reported by a subject receiving the placebo treatment, whose bilirubin concentration increased from 12 μmol/L on the morning of treatment (baseline) to 25 μmol/L 24 h after treatment but decreased to normal 7 days after treatment. The other two possibly related events were reported in the 800- and 1,600-mg groups, and both consisted of one episode of loose stools on the day of treatment.
In phase 2, 67 subjects reported one or more treatment-emergent adverse events: 12 in the 100-mg group, 14 in the 200-mg group, 11 in the 400-mg group, 15 in the 800-mg group, and 15 in the placebo group. Most events were mild, and four subjects reported a moderate event. The most frequent event was headache, which was reported by a total of 19 subjects and by two (400-mg group) to five subjects (100-mg and 200-mg groups) in each of the five groups. The four adverse events that were moderate included two subjects with headache in the 800-mg group, one subject with an elevated CK level in the 400-mg group, and one subject with epilepsy in the placebo group. No subjects discontinued study treatment due to a treatment-emergent adverse event. One subject in the 800-mg group did not take the study medication for 3.5 days due to nausea and diarrhea, which was not considered to have been related to the study drug. Once the subject recommenced treatment, no further treatment-emergent adverse events were reported.
A total of 24 subjects reported events that were considered to be drug-related: eight in the 100-mg group, one in the 200-mg group, two in the 400-mg group, six in the 800-mg group, and seven in the placebo group. The most commonly reported event was flatulence, which was reported by three subjects in the 100-mg groups, one in the 400-mg group, two in the 800-mg group and one in the placebo group. There were no differences in the incidence of treatment-emergent adverse events between active and placebo groups.
Blood pressure, heart rate, and EKG analysis
There was no effect of FM-VP4 on BP or heart rate during phase 1. In phase 2, the mean systolic BP was slightly decreased after 4 weeks of treatment (a maximum average of 4% in the 100-mg group; data not shown), but there was no relationship between this decrease and the dose of FM-VP4. Diastolic BP did not change. All pre- and postdose EKGs were normal in both phases of the trial.
Laboratory analyses
In phase 1, one subject in the placebo group showed a bilirubin concentration increase from 12 μmol/L on the morning of treatment (baseline) to 25 μmol/L 24 h after treatment, as described in “Adverse events.” Seven days after treatment, bilirubin concentration was reduced to 13 μmol/L. There were no other clinically significant changes in hematology, biochemistry, plant sterols, or urinalysis measurements, and there were no differences between treatment groups. One subject who received 100 mg of FM-VP4 showed a decrease in white blood cell (WBC) count from 6.2 × 109/L on the morning of treatment (baseline) to 2.9 × 109/L 24 h after dosing. Seven days after dosing, the WBC count recovered to 7.3 × 109/L. This event was not considered to be clinically significant.
Nine subjects had abnormal laboratory values during phase 2. Four laboratory abnormalities were considered clinically significant and were reported as adverse events. Two subjects showed a decrease of WBC count. One subject in the 800-mg group had a level of 2.2 × 109/L at screening, but those levels were increased to 5.8 × 109/L at the following visit and remained normal during the rest of the trial. This event was not considered clinically significant. A subject in the 100-mg group had a WBC count of 3 × 109/L at screening, and levels fluctuated between 2.8 × 109/L and 4.6 × 109/L during the study. Two subjects showed an increase in CK levels that was greater than five times the ULN. In one of these patients, who was in the 400-mg group, the event was recorded as an adverse event of moderate severity. In the other subject, who was in the placebo group, the event was not recorded as an adverse event, as the patient had performed vigorous exercise the previous day. Both subjects continued with medication, and both had CK levels within normal limits at subsequent measurements. Five subjects showed a decrease in hemoglobin of a least 1.5 g/d (0.93 mmol/L): one each in the 100-mg, 200-mg, and 400-mg groups, and two in the placebo group. However, those decreases occurred only once, and levels were restored to normal at the subsequent visit. Therefore, those changes were not considered clinically significant.
There were no differences between treatment groups in the changes of vitamin A and vitamin E concentrations at week 4 compared with baseline (p=0.32 and p=0.38, respectively; data not shown).
Overall, there were few abnormal laboratory values, and no trends were observed over time. There appeared to be no relationship between laboratory parameters and the dose of FM-VP4 administered.
Efficacy
In phase 1, changes in lipids and lipoproteins were not statistically compared between the seven treatment groups, as only one dose of FM-VP4 was administered.
In the phase 2 run-in period, mean LDL-C levels were 3.94 ± 0.71 mmol/L at 4–8 weeks before baseline, 4.19 ± 0.78 mmol/L halfway through the run-in period, and 4.16 ± 0.76 mmol/L at baseline. The absolute lipoprotein levels at baseline and after 4 weeks of treatment per dose group and the mean percentage change in LDL-C levels are presented in Table 1. The percent changes of LDL-C by visit are also depicted in Fig. 2. ANOVA analyses showed a borderline statistically significant difference in the percentage change in LDL-C and HDL-C, and pairwise comparisons between each active dose and placebo revealed that 400 mg of FM-VP4 reduced LDL-C significantly (p=0.02). In the placebo group, LDL-C increased by 2.7%, and in the 400-mg/day group LDL-C reduced by 6.5%. There were no statistically significant differences in percentage change of LDL-C changes between the other active doses and placebo, and there were no statistically significant differences in percentage change of HDL-C between any of the active doses and placebo (Table 1). When statistical analysis was carried out on a per-protocol basis and subjects who were noncompliant (n=1) or who received statins within 40 days before study treatment (n=5) were excluded, a dose-response in percentage change from baseline in LDL-C was observed: changes in the placebo group and in the 100-, 200-, 400-, and 800-mg/day groups were 3.7%, 0.9%, −4.1%, −6.9%, and −6.2%, respectively. When using these per-protocol data, decreases in the 400- and 800-mg/day groups were significantly different from placebo (p=0.007 and p=0.01, respectively). The percentage change in ApoB at week 4 compared with baseline differed significantly between treatment groups (p 0.007). Absolute changes in ApoB were 0.017, 0.063, −0.058, −0.047, and −0.038 g/L in the placebo, 100-, 200-, 400-, and 800-mg/day group, respectively. There were no differences between treatment groups in ApoAI and Lp(a) changes at week 4 compared with baseline (data not shown).

Mean low-density lipoprotein cholesterol levels during 28 days of treatment with placebo or 100, 200, 400, and 800 mg/day FM-VP4 (disodium ascorbyl campestanol phosphate and disodium ascorbyl sitostanol phosphate) and after 14 days of follow-up
Pharmacokinetics
In the 100- and 200-mg cohorts of phase 1, the majority of plasma samples had DASP and DACP concentrations below detection level. Therefore, the AUC 0→t and the plasma elimination half-live (t1/2) values were not evaluable, and for that reason, those data are not presented. For the groups of 400 mg or higher, the peak DACP level was reached 6–24 h after FM-VP4 administration (tmax), and DASP tmax was reached 12–24 h postdose with the exception of one subject in the 1,600 mg group who had a tmax of 49 h (Table 2). Mean t1/2 of all eligible subjects was 57 h (2–3 days), which ranged from 16 to 134 h for DACP and from 29 to 90 h for DASP.
AUC0→t strongly correlated with the dose (400–2,000 mg) of FM-VP4 for DACP (R 2 = 0.93) and DASP (R 2 = 0.90). However, AUC increased in a lower than dose-proportional manner. Between 400 mg and 2,000 mg, DACP and DASP AUC increased approximately twice and 1.3 times, respectively, when the dose was increased fivefold (Table 2).
In phase 2, trough DACP and DASP concentrations were at or near steady-state levels by day 8 in the subset of subjects per active dose group who were hospitalized and sampled during the first week of dosing (Fig. 3). The mean concentrations for all subjects in each cohort on day 8 were about the same as the mean concentrations on day 28, at the end of the treatment period. At the end of the follow-up period, 14 days after the final dose, DASP was present in the plasma of most or all of the subject plasma samples in the 200-, 400-, and 800-mg groups. DACP was mainly present in plasma samples of most subjects in the 800-mg group. The majority of plasma samples for DACP in the 100- and 200-mg groups and for DASP in the 100-mg group were below detection limit 14 days after follow-up.

Mean trough concentrations of disodium ascorbyl sitostanol phosphate (DASP) and disodium ascorbyl campestanol phosphate (DACP) during 28 days of dosing and 14 days postdosing follow-up. The dots of days 2–5 represent the 16 hospitalized subjects on FM-VP4 (DACP and DASP), whereas the dots of days 8, 28, and 42 represent all 80 subjects on FM-VP4. DASP and DACP were not present in plasma at day 1 (baseline), and DACP concentrations were mostly below detection limit during the first 5 days of intake in the 100-mg group. The mean levels of DASP in the 100-mg group and DACP in the 200-mg group were below the lower limit of quantification (LLOQ) because some of the subjects had levels below the LLOQ, which were regarded as 0 ng/ml
DASP and DACP concentrations increased in a less than dose-proportional manner compared with baseline on days 8 and 28, as well as on day 42, 14 days after treatment.
Plant sterol and stanol concentrations
In phase 1, concentrations of campestanol and sitostanol did not change within 24 h and 7 days after a single dose of FM-VP4. Also, concentrations of campesterol and β-sitosterol were not affected after a single dose of FM-VP4 (data not shown).
In phase 2, sitostanol and campestanol apparently rose in a dose-dependent fashion over the 28-day time period, with the maximal percent change from baseline being 87% for campestanol and 178% for sitostanol in the 800-mg group (Table 3). Campesterol and sitosterol did not change from baseline to day 28 (Table 3).
Discussion
In this study, we showed that a single dose of 100–2,000 mg as well as 4-week treatment in doses of 100–800 mg of FM-VP4 administered to moderate dyslipidemic men was well tolerated and safe. Furthermore, 4-week treatment of FM-VP4 reduced LDL-C levels by 6–7% compared with baseline or by 9–11% compared with placebo.
The main treatment—emergent adverse events were dizziness, headache, and fatigue after a single administration and headache after multiple-dose administration of the drug. All symptoms resolved spontaneously, and subjects receiving placebo also reported these symptoms. As there was no difference in the incidence between active and placebo groups, it is unlikely that the treatment-emergent adverse events were due to FM-VP4.
No drug-related abnormalities could be identified by laboratory safety tests. In phase 1, one subject receiving 100 mg of FM-VP4 showed a decrease in WBC count 24 h after treatment, but this was considered unrelated to the study drug and returned to normal 7 days after treatment. In phase 2, one subject in the 100-mg group and one in the 800-mg group also showed low WBC counts, but these abnormalities were already present at baseline. One subject in the placebo group but also one in the 400-mg group showed increased CK levels at one visit. However, we think this is no reason for concern. First of all, the patient in the 400-mg group had no complaints. Second, the levels had returned to normal by the next visit without medication discontinuation. Because no adverse events have been reported for plant sterols in general [8], we think this single CK increase was likely to be a chance finding. In addition, five subjects from various dose groups showed a hemoglobin decrease at one of the visits. These decreases were nonpersistent, and levels had returned to normal by subsequent visits. No EKG or hemodynamic abnormalities were detected after administration of FM-VP4. Also, no liver or kidney abnormalities were identified. Thus, in terms of safety tests, there was no evidence that treatment with FM-VP4 caused any acute or delayed toxicity.
In phase 1, DACP and DASP were absorbed slowly into plasma, with a tmax of approximately 12 h for both components. Clearance was also rather slow, with the average elimination t1/2 of 57 h. Furthermore, t1/2 from phase 1 can be used to estimate the time to reach steady-state concentration levels by calculating three times t1/2, which is 3 × 57=171 h, or 7.1 days. This estimated time to reach steady state corresponds with phase 2 data that showed a steady state after approximately 8 days, as the mean trough concentration at day 8 was similar to the mean trough concentration after 4 weeks of treatment.
The low plasma concentrations of DACP and DASP suggest low bioavailability of FM-VP4 in humans. This is supported by findings with other plant sterol structures. The bioavailability from 600 mg unesterified soy plant stanols was only 0.04% for sitostanol and 0.15% for campestanol [22], and in another study, the absorption of campestanol from a margarine spread containing 540 mg campestanol as fatty acid esters was 5.5% in healthy subjects [23]. In both studies, absorption of plant sterols was measured by intravenous injection and oral administration of labeled isotopes in the fasting state [22, 23]. As we have not administered labeled FM-VP4 intravenously, we were not able to estimate the bioavailability of FM-VP4 in humans and to investigate whether the esterified ascorbate group affects its absorption. Future studies with FM-VP4 are needed to assess the bioavailability of FM-VP4 in humans.
Repeated doses of FM-VP4 in phase 2 appeared to increase sitostanol levels in plasma by up to 178% and campestanol by 87%, whereas corresponding sterol components did not change. This rise in campestanol and sitostanol appeared to increase with dose but in a less than proportional manner. Furthermore, the difference in increase from baseline between sitostanol and campestanol was consistent with the difference in plasma concentrations for DASP and DACP of approximately 2:1, which was most clear in the 400- and 800-mg groups. We speculate that conversion of DASP and DACP to free stanols may have occurred in vivo or that FM-VP4 alters the metabolism of plant stanols. Nevertheless, the absolute increase in total stanol levels remains relatively small, as the stanol contribution is <5% of total levels (Table 3). Therefore, the clinical relevance of such increases is probably limited.
Based on our data, we may speculate on the mechanism by which FM-VP4 exerts its LDL-C-lowering effect. Plant sterols and stanols are thought to compete with cholesterol for incorporation into mixed micelles, the vehicles that transport sterols to the enterocyte. In the case of FM-VP4, the solubility of plant stanols for the mixed micelles has been improved by the addition of the hydrophilic ascorbyl residue to the hydrophobic campestanol and sitostanol tail through a phosphodiester linkage (FM-VP4). In fact, this new structure allowed self-assembly into micelle structures in aqueous media in the absence of bile salts [24]. Although the assembly of micelles by means of FM-VP4 has never been directly compared with other types of plant sterols, the incorporation of plant stanols and sterols as well as cholesterol into mixed micelle normally requires the addition of bile salts in vitro and in vivo. Furthermore, in animals, FM-VP4 was shown to be more potent than unesterified plant stanols in a direct comparison when administered in the diet [16]. The results of the current clinical trial in humans showed that FM-VP4 lowered LDL-C by 6–7% compared with baseline, whereas the control group showed a 3–4% increase in plasma LDL-C levels. This means that compared with placebo, FM-VP4 showed LDL-C decreases of 9–11% when administered at 400–800 mg/day over 4 weeks. This effect was also reached with plant sterol and stanol esters, but at doses that were two to three times higher than the currently used doses of FM-VP4 [8]. Thus, the higher solubility of FM-VP4 into mixed micelles may lead to a more efficacious competition with cholesterol for incorporation into the micelles, and as a consequence, a lower dose of FM-VP4 is required to realize an equal LDL-lowering effect compared with plant sterol and stanol esters. However, the optimal treatment dose and duration, schedule and formulation for FM-VP4 have yet to be determined. On the other hand, it has also been suggested that, in addition to competition with cholesterol for incorporation into the micelles, plant sterol and stanol esters also may activate liver X receptor (LXR) target genes within the enterocyte [25]. It is unknown whether DACP and DASP activate such genes within the enterocyte or whether there may be systemic effects or additional mechanisms to decrease cholesterol absorption. Further research is needed to elucidate the mechanism by which several types of plant sterol and stanol esters lower LDL-C levels.
In conclusion, this study demonstrated that single and multiple doses of FM-VP4 for 4 weeks are safe and well tolerated by moderately hypercholesterolemic subjects. Furthermore, the higher doses of FM-VP4 significantly reduce LDL-C levels by 6–7% compared with baseline or by 9–11% when compared with placebo. The pharmacokinetics showed that DACP and DASP are absorbed and cleared slowly but that the absolute quantity of drug absorbed is low, as suggested by the low plasma concentrations of DACP and DASP. This study suggests that FM-VP4 merits further investigation as an alternative for treating hyperlipidemia.
References
Cullen P, Assmann G (1999) High risk strategies for atherosclerosis. Clin Chim Acta 286:31–45
Scandinavian Simvastatin Survival Study Group (1994) Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 344:1383–1389
Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E (1996) The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med 335:1001–1009
Heart Protection Study Collaborative Group (2002) MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 360:7–22
Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ (1995) Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med 333:1301–1307
Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, Langendorfer A, Stein EA, Kruyer W, Gotto AM Jr (1998) Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 279:1615–1622
Law M (2000) Plant sterol and stanol margarines and health. BMJ 320:861–864
Katan MB, Grundy SM, Jones P, Law M, Miettinen T, Paoletti R (2003) Efficacy and safety of plant stanols and sterols in the management of blood cholesterol levels. Mayo Clin Proc 78:965–978
Jones PJH, MacDougall DE, Ntanios F, Vanstone CA (1997) Dietary phytosterols as cholesterol-lowering agents in humans. Can J Physiol Pharmacol 75:217–227
Miettinen TA, Gylling H (1997) Sitostanol-ester margarine. In: Yalpani M (ed) New technologies for healthy foods & nutraceuticals. ALT Press, Shrewsbury, pp 71–83
Ramaswamy M, Yau E, Wasan KM, Boulanger KD, Li M, Pritchard PH (2002) Influence of phytostanol phosphoryl ascorbate, FM-VP4, on pancreatic lipase activity and cholesterol accumulation within Caco-2 cells Tc-DTPA and Tc-sulphur colloid as tracers in colonic drug delivery systems using gamma scintigraphy in volunteers. J Pharm Pharm Sci 5:29–38
Wasan KM, Yau E, Boulanger KD, Ramswamy M, Pritchard PH (2003) Effects of disodium ascorbyl phytostanol phosphates (FM-VP4) on cholesterol accumulation within rat intestinal cells. AAPS PharmSci 5:E6
Wasan KM, Peteherych KD, Najafi S, Zamfir C, Pritchard PH (2001) Assessing the plasma pharmacokinetics, tissue distribution, excretion and effects on cholesterol pharmacokinetics of a novel hydrophilic compound, FM-VP4, following administration to rats. J Pharm Pharm Sci 4:207–216
Wasan KM, Najafi S, Peteherych KD, Pritchard PH (2001) Effects of a novel hydrophilic phytostanol analog on plasma lipid concentrations in gerbils. J Pharm Sci 90:1795–1799
Wasan KM, Najafi S, Wong J, Kwong M, Pritchard PH (2001) Assessing plasma lipid levels, body weight, and hepatic and renal toxicity following chronic oral administration of a water soluble phytostanol compound, FM-VP4, to gerbils. J Pharm Pharm Sci 4:228–234
Ebine N, Jia X, Demonty I, Wang Y, Jones PJ (2005) Effects of a water-soluble phytostanol ester on plasma cholesterol levels and red blood cell fragility in hamsters. Lipids 40:175–180
Lukic T, Wasan KM, Zamfir D, Moghadasian MH, Pritchard PH (2003) Disodium ascorbyl phytostanyl phosphate reduces plasma cholesterol concentrations and atherosclerotic lesion formation in apolipoprotein E-deficient mice. Metabolism 52:425–431
Looije NA, Risovic V, Stewart DJ, Debeyer D, Kutney J, Wasan KM (2005) Disodium Ascorbyl Phytostanyl Phosphates (FM-VP4) reduces plasma cholesterol concentration, body weight and abdominal fat gain within a dietary-induced obese mouse model. J Pharm Pharm Sci 8:400–408
Friedewald WT, Levy RI, Fredrickson DS (1972) Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 18:499–502
Ng AW, Lukic T, Pritchard PH, Wasan KM (2004) Development and characterization of liposomal disodium ascorbyl phytostanyl phosphates (FM-VP4). Drug Dev Ind Pharm 30:739–758
Rocci ML Jr, Jusko WJ (1983) LAGRAN program for area and moments in pharmacokinetic analysis. Comput Programs Biomed 16:203–216
Ostlund RE Jr, McGill JB, Zeng CM, Covey DF, Stearns J, Stenson WF, Spilburg CA (2002) Gastrointestinal absorption and plasma kinetics of soy Delta(5)-phytosterols and phytostanols in humans. Am J Physiol Endocrinol Metab 282:E911–E916
Salen G, Xu G, Tint GS, Batta AK, Shefer S (2000) Hyperabsorption and retention of campestanol in a sitosterolemic homozygote: comparison with her mother and three control subjects. J Lipid Res 41:1883–1889
Wasan KM, Choo E, Sivak O, Wallis S, Letchford K, Burt HM, Stewart DJ, Lukic T (2004) Determining the critical micelle concentration of a novel lipid-lowering agent, disodium ascorbyl phytostanyl phosphate (FM-VP4), using a fluorescence depolarization procedure. Drug Dev Ind Pharm 30:725–730
Plat J, Nichols JA, Mensink RP (2005) Plant sterols and stanols: effects on mixed micellar composition and LXR (target gene) activation. J Lipid Res 46:2468–2476
Acknowledgements
We gratefully thank Dr. Richard Koopmans for carefully reading the manuscript and his assistance with respect to the pharmacokinetics, and we thank the volunteers for their enthusiastic participation. The study was funded by Forbes Medi-Tech Inc. The procedures in this study complied with the current laws of the country in which they were performed (the Netherlands).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Vissers, M.N., Trip, M.D., Pritchard, P.H. et al. Efficacy and safety of disodium ascorbyl phytostanol phosphates in men with moderate dyslipidemia. Eur J Clin Pharmacol 64, 651–661 (2008). https://doi.org/10.1007/s00228-008-0462-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-008-0462-1