
Abstract
Mast cell activation disease (MCAD) is a term referring to a heterogeneous group of disorders characterized by aberrant release of variable subsets of mast cell (MC) mediators together with accumulation of either morphologically altered and immunohistochemically identifiable mutated MCs due to MC proliferation (systemic mastocytosis [SM] and MC leukemia [MCL]) or morphologically ordinary MCs due to decreased apoptosis (MC activation syndrome [MCAS] and well-differentiated SM). Clinical signs and symptoms in MCAD vary depending on disease subtype and result from excessive mediator release by MCs and, in aggressive forms, from organ failure related to MC infiltration. In most cases, treatment of MCAD is directed primarily at controlling the symptoms associated with MC mediator release. In advanced forms, such as aggressive SM and MCL, agents targeting MC proliferation such as kinase inhibitors may be provided. Targeted therapies aimed at blocking mutant protein variants and/or downstream signaling pathways are currently being developed. Other targets, such as specific surface antigens expressed on neoplastic MCs, might be considered for the development of future therapies. Since clinicians are often underprepared to evaluate, diagnose, and effectively treat this clinically heterogeneous disease, we seek to familiarize clinicians with MCAD and review current and future treatment approaches.
Similar content being viewed by others


Introduction
Mast cells (MCs, Fig. 1) are immune cells of hematopoietic origin found in all human tissues, especially at the environmental interfaces. They act as both effector and regulatory cells and play a central role in adaptive and innate immunity (Anand et al. 2012; Gri et al. 2012). Their important role in immunological as well as non-immunological processes is reflected by the large number of mediators (>200) including pre-stored ones such as histamine and tryptase as well as numerous mediators synthesized de novo in response to allergic or non-immune triggers such as chemokines and cytokines, by which MCs may influence other cells (Lundequist and Pejler 2011; Ibelgaufts 2016). Their evolved arrays of sensory and response mechanisms engender diverse havoc when MC dysfunction emerges.

May-Grünwald/Giemsa stain of a resting human mast cell and a mast cell following activation-induced degranulation. Note the loss of granule staining. Mast cells obtained from the human bone marrow, magnification 1000×
The umbrella term mast cell activation disease (MCAD; Akin et al. 2010) comprises the full spectrum of primary systemic MC disease, i.e., systemic mastocytosis (SM) which is further divided into several subtypes (Valent et al. 2007; Tables 1 and 2), primary MC activation syndrome (MCAS; Table 3; Molderings et al. 2011a; Hamilton et al. 2011; Valent et al. 2012), and MC leukemia (MCL). Pathogenetically, MCAD denotes a group of polygenic MC disorders (Molderings 2015, 2016) characterized by aberrant release of variable subsets of MC mediators and also an accumulation of either morphologically altered and immunohistochemically identifiable mutated MCs due to MC proliferation (SM and MCL) or morphologically ordinary MCs due to decreased apoptosis (MCAS; Kohno et al. 2005; Aichberger et al. 2009; Karlberg et al. 2010a). According to recent molecular genetic findings (Molderings 2015, 2016; Haenisch et al. 2014; Lasho et al. 2016), the subclasses and clinical subtypes of MCAD do not represent distinct disease entities but should be more accurately regarded as variable presentations of a common generic state of MC dysfunction (Molderings et al. 2007, 2010; Hermine et al. 2008; Akin et al. 2010). Due to both the widespread distribution of MCs and the great heterogeneity of aberrant mediator expression patterns, symptoms can occur in virtually all organs and tissues; hence, the clinical presentation of MCAD is very diverse, sometimes to the even-further-confounding point of presenting opposite abnormalities in different patients (or even in the same patient at different times, or in different sites in the same patient at the same time). While the prevalence of SM in Europeans ranges between 0.3 and 13 per 100,000 (Haenisch et al. 2012; Cohen et al. 2014; van Doormaal et al. 2013), the prevalence of MCAS may be as high as 17 % (in Germany; Molderings et al. 2013a, b).
This review focuses on the current state of drug therapy in SM and MCAS and describes perspectives of promising new approaches for drug treatment. Compounds in various stages of preclinical and clinical development are summarized in tables. We first describe drugs that are currently available and either are used on a regular basis in MCAD therapy or have been used successfully in single MCAD cases. In this context, it should be noted that there is no official guideline for treatment of MCAD.
Treatment options
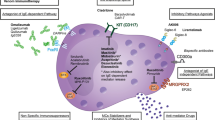
Due to its genetic roots, MCAD generally is regarded as incurable. Recent mutational studies revealed that each patient has an individual pattern of genetic and epigenetic alterations which may affect the intracellular signal transduction pathways and receptive sites involved in sensory perception. As a consequence, mediator formation and release as well as inhibition of apoptosis and/or increase in proliferation are determined by individual genetic and epigenetic conditions (Fig. 2) and represent potential targets for therapy. Hence, there is need of highly personalized therapy for the disease. Unfortunately (with regard to easy detection), most genetic alterations (with a few exceptions such as certain mutations in tyrosine kinase KIT, e.g., KITD816V) do not alter the morphology and immunohistochemistry of the surface of the affected MCs. Thus, in most cases except for patients with the reliably identifiable D816V mutation, it cannot be decided by simple tests whether MCs found in biopsies are genetically altered MCs or physiological MCs.

Scheme of conditions responsible in MCAD for the development of individual phenotypes
First-line treatment options
Step 1 in managing most situations of inappropriate MC activation is identifying the individual patient’s unique triggers (chemical, physical, or otherwise) as precisely as possible and then desensitizing when possible (in truth, rarely) and otherwise practicing avoidance. With respect to drug treatment, only a few clinical therapeutic trials have been conducted in SM (midostaurin, cladribine, masitinib; Table 4), and there have been no therapeutic trials in MCAS yet. Most information about therapeutic effectiveness in MCAD has been found in small case series (Table 4) and single case reports, perhaps unsurprising given the mutational heterogeneity of the disease and thus the heterogeneity of its patterns of clinical presentation and therapeutic responsiveness. Therefore, in the future, it may be helpful to establish an international patient registry in partnership with existing registries so that issues related to molecular and clinical MCAD phenotypes can be adequately addressed. As the primary feature of MCAD is inappropriate MC activation (Molderings et al. 2011a, b; Pardanani 2013; Cardet et al. 2013), mainstays of first-line management are identification and avoidance of triggers plus therapies to control MC mediator production (both primary as well as secondary/reactive; Table 5) as well as their action (Table 6).
Subordinate therapeutic options
Continuous diphenhydramine infusion
Occasional patients suffer nearly continuous anaphylactoid and/or dysautonomic states poorly controlled by intermittently dosed epinephrine, antihistamines, and steroids. As discussed in more detail below, some such patients are particularly triggered by a wide range of medication excipients, making it challenging for them to tolerate trials of any adulterated (non-pure) medications, and yet some modicum of stability is required to pursue medication trials in such patients. Diphenhydramine is a well-tolerated histamine H1 receptor blocker (that among other non-threatening adverse affects can cause dizziness and an increase in appetite) which can quickly suppress MC activation and is used to treat allergic reactions and anaphylaxis. However, its half-life is as short as 1 h (www.drugbank.ca/drugs/DB01075). Intermittently dosed, though, its initial therapeutic serum level rapidly declines to subtherapeutic levels and the patient seesaws into yet another flare. The safety of continuous diphenhydramine infusion was established in trials of the “BAD” regimen (diphenhydramine [Benadryl], lorazepam [Ativan], and dexamethasone) in refractory chemotherapy-induced emesis in adult and pediatric patients (Dix et al. 1999; Jones et al. 2007). In a small series of ten MCAS patients suffering almost continuous anaphylactoid/dysautonomic flares, continuous diphenhydramine infusion at 10–14.5 mg/h appeared effective in most patients at dramatically reducing flare rates and appeared safely sustainable at stable dosing for at least 21 months (Afrin 2015). Stabilization has enabled successful trials of other helpful medications, but no patient has yet successfully stopped continuous diphenhydramine infusion.
Acute and chronic immunosuppressive therapies
Though typically not first-line, acute and chronic immunosuppressive therapies can be considered (Fig. 3; Table 7) and may be particularly appropriate for patients possibly manifesting an autoimmune component of the disease as might be suggested by the presence, for example, of anti-IgE or anti-IgE-receptor antibodies. Glucocorticoids may exert beneficial effects in MCAD, including a decrease in production of stem cell factor (SCF, and possibly other cytokines) and a decrease in MC activation, by various mechanisms which have been extensively reviewed by Oppong et al. 2013. Glucocorticoids at doses >20 mg prednisone equivalent per day are frequently needed to effectively control otherwise refractory acute (and chronic) symptoms. Their chronic toxicity profile is disadvantageous for long-term use, but such toxicities have to be accepted in some cases. The influence of azathioprine, methotrexate, ciclosporine, hydroxyurea, and tamoxifen on MC activity can vary from no to moderate effect depending on individual disease factors. As in therapy of rheumatoid arthritis, azathioprine and methotrexate can be used in daily doses lower than those used in cancer or immunosuppressive post-transplant therapy. Effective MCAD therapy with ciclosporine requires doses as high as those used in transplantation medicine (M. Raithel, personal communication). Methotrexate has to be administered parenterally to be effective (unpublished observation, G.J. Molderings), and in the risk-benefit analysis, a possible non-immunologic histamine release from MCs (Estévez et al. 1996) has to be considered. Hence, use of the compound should be limited to MCAD with methotrexate-sensitive comorbidities (e.g., rheumatoid arthritis and vasculitis).

Suggested treatment options for mast cell activation disease. All drugs should be tested for tolerance in a low single dose before therapeutic use, if their tolerance in the patient is not known from an earlier application. For further details of indication, see text
Recently, the humanized anti-IgE murine monoclonal antibody omalizumab has been described in multiple case reports as safe and effective in MCAD (e.g., Molderings et al. 2011b; Kontou-Fili et al. 2010; Bell and Jackson 2012; Kibsgaard et al. 2014), though a definitive trial has yet to be conducted. Since treatment with omalizumab has an acceptable risk-benefit profile, it should be considered in cases of MCAD resistant to at least a few lines of therapy. The drug’s expense likely consigns it to third-line (or later) treatment (Table 7). If elevated prostaglandin levels induce symptoms such as persistent flushing, inhibition of cyclooxygenases by incremental doses of acetylsalicylic acid (ASA; 50–350 mg/day) may be used with extreme caution, since ASA can induce MC degranulation probably due its chemical property as an organic acid. The leukotriene antagonist montelukast (possibly more effective at twice-daily dosing; personal observation, L.B. Afrin) and the 5-lipoxygenase inhibitor zileuton may be useful adjuvants in people with MCAD, particularly in those with refractory gastrointestinal and urinary symptoms (Tolar et al. 2004; Turner et al. 2012; Akhavein et al. 2012).
Studies of kinase inhibitors, both on-market (e.g., imatinib, nilotinib, dasatinib) and experimental (e.g., midostaurin, masitinib), have yielded variable responses in SM ranging from no response to partial or even complete responses (Fig. 3; Table 8). As with all drugs used in therapy of MCAD, their therapeutic success seems to be strongly dependent on the individual patient, again underscoring the observed mutational heterogeneity of the disease. In formal studies in SM patients, although some kinase inhibitors reduced MC burden as reflected by histological normalization in bone marrow and improved laboratory surrogate markers (e.g., tryptase level in blood), at best only partial improvement of mediator-related symptoms was achieved (Droogendijk et al. 2006; Gotlib et al. 2008; Verstovsek et al. 2008; Vega-Ruiz et al. 2009). There has been repeated suggestion that symptoms in MCAD may be due more to mediator release from normal MCs secondarily activated by pathologically overactive, mutated MCs (Galli and Costa 1995; Rosen and Goetzl 2005; Boyce 2007; Kaneko et al. 2009; Fig. 2 in Molderings et al. 2014), helping to explain why intensity and pattern of symptoms do not correlate with degree of MC proliferation and infiltration (Topar et al. 1998; Hermine et al. 2008; Broesby-Olsen et al. 2013; Erben et al. 2014; Quintás-Cardama et al. 2013). Distinction in pathways in the MC which promote MC proliferation vs. mediator production/release may explain why kinase inhibitors reduce MC burdens and MC-driven symptoms to different degrees (Droogendijk et al. 2006; Gotlib et al. 2008; Verstovsek et al. 2008; Vega-Ruiz et al. 2009; Table 8). However, in some case reports, kinase inhibitors have been significantly effective at relieving symptoms. Thus, in spite of potential serious adverse effects of these drugs, a therapeutic trial may be justified in individual cases at an early stage. Partial and complete responses have been reported with some of these agents in MCAS too (e.g., Afrin 2010, 2011, 2012, 2015; Afrin et al. 2015a). Dosing of the kinase inhibitors in the individual often is considerably lower than how such drugs are dosed for other applications (e.g., imatinib, sunitinib; Afrin et al. 2015a). Possibly due to the causative mutations in multiple genes leading to simultaneous activation of multiple intracellular pathways, multitargeted kinase inhibitors such as midostaurin and sunitinib may be more effective than drugs which selectively downregulate only one intracellular pathway.
In the mastocytosis patient with significant MC burden and/or an aggressive clinical course, cytoreductive drugs are prescribed (Lim et al. 2009; Valent et al. 2010). Unfortunately, effective cytoreductive therapies in SM presently are few in number and typically offer only modest response rates, qualities, and durations. Cytoreductive options include interferon-α and 2-chlorodeoxyadenosine (cladribine, 2-CdA; Fig. 3 and Table 9). Interferon-α is frequently combined with prednisone and is commonly used as cytoreductive therapy for aggressive SM. It ameliorates mastocytosis-related organopathy in a proportion of cases but can be associated with considerable adverse effects (e.g., flu-like symptoms, myelosuppression, depression, hypothyroidism), which may limit its use in MCAD (Simon et al. 2004; Butterfield 2005). PEGylated interferon-α has been shown to be as efficacious as and less toxic than the non-PEGylated form in some myeloproliferative neoplasms, but it has not been specifically studied in MCAD. 2-Chlorodeoxyadenosine is generally reserved for last-choice treatment of patients with aggressive SM who are either refractory or intolerant to interferon-α. Potential toxicities of 2-CdA include significant and potentially prolonged myelosuppression and lymphopenia with increased risk for opportunistic infections.
Last resorts
Polychemotherapy, including intensive induction regimens of the kind used in treating acute myeloid leukemia, as well as high-dose therapy with stem cell rescue, are approaches restricted to rare, selected patients. Allogeneic stem cell transplantation sometimes yields remissions in mastocytosis long thought impermanent (Spyridonidis et al. 2004; Nakamura et al. 2006; Bae et al. 2013; Gromke et al. 2013), though recent data may offer new hope (Ustun et al. 2014).
Investigational drugs
There are several drugs approved for indications other than MCAD which already have been successfully used in isolated cases with MCAD (Table 10). In cases of unsuccessful first- to fourth-line therapy, these compounds may be considered as treatment options.
A variety of drugs have been shown to inhibit MC growth, to decrease MC mediator release, and/or to relieve mediator-induced symptoms in in vitro and in vivo animal models (Table 11). Some of these drugs are approved for certain indications (such as ambroxol, statins, mefloquine, and ruxolitinib) and, thus, may be used (if accessible given financial considerations for some agents) if MCAD patients suffer from both the disorder of indication (e.g., hypercholesterolemia—statins, mucous congestion—ambroxol, polycythemia vera—ruxolitinib) and MCAD. An important question is what the role of the other compounds without approved indications should be in clinical practice. There are several challenges that may hamper the clinical introduction of novel targeted therapies in general. Some of these challenges include inherent problems in the translation of preclinical findings to the clinic, the presence of multiple coactive deregulated pathways in the disease, and questions related to the optimal design of clinical trials (e.g., eligibility criteria and endpoints). In particular, the testing of novel targeted treatment in an isolated fashion may be problematic and may in fact underestimate the effectiveness of these novel compounds. It is reasonable to assume that combination therapy will be the key to target parallel critical pathways.
General considerations on drug treatment of MCAD
Although no biomarkers of symptomaticity or therapeutic response are yet validated, the tolerability and efficacy of most therapies tried in MCAD (starting, and escalating in dosage and composition, cautiously) become clinically evident within 1–2 months. Modest experiments with alternative dosages and/or dosing frequencies are not unreasonable. Therapies clearly shown clinically helpful should be continued; therapies not meeting this high bar should be halted to avoid the troublesome polypharmacy that can easily develop in such patients. With no predictors of response yet available, a cost-based approach to sequencing therapeutic trials in a given patient seems reasonable. It is not even clear yet that medications targeted at mediators found elevated in diagnostic testing (e.g., antihistamines in patients with elevated histamine, non-steroidal anti-inflammatory drugs in patients with elevated prostaglandins, leukotriene inhibitors in patients with elevated leukotrienes) are reliably effective, again perhaps unsurprising given the multitude of MC mediators and the complexity of the signaling networks dysregulated by the multiple mutations in MC regulatory elements present in most MCAD patients. Successful regimens appear highly personalized.
Multiple simultaneous (or nearly so) changes in the medication regimen are discouraged since such can confound identification of the specific therapy responsible for a given improvement (or deterioration). Ineffective or harmful agents should be stopped promptly. Prescribers should be aware that although rapid demonstration of intolerance of a new medication (or a new formulation of a previously well-tolerated medication) often suggests excipient reactivity as further discussed below, some active drug molecules themselves (e.g., cromolyn) sometimes cause an initial symptom flare which usually soon abates. Temporary waiver of gluten-, yeast-, and cow milk protein-containing foods during the initial 3–4 weeks of drug therapy can improve the response rate (Biesiekierski et al. 2011; Rodrigo et al. 2013; own unpublished experiences). When MCAD is suspected, therapies that strongly activate the immune system (e.g., vaccinations with live vaccines or autohemotherapy) must be given with caution (especially if similar therapies were previously already poorly tolerated), as such interventions sometimes dramatically worsen MCAD acutely and/or chronically.
Any drug can induce intolerance symptoms in the individual MCAD patient. In some MCAD patients, the disease creates such remarkable states of not only constitutive MC activation but also aberrant MC reactivity that such patients unfortunately experience a great propensity to react adversely to a wide variety of medication triggers. Those MCAD patients begin demonstrating (either acutely or subacutely) odd/unusual/weird/strange/bizarre/unexpected symptoms soon after beginning new medications. It is very important to note that such patients often demonstrate even a greater propensity to react to medication excipients (i.e., fillers, binders, dyes, preservatives) than to the active ingredients. When the patient tries one or more alternative formulations of a medication with the same active ingredient but sharing as few as possible (preferably none) of the excipients in the offending formulation, the patient may discover the medication to be at least tolerable and perhaps even quite effective. Furthermore, such a scenario obviously provides the patient (and physician and pharmacist) a great opportunity to identify one or more of the specific excipients which are triggering abnormal reactivity in the patient’s dysfunctional MCs, and it is those specific excipients—not the medication as a whole—that should be added to the patient’s allergy list and screened against all present medications being taken by the patient and against all future medications proposed for the patient. An MCAD patient’s physician would be wise to not assume, just because an excipient is very widely used in many medication products and appears innocuous and well tolerated in the vast majority of patients, that the same excipient will necessarily be tolerated well in MCAD patients (unpublished observation of the authors). Sometimes the specificity of the reaction is quite extraordinary. For example, patients who react to wood-based microcrystalline cellulose might tolerate cotton-based microcrystalline cellulose without any difficulty at all, or vice versa. In some cases, the pharmacist is unable to identify alternative commercially available formulations sharing few to none of the excipients in the offending formulation, and in those cases, a compounding pharmacist may need to be engaged to identify/develop a custom-compounded formulation the patient can tolerate. (There can be geographic and financial challenges in accessing compounding pharmacies, though.) Occasionally, MCAD patients may be so remarkably reactive to such a wide range of excipients that they can only tolerate a given medication when provided as pure drug salt, reconstituted in water (without preservatives). Intolerance symptoms can be mediated by IgE antibodies, though this scenario appears to be rare since the symptoms are usually not ameliorated by the anti-IgE monoclonal antibody omalizumab (unpublished observation, G.J. Molderings). Alternatively, they may be mediated by IgG antibodies, raising the question of whether gamma globulin (if itself tolerable) might be a helpful adjunct therapy in such patients (perhaps by directly targeting the MC surface’s IgG receptors or via indirect pathways). Recently, a MC-specific receptor termed MRGPRX2 has been identified which appears to be crucially involved in pseudo-allergic drug reactions (McNeil et al. 2015; Seifert 2015).
Drugs which should not be used in MCAD
Several drugs have the ability to trigger MC mediator release. A compilation of drugs known to be associated with a high risk of release of mediators from MCs is given in Table 12. However, there often are therapeutic alternatives to these drugs (Table 12).
Conclusions and future perspectives
The therapeutic management of individuals with MCAD is complex and requires reviewing the entire spectrum of symptoms. The paucity of randomized, controlled studies makes treatment of refractory disease challenging and requires patience, persistence, and a methodical approach on the parts of both patient and managing provider(s). Delayed control of the symptoms may increase morbidity. Effective therapy often consists simply of antihistamines and MC-stabilizing compounds supplemented with medications targeted at specific symptoms and complications (Table 13). Current treatment options for refractory disease are based mainly on observational studies and case reports. Until larger randomized, controlled trials become available to give more guidance on therapy for refractory disease, clinicians should use the available data in conjunction with their clinical expertise and the adverse effect profile of the available drugs to make treatment decisions. More research is certainly needed to better understand MCAD pathobiology, in particular to determine which deregulated genes contribute to a specific symptom or symptom cluster. The greatest challenge in translational research for the discovery of new rational therapies requires a highly interactive interdisciplinary approach engaging basic science labs and clinicians. Understanding of the key components might hasten the progress of novel treatment for all these devastating MCAD phenotypes.
References
Abdulkadir H, Grootens J, Kjellander M, Hellstram Lindberg E, Nilsson G, Ungerstedt J (2015) Histone deacetylase inhibitor SAHA mediates epigenetic silencing of KIT D816V mutated systemic mastocytosis primary mast cells and selective apoptosis of mutated mast cells. Blood;126: abstract 2834
Adachi S, Maruyama T, Kondo T, Todoroki T, Fukao K (1999) The prevention of postoperative intraperitoneal adhesions by tranilast: N-(3′,4′-dimethoxycinnamoyl)anthranilic acid. Surg Today 29:51–54
Afrin LB (2010) Mast cell activation disorder masquerading as pure red cell aplasia. Int J Hematol 91:907–908
Afrin LB (2011) Polycythemia from mast cell activation syndrome: lessons learned. Am J Med Sci 342:44–49
Afrin LB (2012) Mast cell activation syndrome masquerading as agranulocytosis. Mil Med 177:113–117
Afrin LB (2013) Utility of hydroxyurea in mast cell activation syndrome. Exp Hematol Oncol 2:28
Afrin LB (2015) Utility of continuous diphenhydramine infusion in severe mast cell activation syndrome. Blood 126:5194 abstract
Afrin LB, Molderings GJ (2014) A concise, practical guide to diagnostic assessment for mast cell activation disease. World J Hematol 3:1–17
Afrin LB, Cichocki FM, Patel K, Molderings GJ (2015a) Successful treatment of mast cell activation syndrome with sunitinib. Eur J Haematol 95:595–597
Afrin LB, Pöhlau D, Raithel M, Haenisch B, Dumoulin FL, Homann J, Mauer UM, Harzer S, Molderings GJ (2015b) Mast cell activation disease: an underappreciated cause of neurologic and psychiatric symptoms and diseases. Brain Behav Immun 50:314–321
Aichberger KJ, Gleixner KV, Mirkina I, Cerny-Reiterer S, Peter B, Ferenc V, Kneidinger M, Baumgartner C, Mayerhofer M, Gruze A, Pickl WF, Sillaber C, Valent P (2009) Identification of proapoptotic Bim as a tumor suppressor in neoplastic mast cells: role of KIT D816V and effects of various targeted drugs. Blood 114:5342–5351
Akhavein A, Patel NR, Minoyappa PK, Glover SC (2012) Allergic mastocytic gastroenteritis and colitis: an unexplained etiology in chronic abdominal pain and gastrointestinal dysmotility. Gastroenterol Res Pract 2012:950582
Akin C, Valent P, Metcalfe D (2010) Mast cell activation syndrome: proposed diagnostic criteria. J Allergy Clin Immunol 126:1099–1104
Aldi S, Takano KI, Tomita K, Koda K, Chan NY, Marino A, Salazar-Rodriguez M, Thurmond RL, Levi R (2014) Histamine H4-receptors inhibit mast cell renin release in ischemia/reperfusion via PKCε-dependent aldehyde dehydrogenase type-2 activation. J Pharmacol Exp Ther 349:508–517
Alexandrakis MG, Kyriakou DS, Kempuraj D, Huang M, Boucher W, Seretakis D, Theoharides TC (2003) The isoflavone genistein inhibits proliferation and increases histamine content in human leukemic mast cells. Allergy Asthma Proc 24:373–377
Aman J, van Bezu J, Damanafshan A, Huveneers S, Eringa EC, Vogel SM, Groeneveld AB, Vonk Noordegraaf A, van Hinsbergh VW, van Nieuw Amerongen GP (2012) Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation 126:2728–2738
Anand P, Singh B, Jaggi AS, Singh N (2012) Mast cells: an expanding pathophysiological role from allergy to other disorders. Naunyn Schmiedeberg’s Arch Pharmacol 385:657–670
Baba A, Tachi M, Ejima Y, Endo Y, Toyama H, Matsubara M, Saito K, Yamauchi M, Miura C, Kazama I (2016) Anti-allergic drugs tranilast and ketotifen dose-dependently exert mast cell-stabilizing properties. Cell Physiol Biochem 38:15–27
Babaei S, Bayat M (2012) Effect of pentoxifylline administration on mast cell numbers and degranulation in a diabetic and normoglycemic rat model wound healing. Iran Red Crescent Med J 14:483–487
Bachelet I, Munitz A, Moretta A, Moretta L, Levi-Schaffer F (2005) The inhibitory receptor IRp60 (CD300a) is expressed and functional on human mast cells. J Immunol 175:7989–7995
Bae MH, Kim HK, Park CJ, Seo EJ, Park SH, Cho YU, Jang S, Chi HS, Lee KH (2013) A case of systemic mastocytosis associated with acute myeloid leukemia terminating as aleukemic mast cell leukemia after allogeneic hematopoietic stem cell transplantation. Ann Lab Med 33:125–129
Baek OS, Kang OH, Choi YA, Choi SC, Kim TH, Nah YH, Kwon DY, Kim YK, Kim YH, Bae KH, Lim JP, Lee YM (2003) Curcumin inhibits protease-activated receptor-2 and -4-mediated mast cell activation. Clin Chim Acta 338:135–141
Bairlein M (2010) Characterization of the small molecule kinase inhibitor SU11248 (sunitinib/SUTENT) in vitro and in vivo—towards response prediction in cancer therapy with kinase inhibitors. TU Munich, Munich, Germany, Medical thesis
Barete S, Lortholary O, Damaj G, Hirsch I, Chandesris MO, Elie C, Hamidou M, Durieu I, Suarez F, Grosbois B, Limal N, Gyan E, Larroche C, Guillet G, Kahn JE, Casassus P, Amazzough K, Coignard-Biehler H, Georgin-Lavialle S, Lhermitte L, Fraitag S, Canioni D, Dubreuil P, Hermine O (2015) Long-term efficacy and safety of cladribine (2-CdA) in adult patients with mastocytosis. Blood 126:1009–1016
Bell MC, Jackson DJ (2012) Prevention of anaphylaxis related to mast cell activation syndrome with omalizumab. Ann Allergy Asthma Immunol 108:383–384
Bibi S, Arslanhan MD, Langenfeld F, Jeanningros S, Cerny-Reiterer S, Hadzijusufovic E, Tchertanov L, Moriggl R, Valent P, Arock M (2014) Co-operating STAT5 and AKT signaling pathways in chronic myeloid leukemia and mastocytosis: possible new targets of therapy. Haematologica 99:417–429
Bidri M, Royer B, Averlant G, Bismuth G, Guillosson JJ, Arock M (1999) Inhibition of mouse mast cell proliferation and proinflammatory mediator release by benzodiazepines. Immunopharmacology 43:75–86
Biesiekierski JR, Newnham ED, Irving PM, Barrett JS, Haines M, Doecke JD, Shepherd SJ, Muir JG, Gibson PR (2011) Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol 106:508–514
Böhm A, Sonneck K, Gleixner KV, Schuch K, Pickl WF, Blatt K, Peter B, Herrmann H, Schernthaner GH, Pehamberger H, Rabitsch W, Sperr WR, Valent P (2010) In vitro and in vivo growth-inhibitory effects of cladribine on neoplastic mast cells exhibiting the imatinib-resistant KIT mutation D816V. Exp Hematol 38:744–755
Borzutzky A, Morales PS, Mezzano V, Nussbaum S, Burks W (2014) Induction of remission of frequent idiopathic anaphylaxis with rituximab. AAAAI Meeting 28.2.-04.03, abstract 91
Boyce JA (2007) Mast cells and eicosanoid mediators: a system of reciprocal paracrine and autocrine regulation. Immunol Rev 217:168–185
Breslow RG, Caiado J, Castells MC (2009) Acetylsalicylic acid and montelukast block mast cell mediator-related symptoms during rapid desenesitization. Ann Allergy Asthma Immunol 102:155–160
Broesby-Olsen S, Kristensen T, Vestergaard H, Brixen K, Møller MB, Bindslev-Jensen C, Mastocytosis Centre Odense University Hospital (MastOUH) (2013) KIT D816V mutation burden does not correlate to clinical manifestations of indolent systemic mastocytosis. J Allergy Clin Immunol 132:723–728
Broyd H, Koffer A, Assem ESK (2005) Effect of cyclosporin-A on secretion from intact and permeabilised mast cells. Inflamm Res 54(Supplement 1):7–8
Butterfield JH (2005) Interferon treatment for hypereosinophilic syndromes ans systemic mastocytosis. Acta Haematol 114:26–40
Butterfield JH (2009) Survey of aspirin administration in systemic mastocytosis. Prostaglandins Other Lipid Mediat 88:122–124
Butterfield JH, Chen D (2016) Response of patients with indolent systemic mastocytosis to tamoxifen citrate. Leuk Res 40:10–16
Butterfield JH, Weiler CR (2008) Prevention of mast cell activation disorder-associated clinical sequelae of excessive prostaglandin D(2) production. Int Arch Allergy Immunol 147:338–343
Butterfield JH, Tefferi A, Kozuh GF (2005) Successful treatment of systemic mastocytosis with high- dose interferon-alfa: long-term follow-up of a case. Leuk Res 29:131–134
Cardet JC, Akin C, Lee MJ (2013) Mastocytosis: update on pharmacotherapy and future directions. Expert Opin Pharmacother 14:2033–2045
Carter MC, Robyn JA, Bressler PB, Walker JC, Shapiro GG, Metcalfe DD (2007) Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J Allergy Clin Immunol 119:1550–1551
Casassus P, Caillat-Vigneron N, Martin A, Simon J, Gallais V, Beaudry P, Eclache V, Laroche L, Lortholary P, Raphaël M, Guillevin L, Lortholary O (2002) Treatment of adult systemic mastocytosis with interferon-alpha: results of a multicentre phase II trial on 20 patients. Br J Haematol 119:1090–1097
Caughey GH (2016) Mast cell proteases as pharmacological targets. Eur J Pharmacol 778:44–55
Chan IJ, Kasprowicz S, Tharp MD (2013) Distinct signalling pathways for mutated KIT(V560G) and KIT(D816V) in mastocytosis. Clin Exp Dermatol 38:538–544
Chandesris MO, Damaj G, Canioni D, Brouzes C, Cabaret L, Hanssens K, Durieu I, Durupt S, Besnard S, Beyne-Rauzy O, Launay D, Schiffmann A, Niault M, Ranta D, Agape P, Faure C, Chantepie SP, Daguindau N, Bourgeet P, Dubreuil P, Lortholary O, Hermine O (2014) Treatment of advanced systemic mastocytosis with PKC412: the French compassionate use programme experience and historical comparison. ASH Meeting, abstract 3193
Chatterjee IB, Gupta SD, Majumder AK, Nandi BK, Subramanian N (1975) Effect of ascorbic acid on histamine metabolism in scorbutic guinea-pigs. J Physiol 251:271–279
Church MK, Gradidge CF (1980) Inhibition of histamine release from human lung in vitro by antihistamines and related drugs. Br J Pharmacol 69:663–667
Cikler E, Ersoy Y, Cetinel S, Ercan F (2009) The leukotriene d4 receptor antagonist, montelukast, inhibits mast cell degranulation in the dermis induced by water avoidance stress. Acta Histochem 111:112–118
Clemons A, Vasiadi M, Kempuraj D, Kourelis T, Vandoros G, Theoharides TC (2011) Amitriptyline and prochlorperazine inhibit proinflammatory mediator release from human mast cells: possible relevance to chronic fatigue syndrome. J Clin Psychopharmacol 31:385–387
Cohen SS, Skovbo S, Vestergaard H, Kristensen T, Møller M, Bindslev-Jensen C, Fryzek JP, Broesby-Olsen S (2014) Epidemiology of systemic mastocytosis in Denmark. Br J Haematol 166:521–528
Cooper K, Young J, Wadsworth S, Cui H, diZerega GS, Rodgers KE (2007) Reduction of post-surgical adhesion formation with tranilast. J Surg Res 141:153–161
Corbin AS, Griswold IJ, La Rosée P, Yee KW, Heinrich MC, Reimer CL, Druker BJ, Deininger MW (2004) Sensitivity of oncogenic KIT mutants to the kinase inhibitors MLN518 and PD180970. Blood 104:3754–3757
Correia O, Duarte AF, Quirino P, Azevedo R, Delgado L (2010) Cutaneous mastocytosis: two pediatric cases treated with topical pimecrolimus. Dermatol Online J 16:8
Damaj G, Bernit E, Ghez D, Claisse JF, Schleinitz N, Harlé JR, Canioni D, Hermine O (2008) Thalidomide in advanced mastocytosis. Br J Haematol 141:249–253
De Filippis D, D’Amico A, Iuvone T (2008) Cannabinomimetic control of mast cell mediator release: new perspective in chronic inflammation. J Neuroendocrinol 20(Suppl 1):20–25
Dix S, Cord M, Howard S, Coon J, Belt R, Geller R (1999) Safety and efficacy of a continuous infusion, patient controlled anti-emetic pump to facilitate outpatient administration of high-dose chemotherapy. Bone Marrow Transplant 24:561–566
Droogendijk HJ, Kluin-Nelemans HJC, van Doormaal JJ, Oranje AR, van de Loosdrecht AA, van Daele PLA (2006) Imatinibmesylate in the treatment of systemic mastocytosis: a phase II trial. Cancer 107:345–351
Dueñas-Laita A, Ruiz-Muñoz P, Armentia A, Pinacho F, Martín-Armentia B (2009) Successful treatment of chronic drug-resistant urticaria with alprazolam. J Allergy Clin Immunol 123:504–505
Duffy SM, Lawley WJ, Kaur D, Yang W, Bradding P (2003) Inhibition of human mast cell proliferation and survival by tamoxifen in association with ion channel modulation. J Allergy Clin Immunol 112:965–972
Edwards AM, Hagberg H (2010) Oral and inhaled sodium cromoglicate in the management of systemic mastocytosis: a case report. J Med Case Rep 4:193
Edwards AM, Stevens MT, Church MK (2011) The effects of topical sodium cromoglicate on itch and flare in human skin induced by intradermal histamine: a randomised double-blind vehicle controlled intra-subject design trial. BMC Res Notes 4:47
El-Agamy DS (2012) Anti-allergic effects of nilotinib on mast cell-mediated anaphylaxis like reactions. Eur J Pharmacol 680:115–121
El-Feki G, X Zhou, LC Lau, J Pedersen, AF Walls (2011) Inhibitors of dipeptidyl peptidase I (DPPI) as mast cell stabilising agents: the contribution of DPPI in mast cell activation. J Allergy Clin Immunol 127, Suppl. – Abstracts
Erben P, Schwaab J, Metzgeroth G, Horny HP, Jawhar M, Sotlar K, Fabarius A, Teichmann M, Schneider S, Ernst T, Müller MC, Giehl M, Marx A, Hartmann K, Hochhaus A, Hofmann WK, Cross NC, Reiter A (2014) The KIT D816V expressed allele burden for diagnosis and disease monitoring of systemic mastocytosis. Ann Hematol 93:81–88
Escribano L, Akin C, Castells M, Schwartz LB (2006) Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets 5:61–77
Eskandari N, Bastan R, Peachell PT (2015) Regulation of human skin mast cell histamine release by PDE inhibitors. Allergol Immunopathol (Madr) 43:37–41
Estévez MD, Vieytes MR, Botana LM (1996) Mitoxantrone induces nonimmunological histamine release from rat mast cells. Inflamm Res 45:113–117
Evans E, Gardino A, Hodous B, Davis A, Zhu J, Kohl NE, Lengauer C (2015) Blu-285, a potent and selective inhibitor for hematologic malignancies with KIT exon 17 mutations. ASH Annual Meeting: abstract 568
Evora PR, Simon MR (2007) Role of nitric oxide production in anaphylaxis and its relevance for the treatment of anaphylactic hypotension with methylene blue. Ann Allergy Asthma Immunol 99:306–313
Facci L, Dal Toso R, Romanello S, Buriani A, Skaper SD, Leon A (1995) Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc Natl Acad Sci U S A 92:3376–3380
Finn DF, Walsh JJ (2013) Twenty-first century mast cell stabilisers. Br J Pharmacol 170:23–37
Frenkel A, Roy-Shapira A, Evgeni B, Leonid K, Borer A, Klein M (2015) Life threatening idiopathic recurrent angioedema responding to cannabis. Case Rep Immunol 2015:780824
Friedman BS, Santiago ML, Berkebile C, Metcalfe DD (1993) Comparison of azelastine and chlorpheniramine in the treatment of mastocytosis. J Allergy Clin Immunol 92:520–526
Frieri M, Alling DW, Metcalfe DD (1985) Comparison of the therapeutic efficacy of cromolyn sodium with that of combined chlorpheniramine and cimetidine in systemic mastocytosis. Results of a double-blind clinical trial. Am J Med 78:9–14
Fujimoto T, Nishiyama T, Hanaoka K (2005) Inhibitory effects of intravenous anesthetics on mast cell function. Anesth Analg 101:1054–1059
Fumo G, Akin C, Metcalfe DD, Neckers L (2004) 17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells. Blood 103:1078–1084
Galli SJ, Costa JJ (1995) Mast-cell-leukocyte cytokine cascades in allergic inflammation. Allergy 50:851–862
Gibbs BF, Schmutzler W, Vollrath IB, Brosthardt P, Braam U, Wolff HH, Zwadlo-Klarwasser G (1999) Ambroxol inhibits the release of histamine, leukotrienes and cytokines from human leukocytes and mast cells. Inflamm Res 48:86–93
Giraldo Castellano P, García-Erce JA, Alvarez Alegret R, Arroyo Rubio A, Mayayo Artal P, Vicente Cámara P, Rubio-Félix D, Giralt RM (1998) Interferon alpha and systemic mastocytosis. Analysis of therapeutic efficacy in 6 cases. Rev Clin Esp 198:345–350
Gleixner KV, Mayerhofer M, Cerny-Reiterer S, Hörmann G, Rix U, Bennett KL, Hadzijusufovic E, Meyer RA, Pickl WF, Gotlib J, Horny HP, Reiter A, Mitterbauer-Hohendanner G, Superti-Furga G, Valent P (2011) KIT-D816V-independent oncogenic signaling in neoplastic cells in systemic mastocytosis: role of Lyn and Btk-activation and disruption by dasatinib and bosutinib. Blood 118:1885–1898
Gleixner KV, Peter B, Blatt K, Suppan V, Reiter A, Radia D, Hadzijusufovic E, Valent P (2013) Synergistic growth-inhibitory effects of ponatinib and midostaurin (PKC412) on neoplastic mast cells carrying KIT D816V. Haematologica 98:1450–1457
Gotlib J, George TI, Corless C, Linder A, Ruddell A, Akin C, DeAngelo DJ, Kepten I, Lanza C, Heinemann H, Yin O, Gallagher N, Graubert T (2008) The KIT tyrosine kinase inhibitor midostaurine (PKC412) exhibits a high response rate in aggressive systemic mastocytosis (ASM): interim results of a phase II trial. Blood 110:1039A
Gotlib J, Kluin-Neelemans HC, George TI et al. (2014) Midostaurin (PKC412) demonstrates a high rate of durable responses in patients with advanced systemic mastocytosis: results from the fully accrued global phase 2 CPKC412D2201 trial. Blood;124: ASH Annual Meeting Abstracts; Abstract 636
Gri G, Frossi B, D’Inca F, Danelli L, Betto E, Mion F, Sibilano R, Pucillo C (2012) Mast cell: an emerging partner in immune interaction. Front Immunol 3:120
Gromke T, Elmaagacli AH, Ditschkowski M, Hegerfeldt Y, Koldehoff M, Hlinka M, Ottinger H, Trenschel R, Beelen DW (2013) Delayed graft-versus-mast-cell effect on systemic mastocytosis with associated clonal haematological non-mast cell lineage disease after allogeneic transplantation. Bone Marrow Transplant 48:732–733
Gruson B, Lortholary O, Canioni D, Chandesris O, Lanternier F, Bruneau J, Grosbois B, Livideanu C, Larroche C, Durieu I, Barete S, Sevestre H, Diouf M, Chaby G, Marolleau JP, Dubreuil P, Hermine O, Damaj G (2013) Thalidomide in systemic mastocytosis: results from an open-label, multicentre, phase II study. Br J Haematol 161:434–442
Gurgel JA, Lima-Júnior RC, Rabelo CO, Pessoa BB, Brito GA, Ribeiro RA (2013) Amitriptyline, clomipramine, and maprotiline attenuate the inflammatory response by inhibiting neutrophil migration and mast cell degranulation. Rev Bras Psiquiatr 35:387–392
Hadzijusufovic E, Peter B, Herrmann H, Schuch, Thaiwong T, Yuzbasiyan-Gurkan V, Pickl W, Willmann M, Valent P (2010) Mutations in KIT predict resistance to several TKI (sorafenib, sunitinib, and masitinib) in neoplastic human and canine mast cell lines. Haematologica 95(suppl.2):357 abs. 0857
Haenisch B, Nöthen MM, Molderings GJ (2012) Systemic mast cell activation disease: the role of molecular genetic alterations in pathogenesis, heritability and diagnostics. Immunology 137:197–205
Haenisch B, Fröhlich H, Herms S, Molderings GJ (2014) Evidence for contribution of epigenetic mechanisms in the pathogenesis of systemic mast cell activation disease. Immunogenetics 66:287–297
Hagel AF, Layritz CM, Hagel WH, Hagel HJ, Hagel E, Dauth W, Kressel J, Regnet T, Rosenberg A, Neurath MF, Molderings GJ, Raithel M (2013) Intravenous infusion of ascorbic acid decreases serum histamine concentrations in patients with allergic and non-allergic diseases. Naunyn Schmiedeberg’s Arch Pharmacol 386:789–793
Hagenlocher Y, Kießling K, Schäffer M, Bischoff SC, Lorentz A (2015) Cinnamaldehyde is the main mediator of cinnamon extract in mast cell inhibition. Eur J Nutr 54:1297–1309
Hamilton MJ, Hornick JL, Akin C, Castells MC, Greenberger NJ (2011) Mast cell activation syndrome: a newly recognized disorder with systemic clinical manifestations. J Allergy Clin Immunol 128:147–152
Hantschel O, Rix U, Schmidt U, Bürckstümmer T, Kneidinger M, Schütze G, Colinge J, Bennett KL, Ellmeier W, Valent P, Superti-Furga G (2007) The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc Natl Acad Sci U S A 104:13283–13288
Harvima IT, Levi-Schaffer F, Draber P, Friedman S, Polakovicova I, Gibbs BF, Blank U, Nilsson G, Maurer M (2014) Molecular targets on mast cells and basophils for novel therapies. J Allergy Clin Immunol 134:530–544
Hauswirth AW, Simonitsch-Klupp I, Uffmann M, Koller E, Sperr WR, Lechner K, Valent P (2004) Response to therapy with interferon alpha-2b and prednisolone in aggressive systemic mastocytosis: report of five cases and review of the literature. Leuk Res 28:249–257
Heinrich MC, Joensuu H, Demetri GD, Corless CL, Apperley J, Fletcher JA, Soulieres D, Dirnhofer S, Harlow A, Town A, McKinley A, Supple SG, Seymour J, Di Scala L, van Oosterom A, Herrmann R, Nikolova Z, McArthur AG, Imatinib Target Exploration Consortium Study B2225 (2008) Phase II, open-label study evaluating the activity of imatinib in treating life-threatening malignancies known to be associated with imatinib-sensitive tyrosine kinases. Clin Cancer Res 14:2717–2725
Hennessy B, Giles F, Cortes J, O’brien S, Ferrajoli A, Ossa G, Garcia-Manero G, Faderl S, Kantarjian H, Verstovsek S (2004) Management of patients with systemic mastocytosis: review of M. D. Anderson Cancer Center experience. Am J Hematol 77:209–214
Hermine O, Lortholary O, Leventhal PS, Catteau A, Soppelsa F, Baude C, Cohen-Akenine A, Palmérini F, Hanssens K, Yang Y, Sobol H, Fraytag S, Ghez D, Suarez F, Barete S, Casassus P, Sans B, Arock M, Kinet JP, Dubreuil P, Moussy A (2008) Case-control cohort study of patients’ perceptions of disability in mastocytosis. PLoS One 3:e2266
Hochhaus A, Ottmann OG, Lauber S, Hughes T, Verhoef G, Schwarer AP, Gratwohl A, Rafferty T, Resta D, Gattermann N (2006) A phase II study of nilotinib, a novel inhibitor of c-Kit, PDGFR, and Bcr-Abl, administered to patients with systemic mastocytosis. Blood 108: Abstract 2703 [ASH Annual Meeting Abstracts]
Hochhaus A, Baccarani M, Giles FJ, le Coutre PD, Müller MC, Reiter A, Santanastasio H, Leung M, Novick S, Kantarjian HM (2015) Nilotinib in patients with systemic mastocytosis: analysis of the phase 2, open-label, single-arm nilotinib registration study. J Cancer Res Clin Oncol 141:2047–2060
Hoffmann K, Xifró RA, Hartweg JL, Spitzlei P, Meis K, Molderings GJ, von Kügelgen I (2013) Inhibitory effects of benzodiazepines on the adenosine A(2B) receptor mediated secretion of interleukin-8 in human mast cells. Eur J Pharmacol 700:152–158
Horan RF, Sheffer AL, Austen KF (1990) Cromolyn sodium in the management of systemic mastocytosis. J Allergy Clin Immunol 85:852–855
Ibelgaufts H. (2016.) “Mast Cells” in COPE: cytokines and cells online pathfinder encyclopaedia, Available at http://www.cells-talk.com (accessed March 21, 2016)
Jensen BM, Beaven MA, Iwaki S, Metcalfe DD, Gilfillan A (2008) Concurrent inhibition of KIT- and FcεRI-mediated signaling: coordinated suppression of mast cell activation. J Pharmacol Exp Ther 324:128–138
Jin B, Ding K, Pan J (2014) Ponatinib induces apoptosis in imatinib-resistant human mast cells by dephosphorylating mutant D816V KIT and silencing beta-catenin signaling. Mol Cancer Ther 13:1217–1230
Johnston CS, Martin LJ, Cai X (1992) Antihistamine effect of supplemental ascorbic acid and neutrophil chemotaxis. J Am Coll Nutr 11:172–176
Joks R, Durkin HG (2011) Non-antibiotic properties of tetracyclines as anti-allergy and asthma drugs. Pharmacol Res 64:602–609
Jones E, Koyama T, Ho RH, Kuttesch J, Shankar S, Whitlock JA, Cartwright J, Frangoul H (2007) Safety and efficacy of a continuous infusion, patient-controlled antiemetic pump for children receiving emetogenic chemotherapy. Pediatr Blood Cancer 48:330–332
Kaneko I, Suzuki K, Matsuo K, Kumagai H, Owada Y, Noguchi N, Hishinuma T, Ono M (2009) Cysteinyl leukotrienes enhance the degranulation of bone marrow-derived mast cells through the autocrine mechanism. Tohoku J Exp Med 217:185–191
Karlberg M, Ekoff M, Labi V, Strasser A, Huang D, Nilsson G (2010a) Pro-apoptotic Bax is the major and Bak an auxiliary effector in cytokine deprivation-induced mast cell apoptosis. Cell Death Dis 1:e43
Karlberg M, Ekoff M, Huang DC, Mustonen P, Harvima IT, Nilsson G (2010b) The BH3-mimetic ABT-737 induces mast cell apoptosis in vitro and in vivo: potential for therapeutics. J Immunol 185:2555–2562
Katoh N, Hirano S, Yasuno H (1996) Solitary mastocytoma treated with tranilast. J Dermatol 23:335–339
Kay LJ, Yeo WW, Peachell PT (2006) Prostaglandin E-2 activates EP2 receptors to inhibit human lung mast cell degranulation. Br J Pharmacol 147:707–713
Kempna P, Reiter E, Arocks M, Azzi A, Zingg JM (2004) Inhibition of HMC-1 mast cell proliferation by vitamin E. J Biol Chem 279:50700–50709
Kempuraj D, Castellani ML, Petrarca C, Frydas S, Conti P, Theoharides TC, Vecchiet J (2006) Inhibitory effect of quercetin on tryptase and inetrleukin-6 release, and histidine decarboxylase mRNA transcription by human mast cell-1 cell line. Clin Exp Med 6:150–156
Kettelhut BV, Berkebile C, Bradley D, Metcalfe DD (1989) A double-blind, placebo-controlled, crossover trial of ketotifen versus hydroxyzine in the treatment of pediatric mastocytosis. J Allergy Clin Immunol 83:866–870
Kibsgaard L, Skjold T, Deleuran M, Vestergaard C (2014) Omalizumab induced remission of idiopathic anaphylaxis in a patient suffering from indolent systemic mastocytosis. Acta DermVenereol 94:363–364
Kinney SR, Carlson L, Ser-Dolansky J, Thompson C, Shah S, Gambrah A, Xing W, Schneider SS, Mathias CB (2015) Curcumin ingestion inhibits mastocytosis and suppresses intestinal anaphylaxis in a murine model of food allergy. PLoS One 10:e0132467
Kluin-Nelemans HC, Oldhoff JM, Van Doormaal JJ, Van’t Wout JW, Verhoef G, Gerrits WB, van Dobbenburgh OA, Pasmans SG, Fijnheer R (2003) Cladribine therapy for systemic mastocytosis. Blood 102:4270–4276
Kluin-Nelemans HC, Ferenc V, van Doormaal JJ, van Iperen C, Peters WG, Akin C, Valent P (2009) Lenalidomide therapy in systemic mastocytosis. Leuk Res 33:e19–e22
Knapper S, Cullis J, Drummond MW, Evely R, Everington T, Hoyle C, McLintock L, Poynton C, Radia D (2011) Midostaurin a multi-targeted oral kinase inhibitor in systemic mastocytosis: report of an open-label compassionate use program in the United Kingdom. Blood 118:5145
Koelink PJ, Overbeek SA, Braber S, de Kruijf P, Folkerts G, Smit MJ, Kraneveld AD (2012) Targeting chemokine receptors in chronic inflammatory diseases: an extensive review. Pharmacol Ther 133:1–18
Kohno M, Yamasaki S, Tybolewicz VLJ, Saito T (2005) Rapid and large amount of autocrine IL-3 production is responsible for mast cell survival by IgE in the absence of antigen. Blood 105:2059–2065
Kontou-Fili K, Filis CI, Voulgari C, Panayiotidis PG (2010) Omalizumab monotherapy for bee sting and unprovoked “anaphylaxis” in a patient with systemic mastocytosis and undetectable specific IgE. Ann Allergy Asthma Immunol 104:537–539
Krauth MT, Majlesi Y, Sonneck K, Samorapoompichit P, Ghannadan M, Hauswirth AW, Baghestanian M, Schernthaner GH, Worda C, Muller MR, Sperr WR, Valent P (2006) Effects of various statins on cytokine-dependent growth and IgE-dependent release of histamine in human mast cells. Allergy 61:281–288
Krauth MT, Böhm A, Agis H, Sonneck K, Samorapoompichit P, Florian S, Sotlar K, Valent P (2007) Effects of the CD33-targeted drug gemtuzumab ozogamicin (Mylotarg) on growth and mediator secretion in human mast cells and blood basophils. Exp Hematol 35:108–116
Krug U, Lübbert M, Büchner T (2010) Maintenance therapy in acute myeloid leukemia revisited: will new agents rekindle an old interest? Curr Opin Hematol 17:85–90
Kurosawa M, Amano H, Kanbe N, Igarashi Y, Nagata H, Yamashita T, Kurimoto F, Miyachi Y (1999) Response to cyclosporin and low-dose methylprednisolone in aggressive systemic mastocytosis. J Allergy Clin Immunol 103:S412–S420
Kurzen H. (2009) Neramexane for the treatment of mast cellmediated diseases. Patent application WO2010/069595
Kvasnicka HM, Thiele J, Bueso-Ramos CE, Kamalanabhaiah S, Cortes JE, Kantajian H, Verstovsek S (2014) Changes in activated bone marrow macrophages and mast cells in patients with myelofibrosis following ruxolitinib therapy. ASH Meeting abstract 3184
Laengle UW, Markstein R, Pralet D, Seewald W, Roman D (2006) Effect of GLC756, a novel mixed dopamine D1 receptor antagonist an dopamine D2 receptor agonist, on TNF-alpha release in vitro from activated rat mast cells. Exp Eye Res 83:1335–1339
Laroche M, Livideanu C, Paul C, Cantagrel A (2011) Interferon alpha and pamidronate in osteoporosis with fracture secondary to mastocytosis. Am J Med 124:776–778
Lasho T, Tefferi A, Pardanani A (2010) Inhibition of JAK-STAT signaling by TG101348: a novel mechanism for inhibition of KITD816V-dependent growth in mast cell leukemia cells. Leukemia 24:1378–1380
Lasho T, Finke C, Zblewski D et al. (2016) Concurrent activating KIT mutations in systemic mastocytosis. Br J Haematol 173:153–156
Lau HY, Kam MF (2005) Inhibition of mast cell histamine release by specific phosphodiesterase inhibitors. Inflamm Res 54(Supplement 1):05–06
Lechowski S, Feilhauer K, Staib L, Coëffier M, Bischoff SC, Lorentz A (2013) Combined arginine and glutamine decrease release of de novo synthesized leukotrienes and expression of proinflammatory cytokines in activated human intestinal mast cells. Eur J Nutr 52:505–512
Lee YS, Kim MS, Lee DH, Kwon TH, Song HH, Oh SR, do Yoon Y (2015) Luteolin 8-C-β-fucopyranoside downregulates IL-6 expression by inhibiting MAPKs and the NF-κB signaling pathway in human monocytic cells. Pharmacol Rep 67:581–587
Lieberoth S, Thomsen SF (2015) Cutaneous and gastrointestinal symptoms in two patients with systemic mastocytosis successfully treated with omalizumab. Case Rep Med 2015:903541
Lim KH, Pardanani A, Butterfield JH, Li CY, Tefferi A (2009) Cytoreductive therapy in 108 adults with systemic mastocytosis: outcome analysis and response prediction during treatment with interferon-alpha, hydroxyurea, imatinib mesylate or 2-chlorodeoxyadenosine. Am J Hematol 84:790–794
Lin TY, Bear M, Du Z, Foley KP, Ying W, Barsoum J, London C (2008) The novel HSP90 inhibitor STA-9090 exhibits activity against KIT-dependent and -independent malignant mast cell tumors. Exp Hematol 36:1266–1277
Lin TY, Fenger J, Murahari S, Bear MD, Kulp SK, Wang D, Chen CS, Kisseberth WC, London CA (2010) AR-42, a novel HDAC inhibitor, exhibits biologic activity against malignant mast cell lines via down-regulation of constitutively activated KIT. Blood 115:4217–4225
Lock AD, McNamara CJ, Rustin MH (2015) Sustained improvement in urticaria pigmentosa and pruritus in a case of indolent systemic mastocytosis treated with cladribine. Clin Exp Dermatol 40:142–145
Lundequist A, Pejler G (2011) Biological implications of preformed mast cell mediators. Cell Mol Life Sci 68:965–975
Ma Z, Tovar JP, Kwong KY, Paek D (2010) Pimecrolimus induces apoptosis of mast cells in a murine model of cutaneous mastocytosis. Int Arch Allergy Immunol 153:413–418
Mallet AI, Norris P, Rendell NB, Wong E, Greaves MW (1989) The effect of disodium cromoglycate and ketotifen on the excretion of histamine and N tau-methylimidazole acetic acid in urine of patients with mastocytosis. Br J Clin Pharmacol 27:88–91
Marech I, Patruno R, Zizzo N, Gadaleta C, Introna M, Zito AF, Gadaleta CD, Ranieri G (2014) Masitinib (AB1010), from canine tumor model to human clinical development: where we are? Crit Rev Oncol Hematol 91:98–111
Marquardt DL, Gruber HE, Walker LL (1987) Ribavirin inhibits mast cell mediator release. J Pharmacol Exp Ther 240:145–149
Marton I, Pósfai É, Borbényi Z, Bödör C, Papp G, Demeter J, Korom I, Varga E, Bata-Csörgő Z (2015) Therapeutic challenge during the long-term follow-up of a patient with indolent systemic mastocytosis with extensive cutaneous involvement. Eur Rev Med Pharmacol Sci 19:1607–1609
Matsubara S, Li G, Takeda K, Loader JE, Pine P, Masuda ES, Miyahara N, Miyahara S, Lucas JJ, Dakhama A, Gelfand EW (2006) Inhibition of spleen tyrosine kinase prevents mast cell activation and airway hyperresponsiveness. Am J Respir Crit Care Med 173:56–63
Mattace Raso G, Russo R, Calignano A, Meli R (2014) Palmitoylethanolamide in CNS health and disease. Pharmacol Res 86:32–41
Maurer M, Magerl M, Metz M, Weller K, Siebenhaar F (2013) Miltefosine: a novel treatment option for mast cell-mediated diseases. J Dermatolog Treat 24:244–249
McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, Dong X (2015) Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 519:237–241
Meeran SM, Ahmed A, Tollefsbol TO (2010) Epigenetic targets of bioactive dietary components for cancer prevention and therapy. Clin Epigenetics 1:101–116
Min YD, Choi CH, Bark H, Son HY, Park HH, Lee S, Park JW, Park EK, Shin HI, Kim SH (2007) Quercetin inhibits expression of inflammatory cytokines through attenuation of NF-kappaB and p38 MAPK in HMC-1 human mast cell line. Inflamm Res 56:210–215
Molderings GJ (2015) The genetic basis of mast cell activation disease—looking through a glass darkly. Crit Rev Oncol Hematol 93:75–89
Molderings GJ (2016) Transgenerational transmission of systemic mast cell activation disease-genetic and epigenetic features. Transl Res.
Molderings GJ, Kolck UW, Scheurlen C, Brüss M, Homann J, Von Kügelgen I (2007) Multiple novel alterations in KIT tyrosine kinase in patients with gastrointestinally pronounced systemic mast cell activation disorder. Scand J Gastroenterol 42:1045–1053
Molderings GJ, Meis K, Kolck UW, Homann J, Frieling T (2010) Comparative analysis of mutation of tyrosine kinase KIT in mast cells from patients with systemic mast cell activation syndrome and healthy subjects. Immunogenetics 62:721–727
Molderings GJ, Brettner S, Homann J, Afrin LB (2011a) Mast cell activation disease: a concise practical guide for diagnostic workup and therapeutic options. J Hematol Oncol 4:10
Molderings GJ, Raithel M, Kratz F, Azemar M, Haenisch B, Harzer S, Homann J (2011b) Omalizumab treatment of systemic mast cell activation disease: experiences from four cases. Intern Med 50:1–5
Molderings GJ, Haenisch B, Bogdanow M, Fimmers R, Nöthen MM (2013a) Familial occurrence of systemic mast cell activation disease. PLoS One 8:e76241
Molderings GJ, Haenisch B, Homann J, Wilhelm T, Huber M (2013b) Neue Angriffspunkte der 1-4-Benzodiazepine in der mastzelspezifischen immunsuppressiven Therapie der sytemischen Mastzellüberaktivitätserkrankung. Gastroenterologe 8:170 abstract
Molderings GJ, Homann J, Brettner S, Raithel M, Frieling T (2014) Mast cell activation disease: a concise practical guide for diagnostic workup and therapeutic options. Dtsch Med Wochenschr 139:1523–1534
Moon PD, Lee BH, Jeong HJ, An HJ, Park SJ, Kim HR, Ko SG, Um JY, Hong SH, Kim HM (2007) Use of scopoletin to inhibit the production of inflammatory cytokines through inhibition of the IkB/NF-kB signal cascade in the human mast cell line HMC-1. Eur J Pharmacol 555:218–225
Mousli M, Hugli TE, Landry Y, Bronner C (1994) Peptidergic pathway in human skin and rat peritoneal mast cell activation. Immunopharmacology 27:1–11
Moussy A, Kinet JP (2014) Treatment of mastocytosis with masitinib. US Patent Application
Mühlenberg T, Zhang Y, Wagner AJ, Grabellus F, Bradner J, Taeger G, Lang H, Taguchi T, Schuler M, Fletcher JA, Bauer S (2009) Inhibitors of deacetylases suppress oncogenic KIT signaling, acetylate HSP90, and induce apoptosis in gastrointestinal stromal tumors. Cancer Res 69:6941–6950
Nader MA (2011) Inhibition of anaphylaxis like reaction and mast cell activation by sitagliptin. Int Immunopharmacol 11:1052–1056
Nakamura R, Chakrabarti S, Akin C, Robyn J, Bahceci E, Greene A, Childs R, Dunbar CE, Metcalfe DD, Barrett AJ (2006) A pilot study of nonmyeloablative allogeneic hematopoietic stem cell transplant for advanced systemic mastocytosis. Bone Marrow Transplant 37:353–358
Nolte H, Stahl Skov P (1988) Inhibition of basophil histamine release by methotrexate. Agents Actions 23:173–176
Nurmatov UB, Rhatigan E, Simons FE, Sheikh A (2015) H1-antihistamines for primary mast cell activation syndromes: a systematic review. Allergy 70:1052–1061
Oppong E, Flink N, Cato AC (2013) Molecular mechanisms of glucocorticoid action in mast cells. Mol Cell Endocrinol 380:119–126
Otani IM, Bhagat M, Newbury RO, Dohil R, Broide DH, Aceves SS (2012) The effect of anti-IL-5 therapy on esophageal mastocytosis in pediatric eosinophilic esophagitis. J Allergy Clin Immunol 129:AB202
Paez P, Ryan J, Taruselli M, Ndaw V (2015) Fluvastatin elicits apoptosis in primary and transformed mast cells (INC6P.305). J Immunol 194(Suppl. 1):197.7
Pagano L, Valentini CG, Caira M, Rondoni M, Van Lint MT, Candoni A, Allione B, Cattaneo C, Marbello L, Caramatti C, Pogliani EM, Iannitto E, Giona F, Ferrara F, Invernizzi R, Fanci R, Lunghi M, Fianchi L, Sanpaolo G, Stefani PM, Pulsoni A, Martinelli G, Leone G, Musto P (2008) Advanced mast cell disease: an Italian Hematological Multicenter experience. Int J Hematol 88:483–488
Paivandy A, Calounova G, Zarnegar B, Ohrvik H, Melo FR, Pejler G (2014) Mefloquine, an anti-malaria agent, causes reactive oxygen species-dependent cell death in mast cells via a secretory granule-mediated pathway. Pharmacol Res Perspect 2:e00066
Pan J, Quintas-Cardama A, Kantarjian HM, Akin C, Manshouri T, Lamb P, Cortes JE, Tefferi A, Giles FJ, Verstovsek S (2007) EXEL-0862, a novel tyrosine kinase inhibitor, induces apoptosis in vitro and ex vivo in human mast cells expressing the KIT D816V mutation. Blood 109:315–322
Papaetis GS, Syrigos KN (2009) Sunitinib: a multitargeted receptor tyrosine kinase inhibitor in the era of molecular cancer therapies. BioDrugs 23:377–389
Papayannidis C, Soverini S, Benedittis CD, Abbenante MC, Sartor C, Iacobucci I, Baldazzi C, Ottaviani E, Ferrari A, Guadagnuolo V, Conficoni A, Paolini S, Parisi S, Frabetti F, Piccari S, Grilli S, Lani E, Martinelli G (2014) PKC412 (midostaurin) is safe and highly effective in systemic mastocytosis patients: follow up of a single-center Italian compassionate use. Cancer Res 74:746
Pardanani A (2013) Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol 88:612–624
Pardanani A, Elliott M, Reeder T, Li CY, Baxter EJ, Cross NC, Tefferi A (2003) Imatinib for systemic mast-cell disease. Lancet 362:535–536
Pardanani A, Hoffbrand AV, Butterfield JH, Tefferi A (2004) Treatment of systemic mast cell disease with 2-chlorodeoxyadenosine. Leuk Res 28:127–131
Parikh SA, Kantarjian HM, Richie MA, Cortes JE, Verstovsek S (2010) Experience with everolimus (RAD001), an oral mammalian target of rapamycin inhibitor, in patients with systemic mastocytosis. Leuk Lymphoma 51:269–274
Park TH (2013) The effects of botulinum toxin A on mast cell activity: preliminary results. Burns 39:816–817
Paul C, Sans B, Suarez F, Casassus P, Barete S, Lanternier F, Grandpeix-Guyodo C, Dubreuil P, Palmérini F, Mansfield CD, Gineste P, Moussy A, Hermine O, Lortholary O (2010) Masitinib for the treatment of systemic and cutaneous mastocytosis with handicap: a phase 2a study. Am J Hematol 85:921–925
Peter B, Hadzijusufovic E, Blatt K, Gleixner KV, Pickl WF, Thaiwong T, Yuzbasiyan-Gurkan V, Willmann M, Valent P (2010) KIT polymorphisms and mutations determine responses of neoplastic mast cells to bafetinib (INNO-406). Exp Hematol 38:782–791
Peter B, Gleixner KV, Cerny-Reiterer S, Herrmann H, Winter V, Hadzijusufovic E, Ferenc V, Schuch K, Mirkina I, Horny HP, Pickl WF, Mullauer L, Willmann M, Valent P (2011) Polo-like kinase-1 as novel target in neoplastic mast cells: demonstration of growth-inhibitory effects of siRNA and the Polo-like kinase-1 targeting drug BI 2536. Haematologica 96(5):672–680
Picard M, Giavina-Bianchi P, Mezzano V, Castells M (2013) Expanding spectrum of mast cell activation disorders: monoclonal and idiopathic mast cell activation syndromes. Clin Ther 35:548–562
Purtill D, Cooney J, Sinniah R, Carnley B, Cull G, Augustson B, Cannell P (2008) Dasatinib therapy for systemic mastocytosis: four cases. Eur J Haematol 80:456–458
Quintás-Cardama A, Jain N, Verstovsek S (2011) Advances and controversies in the diagnosis, pathogenesis, and treatment of systemic mastocytosis. Cancer 117:5439–5449
Quintás-Cardama A, Sever M, Cortes J, Kantarjian H, Verstovsek S (2013) Bone marrow mast cell burden and serum tryptase level as markers of response in patients with systemic mastocytosis. Leuk Lymphoma 54:1959–1964
Radojković M, Ristić S, Colović N, Terzić T, Colović M (2011) Response to cladribine in patient with systemic mastocytosis. Vojnosanit Pregl 68:444–446
Randall N, Courville EL, Baughn L, Afrin L, Ustun C (2015) Bosutinib, a lyn/btk inhibiting tyrosine kinase inhibitor, is ineffective in advanced systemic mastocytosis. Am J Hematol 90:E74
Ritter M, El-Nour H, Hedblad MA, Butterfield JH, Beck O, Stephanson N, Holst M, Giscombe R, Azmitia EC, Nordlind K (2012) Serotonin and its 5-HT1 receptor in human mastocytosis. Immunopharmacol Immunotoxicol 34:679–685
Rodrigo L, Perez-Martinez I, Lucendo AJ (2013) Urticaria pigmentosa in a female patient wth celiac disease: response to a gluten-free diet. Allergol Immunopathol (Madr) 41:128–130
Rodrigues JM, Pazin Filho A, Rodrigues AJ, Vicente WV, Evora PR (2007) Methylene blue for clinical anaphylaxis treatment: a case report. Sao Paulo Med J 125:60–62
Rodriguez T, Pfeffer M, Levy BD, Castells M (2011) Lipid mediators in cutaneous and systemic mastocytosis and the impact of 5-LO inhibition. J Allergy Clin Immunol 127(Suppl):AB132
Rosen H, Goetzl EJ (2005) Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol 5:560–570
Ruano I, Gargini R, Izquierdo M (2010) Combination of KIT gene silencing and tocopherol succinate may offer improved therapeutic approaches for human mastocytosis. Br J Haematol 148:59–68
Sagi L, Solomon M, Baum S, Lyakhovitsky A, Trau H, Barzilai A (2011) Evidence for methotrexate as a useful treatment for steroid-dependent chronic urticaria. Acta Derm Venereol 91:303–306
Sandler C, Nurmi K, Lindstedt KA, Sorsa T, Golub LM, Kovanen PT, Eklund KK (2005) Chemically modified tetracyclines induce apoptosis in cultured mast cells. Int Immunohistopharmacol 5:1611–1621
Schittenhelm MM, Akmut F, Illing B, Frey J, Schuster K, Ramachandran A, Kanz L, Kampa-Schittenhelm KM (2014) Gain-of-function KIT mutations sensitize the mutant isoform to the type I tyrosine kinase inhibitor crenolanib: a rationale for the therapeutic use in systemic mastocytosis (SM) and core binding factor leukemias (CBFL). ASH-Meeting abstract 2230
Seifert R (2015) How do basic secretagogues activate mast cells? Naunyn Schmiedeberg’s Arch Pharmacol 388:279–281
Shimoda T, Liang Z, Suzuki H, Kawana S (2010) Inhibitory effects of antipsychotic and anxiolytic agents on stress-induced degranulation of mouse dermal mast cells. Clin Exp Dermatol 35:531–536
Sido B, Dumoulin FL, Homann J, Hertfelder HJ, Bollmann M, Molderings GJ (2014) Surgical interventions in patients with mast cell activation disease. Aspects relevant for surgery using the example of a cholecystectomy. Chirurg 85:327–333
Siebenhaar F, Förtsch A, Krause K, Weller K, Metz M, Magerl M, Martus P, Church MK, Maurer M (2013) Rupatadine improves quality of life in mastocytosis: a randomized, double-blind, placebo-controlled trial. Allergy 68:949–952
Simhadri VR, Andersen JF, Calvo E, Choi SC, Coligan JE, Borrego F (2012) Human CD300a binds to phosphatidylethanolamine and phosphatidylserine, and modulates the phagocytosis of dead cells. Blood 119:2799–2809
Simon J, Lortholary O, Caillat-Vigneron N, Raphael M, Martin A, Briere J, Barete S, Hermine O, Casussus P (2004) Interest of interferon alpha in systemic mastocytosis. The french experience and review of the literature. Pathol Biol 52:294–299
Soter NA, Austen KF, Wasserman SI, Soter NA, Austen KF, Wasserman SI (1979) Oral disodium cromoglycate in the treatment of systemic mastocytosis. N Engl J Med 301:465–469
Spirkoski J, Melo FR, Grujic M, Calounova G, Lundequist A, Wernersson S, Pejler G (2012) Mast cell apoptosis induced by siramesine, a sigma-2 receptor agonist. Biochem Pharmacol 84:1671–1680
Spyridonidis A, Thomas AK, Bertz H, Zeiser R, Schmitt-Graff A, Lindemann A, Waller CF, Finke J (2004) Evidence for a graft-versus-mast-cell effect after allogeneic bone marrow transplantation. Bone Marrow Transplant 34:515–519
Strati P, Kantarjian H, Ravandi F, Nazha A, Borthakur G, Daver N, Kadia T, Estrov Z, Garcia-Manero G, Konopleva M, Rajkhowa T, Durand M, Andreeff M, Levis M, Cortes J (2015) Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am J Hematol 90:276–281
Suzuki-Nishimura T, Sano T, Uchida MK (1989) Effects of benzodiazepines on serotonin release from rat mast cells. Eur J Pharmacol 167:75–85
Tachibana M, Wada K, Katayama K, Kamisaki Y, Maeyama K, Kadowaki T, Blumberg RS, Nakajima A (2008) Activation of peroxisome proliferator-activated receptor gamma suppresses mast cell maturation involved in allergic diseases. Allergy 63:1136–1147
Tanaka A, Konno M, Muto S, Kambe N, Morii E, Nakahata T, Itai A, Matsuda H (2005) A novel NF-kappa B inhibitor, IMD-0354, suppresses neoplastic proliferation of human mast cells with constitutively activated c-Kit receptors. Blood 105:2324–2331
Tang C, Lan C, Wang C, Liu R (2005) Amelioration of the development of multiple organ dysfunction syndrome by somatostatin via suppression of intestinal mucosal mast cells. Shock 23:470–475
Tanimoto A, Ogawa Y, Oki C, Kimoto Y, Nozawa K, Amano W, Noji S, Shiozaki M, Matsuo A, Shinozaki Y, Matsushita M (2015) Pharmacological properties of JTE-052: a novel potent JAK inhibitor that suppresses various inflammatory responses in vitro and in vivo. Inflamm Res 64:41–51
Tefferi A, Li CY, Butterfield JH, Hoagland HC (2001) Treatment of systemic mast-cell disease with cladribine. N Engl J Med 344:307–309
Tolar J, Tope WD, Neglia JP (2004) Leukotriene-receptor inhibition for the treatment of systemic mastocytosis. N Engl J Med 350:735–736
Topar G, Staudacher C, Geisen F, Gabl C, Fend F, Herold M, Greil R, Fritsch P, Sepp N (1998) Urticaria pigmentosa: a clinical, hematopathologic, and serologic study of 30 adults. Am J Clin Pathol 109:279–285
Trojan TD, Khan DA (2012) Calcineurin inhibitors in chronic urticaria. Curr Opin Allergy Clin Immunol 12:412–420
Turner PJ, Kemp AS, Rogers M, Mehr S (2012) Refractory symptoms successfully treated with leukotriene inhibition in a child with systemic mastocytosis. Pediatr Dermatol 29:222–223
Uchida K, Mitsui M, Kawakishi S (1989) Monooxygenation of N-acetylhistamine mediated by L-ascorbate. Biochim Biophys Acta 991:377–379
Ustun C, Reiter A, Scott BL, Nakamura R, Damaj G, Kreil S, Shanley R, Hogan WJ, Perales MA, Shore T, Baurmann H, Stuart R, Gruhn B, Doubek M, Hsu JW, Tholouli E, Gromke T, Godley LA, Pagano L, Gilman A, Wagner EM, Shwayder T, Bornhäuser M, Papadopoulos EB, Böhm A, Vercellotti G, Van Lint MT, Schmid C, Rabitsch W, Pullarkat V, Legrand F, Yakoub-Agha I, Saber W, Barrett J, Hermine O, Hagglund H, Sperr WR, Popat U, Alyea EP, Devine S, Deeg HJ, Weisdorf D, Akin C, Valent P (2014) Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J Clin Oncol 32:3264–3274
Vaali K, Lappalainen J, Lin AH, Mäyränpää MI, Kovanen PT, Berstad A, Eklund KK (2012) Imatinib mesylate alleviates diarrhea in a mouse model of intestinal allergy. Neurogastroenterol Motil 24:e325–e335
Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, Marone G, Nuñez R, Akin C, Sotlar K, Sperr WR, Wolff K, Brunning RD, Parwaresch RM, Austen KF, Lennert K, Metcalfe DD, Vardiman JW, Bennett JM (2001) Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res 25:603–625
Valent P, Akin C, Escribano L, Födinger M, Hartmann K, Brockow K, Castells M, Sperr WR, Kluin-Nelemans HC, Hamdy NA, Lortholary O, Robyn J, van Doormaal J, Sotlar K, Hauswirth AW, Arock M, Hermine O, Hellmann A, Triggiani M, Niedoszytko M, Schwartz LB, Orfao A, Horny HP, Metcalfe DD (2007) Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Investig 37:435–453
Valent P, Sperr WR, Akin C (2010) How I treat patients with advanced systemic mastocytosis. Blood 116:5812–5817
Valent P, Akin C, Arock M, Brockow K, Butterfield JH, Carter MC, Castells M, Escribano L, Hartmann K, Lieberman P, Nedoszytko B, Orfao A, Schwartz LB, Sotlar K, Sperr WR, Triggiani M, Valenta R, Horny HP, Metcalfe DD (2012) Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Arch Allergy Immunol 157:215–225
van Doormaal JJ, Idema IG, de Monchy JG, Breukelman H, Keyzer JJ, Doorenbos H (1986) Effects of isoprenaline and terbutaline on urinary excretion of histamine and its two main metabolites in systemic mastocytosis. Agents Actions 18:269–272
van Doormaal JJ, Arends S, Brunekreeft KL, et al. (2013) Prevalence of indolent systemic mastocytosis in a Dutch region. J Allergy Clin Immunol 131:1429–1431
Vega-Ruiz A, Cortes JE, Sever M, Manshouri T, Quintás-Cardama A, Luthra R, Kantarjian HM, Verstovsek S (2009) Phase II study of imatinibmesylate as therapy for patients with systemic mastocytosis. Leuk Res 33:1481–1484
Verstovsek S, Tefferi A, Cortes J, O’Brien S, Garcia-Manero G, Pardanani A, Akin C, Faderl S, Manshouri T, Thomas D, Kantarjian H (2008) Phase II study of dasatinib in Philadelphia chromosome-negative acute and chronic myeloid diseases, including systemic mastocytosis. Clin Cancer Res 14:3906–3915
Vrugt B, Wilson S, Bron A, Shute J, Holgate ST, Djukanovic R, Aalbers R (2000) Low-dose methotrexate treatment in severe glucocorticoid-dependent asthma: effect on mucosal inflammation and in vitro sensitivity to glucocorticoids of mitogen-induced T-cell proliferation. Eur Respir J 15:478–485
Welch EA, Alper JC, Bogaars H, Farrell DS (1983) Treatment of bullous mastocytosis with disodium cromoglycate. J Am Acad Dermatol 9:349–353
Weller K, Artuc M, Jennings G, Friedrichson T, Guhl S, Dos Santos RV, Sünder C, Zuberbier T, Maurer M (2009) Miltefosine inhibits human mast cell activation and mediator release both in vitro and in vivo. J Invest Dermatol 129:496–498
Weng Z, Zhang B, Asadi S, Sismanopoulos N, Butcher A, Fu X, Katsarou-Katsari A, Antoniou C, Theoharides TC (2012) Quercetin is more effective than cromolyn in blocking human mast cell cytokine release and inhibits contact dermatitis and photosensitivity in humans. PLoS One 7:e33805
Weng Z, Patel AB, Panagiotidou S, Theoharides TC (2015) The novel flavone tetramethoxyluteolin is a potent inhibitor of human mast cells. J Allergy Clin Immunol 135:1044–1052
Worobec AS, Kirshenbaum AS, Schwartz LB, Metcalfe DD (1996) Treatment of three patients with systemic mastocytosis with interferon alpha-2b. Leuk Lymphoma 22:501–508
Yacoub A, Prochaska L (2016) Ruxolitinib improves symptoms and quality of life in a patient with systemic mastocytosis. Biomark Res 4:2
Yamaguchi S, Okada Y, Matsunaga Y, Nambu F (2016) Effect of ONO-4053 on FcεRI stimulated mast cell activation. J Allergy Clin Immunol 137:AB77
Yamaki K, Yoshino S (2012) Tyrosine kinase inhibitor sunitinib relieves systemic and oral antigen-induced anaphylaxes in mice. Allergy 67:114–122
Yang Y, Lu JY, Wu X, Summer S, Whoriskey J, Saris C, Reagan JD (2010) G-protein-coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology 86:1–5
Yoshida C, Takeuchi M, Tsuchiyama J, Sadahira Y (2009) Successful treatment of KIT D816V-positive, imatinib-resistant systemic mastocytosis with interferon-alpha. Intern Med 48:1973–1978
Zachariae H, Herlin T, Larsen PO (1981) Oral disodium cromoglycate in mastocytosis. Acta Derm Venereol 61:272–273
Zen M, Canova M, Campana C, Bettio S, Nalotto L, Rampudda M, Ramonda R, Iaccarino L, Doria A (2011) The kaleidoscope of glucorticoid effects on immune system. Autoimmun Rev 10:305–310
Zhang T, Finn DF, Barlow JW, Walsh JJ (2016) Mast cell stabilisers. Eur J Pharmacol 778:158–168
Acknowledgments
The publication of this article was financially supported by the Förderclub Mastzellforschung e.V.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Molderings, G.J., Haenisch, B., Brettner, S. et al. Pharmacological treatment options for mast cell activation disease . Naunyn-Schmiedeberg's Arch Pharmacol 389, 671–694 (2016). https://doi.org/10.1007/s00210-016-1247-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-016-1247-1