
Abstract
The clinical development of selective alpha-7 nicotinic acetylcholine receptor (α7 nAChR) agonists has hitherto been focused on disorders characterized by cognitive deficits (e.g., Alzheimer’s disease, schizophrenia). However, α7 nAChRs are also widely expressed by cells of the immune system and by cells with a secondary role in pathogen defense. Activation of α7 nAChRs leads to an anti-inflammatory effect. Since sterile inflammation is a frequently observed phenomenon in both psychiatric disorders (e.g., schizophrenia, melancholic and bipolar depression) and neurological disorders (e.g., Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis), α7 nAChR agonists might show beneficial effects in these central nervous system disorders. In the current review, we summarize information on receptor expression, the intracellular signaling pathways they modulate and reasons for receptor dysfunction. Information from tobacco smoking, vagus nerve stimulation, and cholinesterase inhibition is used to evaluate the therapeutic potential of selective α7 nAChR agonists in these inflammation-related disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alpha-7 nicotinic acetylcholine receptors (α7 nAChRs) are expressed in the central nervous system (CNS) and are thought to play a role in a wide variety of psychiatric and neurological disorders. Peripheral and CNS immune cells strongly express α7 nAChRs; activation of α7 nAChRs on these cells has been shown to suppress inflammatory processes. Since inflammation is involved in several psychiatric disorders, as well as in basically all neurological disorders, specific α7 nAChR agonists could display therapeutic effects. In the current review, we summarize information on receptor expression, the intracellular signaling pathways modulated by these receptors, reasons for receptor dysfunction, clinical evidence for altered α7 nAChRs, and we conclude with a discussion about potential indications for selective α7 nAChRs agonists.
Expression of α7 nAChR
Distinct from most nicotinic acetylcholine receptors (nAChR), the α7-subtype mainly forms homomeric, rather than heteromeric pentamers. In the central nervous system, including the human brain, such nicotinic-α7 homomeric pentamers are expressed by pyramidal and interneurons [1–3]. Apart from neurons, immature (doublecortin positive) granule cells [4], astrocytes [5–7], and microglia cells [8–13] also express the α7 nAChR. Finally, NG2-positive cells (these are oligodendrocyte precursors) also express α7 nAChRs [5, 14]. The dogma that α7 nAChRs exclusively assemble as homomeric pentamers was recently overturned by the discovery that α7 subunits also form heteromeric pentameric ion channels with β2 subunits [15]. Such heteromeric α7β2 channels were found on cholinergic projection neurons in mouse and human basal forebrain [15, 16]. Concerning function, activation of α7 nAChRs results in strong calcium and sodium influxes, which in the case of a presynaptic location facilitates neurotransmitter release [17, 18]. A postsynaptic localization on parvalbumin-positive GABA neurons suggests a role in synchronized oscillatory output of pyramidal neurons.
Outside the brain, the receptor is expressed on several cell types of the immune system. This includes monocytes [19–21], dendritic cells [22], macrophages [23–26], T-cells [27, 28], and B-cells [29, 30]. The receptor has also been identified on additional cell types known to play a role in the host’s defense against pathogenic organisms. Examples are the microvascular endothelium [31], keratinocytes [32, 33], placenta [34], bronchial epithelial cells [35], platelets [36], adipocytes [37], and synoviocytes [38]. During wound healing, expression of α7 nAChRs is transiently observed on fibrocytes and myofibroblasts in the wound zones [39]. Completing this list, α7 nAChRs were detected in mouse testes, in mouse and human sperm (where they modify sperm motility [40]), in rat superior cervical ganglia [41], and in group-IV muscle afferent neurons [42].
Not unexpectedly, numerous cell lines endogenously express α7 nAChR too. This includes rat PC12-cells [43], human SH-SY5Y neuroblastoma cells [44, 45], mouse RAW264.7 [46, 47], the human leukemic T-cell line MOLT3 [48], the human monocyte U937 cell line [20, 49], and immortalized human T-lymphocyte ‘Jurkat’ cells [27]. Recombinant expression has also been achieved in cell lines that endogenously express the chaperones required for α7 nAChR expression, e.g., SH-EP1 cells [50], SH-SY5Y [51], or GH3 cells [52].
With respect to the above-listed localizations of α7 nAChR, a word of caution is needed, since some expression studies are confounded by the use of tools that also detect a distinct, duplicated α7-like protein ‘dupα7’ [26, 53], by the use of antibodies which recognize cross-reacting epitopes [54] or by the use of the non-selective radioligand MLA (besides α7 nAChRs this compound also binds to nicotinic α3/α6β2β3* receptors, see [55]).
In addition to expression on the cell surface, an intracellular localization of α7 nAChRs has been observed in brain mitochondria [56]. In this organelle, the α7 nAChR may assemble with β2 subunits where it presumably influences pore formation and cytochrome-c release [57].
Intracellular signaling pathways
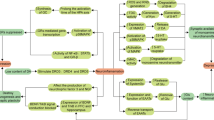
In mouse hippocampal neurons, α7 nAChRs are characterized by rapid activation and desensitization. The fractional calcium current (P f) has been assessed and found to be 6.1 [58]. This indicates that in neurons α7 nAChRs have a special role in the modulation of intracellular calcium, enabling substantial calcium entry at resting or hyperpolarized membrane potential, which is different compared to other nAChRs, but similar to NMDA receptors at depolarized membrane potential. However, thus far, it is not known if this also holds true for α7 nAChRs in other cells (e.g., immune cells). The intracellular pathways following α7 nAChR activation in non-neuronal cells involve calcium influx through the channel pore, which then triggers calcium-induced calcium release from ryanodine- and IP3-dependent stores [10, 28, 59]. Further long-lasting intracellular pathways involve activation of intracellular phosphatases and kinases, which trigger signaling events independent of an ion flux (which implies that the α7 nAChR acts as metabotropic receptor) [60]. For instance, activation of α7 nAChRs leads to stimulation of adenylate cyclase-1 and thus to increases in cAMP levels [61]. This in turn stimulates protein kinase A (PKA), which may result in further signaling events such as CREB activation [61] and GSK3 inhibition [62]. Activation of the α7 nAChR on non-neuronal cells inhibited TLR3-, TLR4- or TLR9-induced transcription and release of inflammatory cytokines [9, 19, 21, 34, 47]. One of the intracellular signaling cascades described in this context is a pathway that involves JAK2-mediated tyrosine-phosphorylation of the p85 subunit of PI3 K, activation of Akt and CREB, and subsequent inhibition of (or competition with) NFκB [20, 25, 49, 63] (see Fig. 1). Egea and colleagues emphasize that this pathway furthermore leads to activation of the transcription factor Nrf2, which is important for transcription of numerous anti-oxidative proteins and for the induction of an anti-inflammatory phenotype of microglia cells [64]. Alternatively, downstream signaling towards NFκB may involve JAK2 activation of STAT3 [65–68] (see Fig. 1). Finally, activation of α7 nAChRs can result in inhibition of p38 MAP-kinase [8, 10, 19]. A functional consequence of this latter pathway is inhibition of the release of inflammatory mediators like TNFα and HMGB1 [8, 10, 19].

Schematic anti-inflammatory signaling pathways activated by nAChR α7. Stimulation of nAChR α7 activates Jak2 leading to inhibition of NFκB and GSK3 but also to CREB activation. A separate signaling cascade involves activation of PKA and AKT enabling the nuclear translocation of Nrf2 (NFE2L2), which drives expression of HMOX1 (HO-1). This pathway elicits potent anti-inflammatory and neuroprotective effects
The anti-inflammatory activity of α7 nAChR stimulation
As early as 1998, Sugano et al. [49] described that nicotine displayed an anti-inflammatory activity involving inhibition of NFκB-signaling. Following this observation, it was shown that the receptor responsible for this response was the α7 nAChR subtype [20, 23, 24, 69, 70]. Moreover, it was shown that the anti-inflammatory effect of electrical stimulation of the vagus nerve was also mediated by the α7 nAChR [23, 24, 69]. Notably, after splenectomy the beneficial effects of vagus nerve stimulation were lost [71], but some controversy still exists about the exact localization of the α7 nAChRs involved in the response to vagus nerve stimulation. The vagus nerve is supposed to activate the celiac ganglion, which is the origin of the adrenergic splenic nerve. According to one scenario, the splenic nerve releases noradrenaline onto memory T-cells (CD4+ CD44high, CD62Llow), resulting in synthesis and release of acetylcholine that activates α7 nAChRs on spleen-macrophages [69, 72, 73]. The alternative proposal assumes that the α7 nAChRs are localized postsynaptically in the celiac ganglion. This view is supported by data showing that postganglionic stimulation of the splenic nerve still results in an anti-inflammatory response, even in mice with a genetic deletion of the α7 nAChR [74]. However, in a critical review Martelli [75] proposes that efferent vagus nerve stimulation achieves its anti-inflammatory effect via a non-neuronal (humoral) pathway. It is evident that this is an area that is still very much in development. Interestingly, activation of glucocorticoid receptors increases the expression of α7 nAChRs [76]. This implies that the anti-inflammatory activity of glucocorticoids may include a nicotinergic mechanism. Overall, given the expression pattern, the cellular signaling cascades, and information from vagus nerve stimulation, the data strongly indicate that activation of the α7 nAChR suppresses the responsiveness of the immune system.
Surprisingly, whereas the activity of positive allosteric modulators (PAMs) in animal models of learning/memory or on evoked potentials is well documented (see [77] for a recent review), there is a paucity of reports on their anti-inflammatory activity. In this context, PNU-120596 has been shown to attenuate TNFα and IL-6 in a rodent model of inflammatory pain. These findings have been recently corroborated with a different molecule (“PAM-2,” [78]). Along the same lines several reports have shown that α7 nAChR PAMs reduce brain injury and improve neurological function after focal cerebral ischemia in rats [79–81].
Notably, the anti-inflammatory response to α7 nAChR stimulation also occurs in the brain. Thus, activation of α7 nAChRs is known to alter the phenotype of both macrophages and microglia from an M1-like to an M2-like phenotype [11, 19, 82, 83]. Consequently, any dysfunction in the α7 nAChR and its signaling processes could tip the balance towards more inflammation.
Reasons for dysfunction of the α7 nAChR
Dysfunction of α7 nAChRs may have a variety of causes. For adequate membrane insertion, the α7 nAChR has to be assembled first as a pentamer and is thereafter shuttled to the plasma membrane. This process requires several chaperone molecules such as RIC3, SLURP1, Lynx1, EPHB2, or PICK1, and evidently, their dysfunction could affect receptor level and function [1, 33, 48, 84–88]. In addition, transcription of the α7 nAChR can be diminished by heterozygotic or homozygotic 15q13.3 microdeletions [89], by methylation of the promotor [90] or by MeCP2 (methyl CpG binding protein 2) dysfunction [91].
Moreover, activity of the α7 nAChR is modified by phosphorylation. An as yet undefined tyrosine-phosphorylated protein was shown to inhibit the functional activity of the α7 nAChR, whereas tyrosine kinase inhibition by genistein enhanced surface expression of the α7 nAChR [35, 37, 84, 92] or its function [51]. Serine-phosphorylation of the α7 nAChR can influence function too. It has been reported that activation of D1/D5 dopamine receptors attenuated α7 nAChR currents via PKA-mediated phosphorylation of the serine residue 365 in the M3–M4 cytoplasmic loop of the channel [93]. Other possibilities have been proposed as well. For instance, diminished signaling of α7 nAChR might result from increased levels of the purported endogenous α7 nAChR inhibitor, kynurenic acid [94], though conflicting data have been reported too [95]. Also cholinergic input may be dysfunctional [69], or the signaling pathway downstream of the receptor may be altered [27, 66, 92]. Furthermore, dysfunction of the α7 nAChR could result from the presence of the CHRFAM7-gene. This gene encodes a dominant negative variant of the α7 nAChR [26, 96] and is unique for humans [26, 97]. Transcripts, often described as ‘dupα7’, have been found in the promyelocytic leukemia cell line HL-60 [26], monocyte cell lines (THP1, U937, and Mono-Mac6 [98]), and neuroblastoma cell lines (SH-SY5Y, IMR32 [98]). High levels of dupα7 transcripts have also been described in native immune cells such as peripheral blood monocytic cells, lymphocytes and synoviocytes, and in lower amounts also in human brain tissue [7, 26, 53].
Rapid metabolism of acetylcholine and receptor desensitization
Cholinergic innervation in the brain arises mainly from two sources. Cholinergic neurons from the medial septal nucleus innervate the hippocampus [99], whereas cholinergic nerves from the basal forebrain (including the nucleus basalis Meynert) project to the cortex, amygdala, caudate nucleus, putamen, and thalamus [100]. Acetylcholine locally released by cholinergic neurons is assumed to cross the synaptic cleft despite effective catabolism by cholinesterases [101]. However, it is less clear whether volume transmission to extrasynaptic neuronal sites is sufficiently high to lead to relevant α7 nAChR stimulation. Outside the brain, high levels of cholinesterases are found in the blood and therefore it has been questioned whether α7 nAChRs on, for example, circulating immune cells will be exposed to levels of acetylcholine that are sufficient for activation [102]. Further doubts about the physiological relevance of α7 nAChRs relate to their rapid desensitization [101, 103, 104]. However in the latter case, there are two counter-arguments. In the first place, α7 nAChRs not only rapidly desensitize, they also quickly recover [105]. The second argument is that intracellular effects are presumably more sustained and therefore could outlast receptor desensitization. Vijayaraghavan et al. [6] provide yet one more piece of information, which, in addition, addresses the issue of volume transmission. The authors found that extracellular levels of choline-acetyl transferase (ChAT) are actively regulated. This means that, despite ongoing extracellular cholinesterase-activity, extracellular acetylcholine is permanently resynthesized. As a consequence, acetylcholine may act over long distances from its site of release. Importantly, acetylcholine is not only synthesized and released by neurons but also by several non-neural cells. Frequently these cells express both ChAT and α7 nAChRs, and as a result, an intrinsic paracrine loop is formed [102]. Examples are bronchial epithelial cells [35], lymphocytes [6, 28], human neuronal stem cells [6], and astrocytes [6, 106]. As a last argument in favor of a physiological relevance of extrasynaptic α7 nAChRs, it should be mentioned that these receptors are also activated by choline, the precursor and hydrolytic split product of acetylcholine [46, 67, 81, 107]. In summary, extrasynaptic α7 nAChRs will be readily activated under physiological conditions.
Diseases in which treatment with α7 nAChR agonists could be useful
Since α7 nAChRs are expressed on interneurons, presynaptically on glutamatergic neurons, and on neuronal progenitor cells, the logical prediction was that α7 nAChR agonists could provide a beneficial influence on cognitive function. Consequently, the clinical testing of α7 nAChR agonists has focused on disorders with profound cognitive dysfunction (i.e., schizophrenia, Alzheimer’s disease). The fact that nicotinic α7 receptors are also strongly expressed by cells of the immune system, including those of the brain innate immune system, has often been (but not always, [64, 108]) overlooked. With this in mind, in the following sections we will reflect on the therapeutic use of selective α7 nAChR agonists in psychiatric and neurological disorders.
Depression
Inflammation and exposure to stress are generally recognized as strong proximal factors for the development of depression symptoms [109]. Many kinds of stress, but in particular those involving threat, loss, entrapment, and humiliation, lead to activation of the immune system [109–112]. This has been conceptualized as an anticipatory response by the innate immune system to prepare for physical injury [113]. Phagocytic cells such as macrophages and microglia are essential components of the innate immune system and contribute to the host’s defense against invading microorganisms. Notably, phagocytes from patients with depression are hyper-responsive [114–116]. Upon activation, these cells produce cytotoxic compounds like nitric oxide and oxygen radicals [117, 118]. These reactive oxygen radicals formed during the ‘respiratory burst’ may irreversibly oxidize tetrahydrobiopterin [119]. Since tetrahydrobiopterin is an essential cofactor for the production of dopamine, noradrenaline, and serotonin, the heightened activity of microglial cells may ultimately lead to a reduction in central monoamine levels (see Fig. 2). This could be the underlying cause of low mood and anhedonia in depression (Kalkman and Feuerbach, in preparation). As outlined above, α7 nAChRs are expressed both on macrophages and microglia, and their activation leads to anti-inflammatory effects. In particular, activation of α7 nAChRs might lead to a shift in microglia phenotype from M1-like (activated for anti-microbial activity) to M2-like (resolution, removal of debris) [10, 11, 13, 83, 120], while a similar process has been described in the periphery. Stimulation of α7 nAChRs attenuates macrophage responsiveness and diminishes release of cytokines [23, 46, 47, 66] and therefore will limit the negative impact of inflammation on tetrahydrobiopterin metabolism.

Microglia cells play a role in eradication of invading microorganisms and in removal of debris. During these processes, the cells adapt specialized phenotypes (M1 and M2-like, respectively). During the respiratory burst, labile oxygen products are formed that oxidize microbial proteins and nucleic acids, but also oxidize the essential cofactor for monoamine synthesis, tetrahydrobiopterin. Sterile neuroinflammation is a common observation in neurological and psychiatric disorders. Acetylcholine, acting via α7 nAChRs, promotes M2-polarization. As such, it reduces neuroinflammation, while promoting phagocytosis. M2-polarized microglia cells not only produce neurotrophins and anti-inflammatory cytokines, they also effectively phagocytose and catabolize Aβ. α7 nAChR agonists are expected to improve neurological and psychiatric disorders via inhibition of neuroinflammation, to restore tetrahydrobiopterin levels (improve mood symptoms) and to provide neuroprotection
Activation of α7 nAChRs in the brain has been shown to activate protein kinase A (PKA) via a mechanism involving calcium-dependent activation of adenylatecyclase-1 and, consequently, an increase in cAMP levels [61]. PKA is one of the kinases that increase Ser9-phosphorylation of GKS3β, which results in inhibition of the kinase activity. PKA also fosters Ser133-phosphorylation of the transcription factor CREB. Ser133-P-CREB is a substrate for GSK3β, which subsequently results in rapid catabolism of CREB and thus termination of CREB signaling. CREB competes with NFκB for binding to CREB-binding protein (CBP), and therefore limits the inflammatory NFκB signal. Activation of α7 nAChRs in the brain may thus lead to an anti-inflammatory activity. Increased PKA activity, increased Ser9-phosphorylation of GSK3β, and increased Ser133-phosphorylation of CREB have all been observed in mouse brain after chronic treatment with the α7 nAChR agonist A582941 [62]. Notably, Ser9-phosphorylation of GSK3β following A582941 treatment was absent in mice with a genetic deletion of the α7 nAChR [121].
Vagus nerve stimulation, which reduces inflammation [23, 66, 69, 122], has been approved for treatment of drug-resistant depression in humans [123]. In long-term naturalistic studies, significant improvements and increasing remission rates were noted in patients with refractory depression [124–126]. Oxytocin (reported to display antidepressant activity [127]) increases the excitability of central vagal neurons in rats [128] and inhibits LPS-induced release of inflammatory cytokines in healthy human subjects [129]. The antidepressant activity of oxytocin could thus be a consequence of stimulation of the cholinergic anti-inflammatory pathway. In conclusion, there is accumulating evidence that α7 nAChR agonists could possess antidepressant activity, although none of the compounds that were in clinical development has been tested in this indication.
Schizophrenia
Symptoms of schizophrenia are commonly divided into three domains, namely positive symptoms (delusions, hallucinations), negative symptoms (social withdrawal, anhedonia), and cognitive deficits (learning and memory deficits, alogia). A history of maternal and prenatal infections, prior hospitalization for severe infection, and autoimmune comorbidity represent major risk factors for schizophrenia [130]. These conditions, which are linked to the elevation of pro-inflammatory cytokines has led to the formulation of the “prenatal cytokine hypothesis” [131]. This hypothesis proposes that early alterations in the peripheral and central innate immune system disrupt normal development and maturation of neuronal systems during the juvenile and early adult stages of life, affecting processes such as myelination, synaptic pruning, and neuronal modeling [131]. Results from genetic studies are consistent with such a process, as confirmed genetic risk factors for schizophrenia include mutations in genes involved in immune function (e.g., HLA-C and HLA-DRA) and synaptic pruning [132, 133]. The first episode of psychosis is often associated with microglia activation [130, 134, 135], elevated levels of pro-inflammatory cytokines in the CSF [136–138], and a significant loss in white matter volume (reviewed in [136]). It is assumed that cytokines, chemokines, prostaglandins, and reactive oxygen products released by activated microglia generate a toxic milieu for oligodendrocytes (leading to white matter loss) and neurites (gray matter loss) [130, 136, 139, 140]. The severity of negative symptoms correlates with the diminution of white matter [130].
The impetus to develop α7 nAChR agonists was based on two sets of clinical information. First, the early literature reported extremely high values for the prevalence of smoking in schizophrenia patients (see [141]). This information was supplemented by data from radioligand binding experiments investigating brain tissue of deceased schizophrenia patients (for summary see [100]). Using [125I]-α-bungarotoxin (α-btx) as radioligand, a reduction in labeling of the α7 nAChR was found in the dentate gyrus [142], the reticular nucleus of the thalamus [143], the frontal cortex [144], and the cingulate cortex [1]. A vast body of genetic literature supports the contention that the functionality of the α7 nAChR in schizophrenia is diminished. CHRNA7, the gene encoding the α7 nAChR, is located on 15q14, a chromosomal area that is linked to genetic transmission of schizophrenia [145]. This area is also subject to deletion copy number variations (CNVs,) [146, 147]. Decreases in α-btx binding observed in schizophrenia patients could be due to an increased expression and insertion of dupα7, since heteromeric dupα7/α7 nAChRs do not bind this radioligand [84, 148]. Notably, decreased α-btx binding is unlikely to be explained by differences in smoking behavior [1]. Polymorphisms in the CHRNA7 promoter that decrease gene-transcription are also associated with schizophrenia [149]. A 2 bp deletion allele in CHRFAM7A is frequent in Caucasians (42 %) and less in African-Americans (14 %) [150]. This 2 bp deletion form of CHRFAM7A is an even stronger inhibitor of α7 nAChR than wild-type CHRFAM7A and consistent with this, several studies support an association of the 2 bp deletion in the CHRFAM7A gene with schizophrenia and bipolar disorder (summarized by Sinkus et al. [150]). Finally, CNVs occur in the CHRNA7 and in CHRFAM7A, and deletions were strongly associated with schizophrenia. Interestingly, this was especially the case when the CHRNA7 was deleted, while CHRFAM7A was present [150].
The pathophysiological consequence of the diminished functionality was mainly sought in the cognitive domain. The prediction that α7 nAChR agonists would improve cognition in schizophrenia was tested clinically with a series of development compounds (for a recent reviews, see [100, 151, 152]). Since the nicotine α7 receptor and the 5HT3 receptor are phylogenetically very similar, several developmental α7 nAChR agonists lack selectivity and block the 5HT3 receptor concomitantly [108]. Encenicline (EVP6124), which is a mixed α7 nAChR agonist/5HT3 antagonist, showed significant clinical improvement on PANSS cognitive impairment domain and also for the PANSS negative scale [153]. Another mixed α7 nAChR agonist and 5HT3 antagonist tropisetron significantly reduced PANSS total and the negative symptom subscale with increasing treatment time [154]. Further, a mixed α7 agonist/5HT3 antagonist RG3487 (also known as MEM3454), was reported to improve negative symptoms and depression ratings [155]. It remains unclear if the beneficial effect on negative symptom ratings is due to α7 nAChR activation or to 5HT3 blockade, as both a selective α7 nAChR agonist (TC-5619) and a selective 5HT3 antagonist (ondansetron) improved negative symptoms [156, 157]. DMXB-A (GTS-21) [158], a α7 nAChR agonist with additional inhibitory activity at α4β2 nAChRs [159], improved alogia and anhedonia ratings in the scale for assessment of negative symptoms. Remarkably, α7 nAChR-positive allosteric modulators (galantamine, galantamine plus choline, and JNJ39393406) were completely inactive [100]. Taken together, these data show that α7 nAChR activation did not improve positive symptoms, whereas a beneficial effect against negative symptoms was observed repeatedly. The clinical evidence that α7 nAChR activation leads to improvement of cognitive dysfunction in schizophrenia still remains somewhat equivocal [100, 160], although the data for encenicline are encouraging [153, 161]. It is noteworthy that the clinical development of several mixed nAChR α7 agonist/5HT3 antagonist compounds (e.g., RG3487, tropisetron) in schizophrenia has been halted [152]. This may be related to side effects associated with 5HT3 receptor blockade (e.g., constipation, arrhythmias) (see [108, 162, 163]). Currently, three α7 nAChR agonists remain in clinical development (AQW051, encenicline, and GTS-21) [152].
The observation that nAChR α7 agonists improve negative symptoms is remarkable. It can be speculated that stimulation of nAChRs α7 counteracts microglia activation, such that white and gray matters are exposed less to the toxic microglia products. In this respect it is noteworthy that minocycline, a compound which suppresses microglia activation, also specifically improved negative symptoms (for review see [164]). Negative symptoms, like symptoms of melancholic depression, might reflect distinct behavioral consequences of central inflammation. However, it could be that negative symptoms are not distinct and in fact reflect aspects of melancholic depression, or that rating scales for negative symptoms do not discriminate between depression symptoms and negative symptoms [155]. Since microglia activation occurs early in the disease process (i.e., during, or even before, the first period of psychosis), early intervention with compounds that limit microglia activation (nAChR α7 agonists, minocycline) might be appropriate.
Bipolar disorder
The cardinal features of bipolar disorder are recurrent episodes of depression and hypomania, whereas the latter includes euphoric or irritable mood, mental and behavioral over-activity, as well as decreased need for sleep and involvement in risky activities [165]. As for major depressive disorder, diseases and habits that are associated with peripheral inflammation (diabetes, obesity, cardiovascular disease, smoking, and alcohol abuse) are recognized risk factors for bipolar disorder (reviewed in [166–168]). And in further similarity to major depression, childhood trauma is a strong predictor for appearance of bipolar disorder later in life [169]. Increases in serum levels of inflammation markers have been observed during manic, depressed and even euthymic phases [170–173]. Bipolar disorder shares genetic risk loci with schizophrenia and with major depression [171, 174]. There is evidence for microglia activation in the brain of bipolar disorder patients: first, higher levels of IL1β, IL1R, MYD88, and iNOS have been found in a post-mortem study in the frontal cortex [175]. Second, microglia-derived inflammation markers MCP1 and YKL40 were increased in CSF [176], and third, microglia activation was detected by PK11195 imaging [177]. The inflammatory milieu in the brain is probably responsible for atrophy, volumetric changes, cognitive decline, and symptom worsening [167, 175]. The same genetic polymorphisms, copy number variations and alteration in the pseudo-gene CHRFAM7A, which are presumed to diminish the functionality of the nAChR α7 receptor in schizophrenia, have also been detected in bipolar disorder [161, 178].
The neurodevelopmental consequences of diminished α7 nAChR signaling may be inferred from studying CHRNA7 knockout mice. It has been reported that nicotine administration to these animals led to a longer period of elevated extracellular dopamine levels in the nucleus accumbens than in control mice [179]. Excessive dopamine signaling via D2 receptors in the striatum causes an activation of GSK3β via a multiprotein complex involving the D2 receptor, β-arrestin, Akt, GSK3β, and PP2A [180, 181], while active GSK3β results in diminished long-term potentiation (LTP) [182–184]. Unexpected rewarding outcomes result in dopamine release in the striatum, whereas unpredicted negative outcomes result in a strong reduction in dopamine output [185]. Such a dopamine “dip” improves avoidance learning (via D2 receptor hypo-stimulation) by decreasing GSK3β activity, and thus promoting LTP. In contrast, hyper-stimulation of D2 receptors, and activation of GSK3β, both result in a diminished learning from incorrect reward predictions. If we assume that the α7-p85-Akt-GSK3β pathway is activated in striatal neurons (as it is in immune cells), dysfunction of the α7 nAChR would result in reduced inhibitory GSK3β-phosphorylation, in poorer learning from unrewarding conditions, and eventually in more (i.e., less suppressed) risk-taking behavior. Taken together, dysfunction of the α7 nAChR would increase both depression-risk and hypomania symptoms. The prediction that selective α7 nAChR agonists might counteract these symptoms remains to be tested.
Autism spectrum disorder
Autism is a general term for a group of neurodevelopmental disorders characterized by difficulties in social interaction, verbal and non-verbal communication, and repetitive behaviors. Several distinct subtypes have been identified, and these are often combined under the umbrella diagnosis “autism spectrum disorder” (ASD). Certain genetic copy number variations, which can be either inherited or occur de novo, exert a profound effect on brain development and lead to syndromes that fit the ASD diagnosis. The 15q13.3 microdeletion syndrome is such an example. Gillentine and Schaaf [147] collected all published cases of heterozygous 15q13.3 deletions and calculated that nearly half of them displayed cognitive deficits, while seizures and symptoms of ASD were noted in 26 and 21 % of these cases, respectively. It is not clarified by which mechanism heterozygosity for the α7 nAChR could lead to ASD. It is known, however, that the α7 nAChR is essential for the formation of NMDA synapses [86], and that the absence of functional α7 nAChRs results in perturbation of NMDA neurotransmission at glutamatergic synapses [186]. The EphB2 receptor is involved in the process by which α7 nAChRs enhance formation of NMDA synapses [86, 187] and, interestingly, also dysfunction of the EphB2-receptor has been recognized as a risk factor for autism spectrum disorder [188].
Rett syndrome is another distinct genetic form of autism. In the majority of cases, Rett syndrome is caused by a mutation in the MeCP2 gene; the functional consequence of this mutation is a severe reduction in the expression of α7 nAChRs [91]. Yasui and colleagues have therefore proposed to use α7 nAChR agonists for the treatment of Rett syndrome.
Although the pathology seen after partial deletion or dysfunction of the α7 nAChR provides support for its physiological role, it also creates a dilemma for pharmacotherapy with α7 agonists. Such compounds can only work when the receptor is present and functional. A study by Le Pichon and colleagues exemplifies this [189]. The authors created lymphoblastoid cell lines from a patient with a homozygous 15q13.3 microdeletion, his heterozygous parents, and from several further family members and age-matched controls. The lymphoblast cells were treated with lipopolysaccharide, which provoked the expected increase in TNFα release. TNFα release was suppressed by nicotine (which in turn could be blocked by the α7 nAChR antagonist α-btx) in all control and hemizygous deletion cell lines, but notably not in lymphoblasts from the index patient [189]. Patients with a strongly dysfunctional or absent α7 nAChR will not benefit from a α7 nAChRs agonist. It is reasonable to assume that cases of moderate hypoactivity of the α7 nAChR will exist among the wide spectrum of autism disorders, but the challenge will be to identify a biomarker to enable their selection. The current clinical evidence for a therapeutic effect of an increase in cholinergic signaling in ASD is quite limited. It is based on a few open label studies, case reports and a single small-size double-blind augmentation study with a cholinesterase-inhibitor [190].
In a subset of autism patients, there is evidence for an increase in allergic inflammation [191] and for mast cell activation [192]. These observations hint to an increased TH2 polarization (perhaps owing to a helminth infection). An infection with a helminth would skew the differentiation of helper T-cell towards the TH2 phenotype. IL4 and IL5, cytokines produced by TH2 cells [193], promote macrophage/microglia M2A polarization and mast cell activation [194–196]. Indeed, Gupta et al. [197], recently reported that the expression pattern of microglia genes in autism cases was characteristic for an increased M2A phenotype. Treatment with α7-agonists is expected to further increase M2 polarization, and thus could accentuate the autism-phenotype. According to this line of reasoning, the treatment with α7 nAChR agonists might be contra-indicated in at least some forms of autism spectrum disorder.
Multiple sclerosis
Neuroinflammation represents a key aspect of the neurological disorders that will be discussed in this and following sections. Multiple sclerosis (MS) is an autoimmune disease that affects axonal nerve transmission in peripheral, lumbal, and central nerves, which as first symptoms often results in motor and sensory problems. Depression is a common comorbidity [198]. The precise sequence of events that leads to MS in patients is not known [199], but activation of lymphocytes, macrophages, dendritic cells, and microglia are known to occur early in the disease [200]. α7 nAChR activation can lead to inhibition of lymphocyte proliferation [28, 201] and to inhibition of macrophage and microglia activation [8, 11, 24, 25, 46]. Stimulation of α7 nAChR on endothelial cells furthermore limits the extravasation of leukocytes during inflammation [31], although it is not known if this is also true for the blood–brain barrier. Together, these findings suggest that α7 nAChR agonists might be therapeutically useful in multiple sclerosis. Indeed, nicotine was shown to inhibit experimental MS in rodents [83, 201], but to our knowledge, selective α7 nAChR agonists remain to be investigated in human MS.
Parkinson’s disease
The motor symptoms of Parkinson’s disease (PD) are ascribed to degeneration of dopaminergic neurons in the substantia nigra pars compacta. Other important symptoms are anxiety and depression, as well as memory loss, confusion, and dementia. In this respect, it is noteworthy that not only dopamine neurons perish but also the cholinergic system. Degeneration of the basal forebrain cholinergic system occurs early in the disease process and precedes the dementia symptoms [202, 203]. In fact, in PD-dementia the levels of cerebral ChAT are reduced to levels below those seen in Alzheimer’s disease [204], whereas cholinesterase inhibition improves dementia in Parkinson’s disease [205].
A large number of epidemiological studies (for a recent summary see [120]) have consistently shown that smoking is associated with a lower incidence of PD. Data from preclinical models of Parkinson’s disease indicate that nicotine and selective α7 nAChR agonists reduce microglia activation and neuroinflammation, and prevent nigro-striatal dopamine-neuronal loss [206, 207]. α7 nAChRs expressed on astrocytes may also contribute to the neuroprotective effect, since activation of these receptors suppressed astroglial apoptosis induced by oxidative stress and preserved neurotrophic factor supply by glial cells [208]. An alternative suggestion is that nicotine reduces PD symptoms owing to its stimulant effect on nigro-striatal dopamine release. This nicotine-induced dopamine release is mediated primarily via α6β2-containing receptors localized on dopaminergic neurons. These neurons are particularly vulnerable to damage and in models of nigro-striatal damage, the α6β2-receptor numbers are significantly reduced [209]. For this reason, it is rather unlikely that the indirect dopamine release induced by nicotine acting on α6β2 receptors plays a significant role in the positive effect of smoking in PD. Post-mortem studies of brains from patients with Parkinson’s disease provide convincing evidence for neuroinflammation in the pars compacta, with increases in neurotoxic cytokines, microglia activation, and lymphocyte infiltration (reviewed by Hirsch and Hunot [210]).
Depression occurs in about 35 % of PD-patients [211] and might develop in the premotor stage of the disease [211, 212]. Consistent with theories about microglia activation as contributor to depression [213, 214], depressive symptoms may be considered as an early indicator of Parkinson’s disease. The protective effect that smoking exerts on PD could thus relate to α7 nAChR-mediated anti-inflammatory activity of nicotine [11]. This would imply that selective α7 nAChR agonists could be effective as prophylactic treatment for Parkinson’s disease. In this respect, it is worthwhile to mention that in MPTP-treated laboratory animals, the density of α7 nAChR increases (which is in contrast to α6β2-containing nAChRs, see above) [215]. The α7 nAChR agonist AQW051 [216] has been tested in patients with established Parkinson’s disease (ClinicalTrials.gov), but results remain to be published.
Alzheimer’s disease
One of the functions of microglia is to remove debris [217]. Microglia cells phagocytose and subsequently degrade Aβ, and thereby promote the removal of Aβ from the brain [218]. As mentioned before, microglia cells assume different phenotypes. With aging, microglia shift their morphology to a pro-inflammatory state and presumably lose their ability to phagocytose [219]. Activation of α7 nAChR expressed on microglia alters the phenotype and promotes phagocytosis and metabolism of Aβ [12]. Based on these data, one would expect that nicotine (smoking) and cholinesterase inhibitors would diminish Aβ load and improve Alzheimer disease. In transgenic animals, overexpressing Aβ beneficial effects of nicotine or ChE-I were indeed observed, while knock out of the α7 nAChR worsened pathology [12, 62, 220]. In patients with Alzheimer’s disease, cholinesterase inhibitors limit the cognitive deficits early in the course of the disease, but dosing and efficacy are limited by cholinergic (in particular muscarinic) side effects. For this reason, it is expected that α7 nAChR agonists might reach a similar therapeutic efficacy, but with less side effects. In a recent meta-analysis of prospective cohort studies investigating the effect of smoking on dementia and Alzheimer’s disease, smoking increased risk for dementia and AD [221]. This meta-analysis study included 37 studies with a total of almost 1 million patients and the statistical power was sufficient to investigate the influence of age. The increase in dementia risk was observed in the age group between 65 and 75, whereas the interpretation of results from patients older than 75 was hampered by a survival effect. Importantly, the increase in dementia or AD risk was not significant for smokers under 65 [221]. This result is clearly different from Parkinson’s disease, where smoking tendentially caused more benefit than harm [222]. Why would this be? The answer may lie in the special interaction of Aβ with α7 nAChRs. Parri et al. [223] have recently reviewed this in great detail. The fact that Aβ binds to α7 nAChRs has been observed and confirmed in numerous experimental settings, including post-mortem AD brain; however, it is unclear if this interaction results in inhibition or in stimulation of the receptor, and whether this interaction is reversible by agonists and antagonists. An intra-subunit allosteric binding pocket within the transmembrane domain of the α7 nAChR has been described as mechanism for non-competitive antagonism by Aβ (summarized by Parri et al. [223]). A further complication with relevance to AD pathology is the observation that cholinergic neurons in the basal forebrain express a heteromeric α7β2 isoform [224, 225]. Liu et al. [224] have reported that the α7β2 receptor is particularly sensitive to Aβ, since concentrations as low as 1 nM inhibited the functional responses to choline. They furthermore noted that inhibition was strongest with Aβ in its oligomeric form, followed by fibrillar Aβ, whereas monomeric Aβ was inactive. It is not clear why blockade of the α7β2 receptor is neurotoxic to cholinergic neurons, and if microglia cells from basal forebrain structures express this heteromeric nicotinic receptor. Nevertheless, one can begin to sketch a positive feedback process where aging causes polarization of microglia towards a phenotype that is less effective in phagocytosing and degrading Aβ. An overload of oligomeric Aβ in the extracellular space will block the α7β2 nicotinic receptor on basal forebrain cholinergic neurons, and these die as a consequence. Less acetylcholine leads to a reduction in α7 nAChR stimulation of microglia cells, which results in further loss of their phagocytic capacity. Diminished clearance of Aβ ultimately leads to extracellular precipitates, which presumably are a further trigger to microglia recruitment, inflammatory processes, and further toxicity to cholinergic neurons (see Fig. 3). In this process, an early intervention with nicotine, cholinesterase inhibitors, and in principle, α7 nAChR agonists may delay the start of the vicious circle. However, later in the process the availability of α7 nAChRs will be reduced by the negative interaction with the accumulating oligomeric Aβ. This would explain why the toxic effects of smoking become dominant over the neuroprotective effects in Alzheimer’s disease, but not (or less) in Parkinson’s disease. If the above-sketched vicious circle is correct, treatment with selective α7 nAChR agonists would be useful as prophylaxis, but less so for treatment of established severe AD-dementia.

During aging the polarization of microglia gradually shifts towards the M1 phenotype. Diminution of M2 polarization presumably has negative consequences for Aβ catabolism. When extracellular Aβ levels increase, several positive feedback loops are triggered that ultimately lead to the demise of cholinergic neurons. Activation of α7 nAChRs counteracts the loss of the M2 phenotype. α7 nAChR agonists may furthermore compete with Aβ at (mitochondrial?) nicotinic α7β2 receptors. Treatment with α7 nAChR agonists might therefore delay the demise of cholinergic neurons, and thus delay the onset of dementia
Discussion
From a historical perspective, selective α7 nAChR agonists have been targeted for cognitive deficits associated with schizophrenia (for recent reviews see [151, 152, 226]) and dementia in Alzheimer’s disease [227, 228]. Where investigated, the dose–response relationships for the pro-cognitive effects of α7 nAChR agonists usually display an inverted U-shape, indicating that higher doses are less effective than certain lower doses [142, 229–232]. Although the exact mechanism behind this profile remains speculative, it has been frequently noted that high concentrations of agonist cause receptor desensitization and suppression of functional responses [103, 233]. For instance, in in vitro experiments in oocytes, high concentrations of α7 nAChR agonists desensitized the receptor and blocked the calcium influx to the endogenous agonist acetylcholine [234]. Similar inhibitory effects on calcium influx were observed with synthetic α7 nAChR agonists at high concentrations [235]. Remarkably though, at lower concentrations a number of α7 nAChR agonists actually may potentiate the acetylcholine-induced response [236]. Estimations of brain levels at which α7 nAChR agonists evoke cognition-enhancing effects are in the range where they potentiate the acetylcholine-induced calcium influx in oocytes. It is therefore reasonable to assume that the pro-cognitive effect of α7 nAChR agonists is due to an enhancement of the acetylcholine-evoked response (known as the “co-agonist hypothesis,” see [236, 237]). This assumption is supported by preclinical data demonstrating an additive pro-cognitive effect of donepezil and the α7 nAChR agonist encenicline [236]. Although not yet reported, a similar co-agonist effect may occur at α7 receptors expressed by immune cells. If true, this would imply that low doses of a α7 nAChR agonist would be sufficient to enhance the anti-inflammatory response to the endogenous agonists (choline and/or acetylcholine).
In reviews dealing with potential indications of selective α7 nAChR agonists, the anti-inflammatory activity has never taken central stage (at least up to recently, see [64, 108]). The role that inflammation plays in the pathophysiology of psychiatric disorders is, however, well recognized [130, 140, 167, 171, 176, 238]. In the current review, we argue that several alternative indications for α7 agonists may be delineated from their effect on inflammation. Treatment with α7 nAChRs agonist may result in inhibition of the pro-inflammatory enzyme, GSK3β. In this respect, the treatment with α7-agonists resembles lithium treatment. This similarity may hold true not only for an indication like bipolar disorder (improvement in both manic and depressive symptoms) but also for suicide and neurological disorders. Suicide is a major cause of death in depression, bipolar disorder, and schizophrenia. Clinical data have accumulated which indicate that inflammation and microglia M1-polarization contribute to the pathophysiology of suicide independent from the underlying psychiatric disease [239–242]. Diverse pro-inflammatory mechanisms such as autoimmunity, neurotropic pathogens, stress, or traumatic brain injury have been documented in suicidal patients [243]. Since α7 nAChR stimulation in immune cells can result in GSK3β inhibition, and since the GSK3-inhibitor, lithium [244], is a recognized anti-inflammatory [245] and anti-suicidal compound [246, 247], one may propose the use of nicotine α7-agonists for prevention of suicide. Chronic treatment with lithium by virtue of its GSK3β inhibitory effect also ameliorated the disease processes in preclinical models of multiple sclerosis [248], Alzheimer [249–251], and Parkinson’s disease [252–254]. It should be noted though that GSK3β inhibition by lithium in these models is just a symptomatic treatment and does not stop the underlying pathological processes. Consequently, once the pharmacotherapy is interrupted, the disease is likely to return. Nevertheless, treatment with a α7 nAChR agonist might constitute a safe alternative to lithium and one could propose the use of α7 nAChR agonists for these neurological disorders.
In contrast to lithium treatment, the beneficial effect of α7 nAChR stimulation is lost when the receptor is defective (or missing all together). An example was presented in the autism section, and also in late-stage Alzheimer’s disease, the nicotine receptor owing to the interaction with the β-amyloid protein may become severely dysfunctional. One potential explanation for the lack of robust effects of α7 nAChR agonists on cognitive function in schizophrenia is the rapid desensitization of α7 nAChRs. This is a general concern and applies to any α7 nAChR agonist indication. A simple biomarker test for quantification of α7 nAChR stimulation on an inflammation read-out would thus be highly desirable. Perhaps the receptor desensitization of α7 nAChRs may be less of an issue for inflammation-related indications. In this context, it is remarkable that basically all α7 agonists were effective against negative symptoms of schizophrenia (while effects on cognition were equivocal). We would also expect that the beneficial effects of smoking and cholinesterase-inhibitor-treatment would be absent if α7 nAChRs would be desensitized for most of the time. However, it must be admitted that a thorough investigation of the concentration/response relationship for the anti-inflammatory effect of any of the selective α7 nAChR agonists remains to be determined.
Since epidemiological data suggest that smoking exerts a protective effect in Parkinson’s disease, α7 nAChR agonists might be a preferable alternative to cholinesterase inhibition and smoking. Registration studies for prophylactic indications such as in Parkinson’s or Alzheimer’s disease are however difficult, long lasting, and expensive, and it is unlikely that companies will invest in these indications without the opportunity for registration in an acute disorder. What could be the pioneer indication for α7 nAChR agonists? Based on clinical information from vagus nerve stimulation treatment, depression would be a logical choice. Unfortunately, depression studies generally suffer from a high placebo response, making also the indication ‘depression’ less attractive.
We currently favor l-DOPA-induced dyskinesia as possible ‘pioneer indication.’ It has been published that two chemically distinct α7 nAChR agonists, ABT-107 and AQW051, suppress l-DOPA-induced dyskinesias in (MPTP-treated) Parkinsonian monkeys [255, 256]. The precise mechanism by which this response is brought about remains unknown; however, again it may involve GSK3 inhibition, since low-dose lithium was also recently shown to be active in a similar Parkinson model in mice [254]. The beneficial effect of α7 nAChR agonists may involve attenuation of MPTP-induced neuroinflammation and protection dopamine neurons in the substantia nigra pars compacta [206]. Thus, efficacy for α7 nAChR agonists in clinical trials of l-DOPA-induced dyskinesias could serve as a clinical entry point to pave the way for indications where longer treatment regimens are warranted.
References
Marutle A, Zhang X, Court J, Piggott M, Johnson M, Perry R, Perry E, Nordberg A (2001) Laminar distribution of nicotinic receptor subtypes in cortical regions in schizophrenia. J Chem Neuroanat 22:115–126
Hurst R, Rollema H, Bertrand D (2013) Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther 137:22–54. doi:10.1016/j.pharmthera.2012.08.012
Dineley KT, Pandya AA, Yakel JL (2015) Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci 36:96–108. doi:10.1016/j.tips.2014.12.002
John D, Shelukhina I, Yanagawa Y, Deuchars J, Henderson Z (2015) Functional alpha7 nicotinic receptors are expressed on immature granule cells of the postnatal dentate gyrus. Brain Res 1601:15–30. doi:10.1016/j.brainres.2014.12.041
Shen JX, Yakel JL (2012) Functional alpha7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J Mol Neurosci 48:14–21. doi:10.1007/s12031-012-9719-3
Vijayaraghavan S, Karami A, Aeinehband S, Behbahani H, Grandien A, Nilsson B, Ekdahl KN, Lindblom RP, Piehl F, Darreh-Shori T (2013) Regulated extracellular choline acetyltransferase activity—the plausible missing link of the distant action of acetylcholine in the cholinergic anti-inflammatory pathway. PLoS One 8:e65936. doi:10.1371/journal.pone.0065936
Sinkus ML, Graw S, Freedman R, Ross RG, Lester HA, Leonard S (2015) The human CHRNA7 and CHRFAM7A genes: a review of the genetics, regulation, and function. Neuropharmacology 96:274–288. doi:10.1016/j.neuropharm.2015.02.006
Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR, Tan J (2004) Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem 89:337–343. doi:10.1046/j.1471-4159.2004.02347.x
De Simone R, Ajmone-Cat MA, Carnevale D, Minghetti L (2005) Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J Neuroinflamm 2:4. doi:10.1186/1742-2094-2-4
Suzuki T, Hide I, Matsubara A, Hama C, Harada K, Miyano K, Andra M, Matsubayashi H, Sakai N, Kohsaka S, Inoue K, Nakata Y (2006) Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res 83:1461–1470. doi:10.1002/jnr.20850
Park HJ, Lee PH, Ahn YW, Choi YJ, Lee G, Lee DY, Chung ES, Jin BK (2007) Neuroprotective effect of nicotine on dopaminergic neurons by anti-inflammatory action. Eur J Neurosci 26:79–89. doi:10.1111/j.1460-9568.2007.05636.x
Takata K, Kitamura Y, Saeki M, Terada M, Kagitani S, Kitamura R, Fujikawa Y, Maelicke A, Tomimoto H, Taniguchi T, Shimohama S (2010) Galantamine-induced amyloid-{beta} clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J Biol Chem 285:40180–40191. doi:10.1074/jbc.M110.142356
Parada E, Egea J, Buendia I, Negredo P, Cunha AC, Cardoso S, Soares MP, Lopez MG (2013) The microglial alpha7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid Redox Signal 19:1135–1148. doi:10.1089/ars.2012.4671
Velez-Fort M, Audinat E, Angulo MC (2009) Functional alpha 7-containing nicotinic receptors of NG2-expressing cells in the hippocampus. Glia 57:1104–1114. doi:10.1002/glia.20834
Liu Q, Huang Y, Shen J, Steffensen S, Wu J (2012) Functional alpha7beta2 nicotinic acetylcholine receptors expressed in hippocampal interneurons exhibit high sensitivity to pathological level of amyloid beta peptides. BMC Neurosci 13:155. doi:10.1186/1471-2202-13-155
Moretti M, Zoli M, George AA, Lukas RJ, Pistillo F, Maskos U, Whiteaker P, Gotti C (2014) The novel alpha7beta2-nicotinic acetylcholine receptor subtype is expressed in mouse and human basal forebrain: biochemical and pharmacological characterization. Mol Pharmacol 86:306–317. doi:10.1124/mol.114.093377
Frazier CJ, Strowbridge BW, Papke RL (2003) Nicotinic receptors on local circuit neurons in dentate gyrus: a potential role in regulation of granule cell excitability. J Neurophysiol 89:3018–3028. doi:10.1152/jn.01036.2002
Yakel JL (2012) Nicotinic ACh receptors in the hippocampus: role in excitability and plasticity. Nicotine Tob Res 14:1249–1257. doi:10.1093/ntr/nts091
Hamano R, Takahashi HK, Iwagaki H, Yoshino T, Nishibori M, Tanaka N (2006) Stimulation of alpha7 nicotinic acetylcholine receptor inhibits CD14 and the toll-like receptor 4 expression in human monocytes. Shock. 26:358–364. doi:10.1097/01.shk.0000228168.86845.60
Yoshikawa H, Kurokawa M, Ozaki N, Nara K, Atou K, Takada E, Kamochi H, Suzuki N (2006) Nicotine inhibits the production of proinflammatory mediators in human monocytes by suppression of I-kappaB phosphorylation and nuclear factor-kappaB transcriptional activity through nicotinic acetylcholine receptor alpha7. Clin Exp Immunol 146:116–123. doi:10.1111/j.1365-2249.2006.03169.x
Rosas-Ballina M, Goldstein RS, Gallowitsch-Puerta M, Yang L, Valdes-Ferrer SI, Patel NB, Chavan S, Al-Abed Y, Yang H, Tracey KJ (2009) The selective alpha7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol Med 15:195–202. doi:10.2119/molmed.2009.00039
Sato K, Nagayama H, Tadokoro K, Juji T, Takahashi TA (1999) Extracellular signal-regulated kinase, stress-activated protein kinase/c-Jun N-terminal kinase, and p38mapk are involved in IL-10-mediated selective repression of TNF-alpha-induced activation and maturation of human peripheral blood monocyte-derived dendritic cells. J Immunol 162:3865–3872
Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405:458–462. doi:10.1038/35013070
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421:384–388. doi:10.1038/nature01339
Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L (2004) Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 10:1216–1221. doi:10.1038/nm1124
de Lucas-Cerrillo AM, Maldifassi MC, Arnalich F, Renart J, Atienza G, Serantes R, Cruces J, Sanchez-Pacheco A, Andres-Mateos E, Montiel C (2011) Function of partially duplicated human alpha7 nicotinic receptor subunit CHRFAM7A gene: potential implications for the cholinergic anti-inflammatory response. J Biol Chem 286:594–606. doi:10.1074/jbc.M110.180067
Razani-Boroujerdi S, Boyd RT, Davila-Garcia MI, Nandi JS, Mishra NC, Singh SP, Pena-Philippides JC, Langley R, Sopori ML (2007) T cells express alpha7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J Immunol 179:2889–2898
De Rosa MJ, Dionisio L, Agriello E, Bouzat C, Esandi Mdel C (2009) Alpha 7 nicotinic acetylcholine receptor modulates lymphocyte activation. Life Sci. 85:444–449. doi:10.1016/j.lfs.2009.07.010
Skok MV, Kalashnik EN, Koval LN, Tsetlin VI, Utkin YN, Changeux JP, Grailhe R (2003) Functional nicotinic acetylcholine receptors are expressed in B lymphocyte-derived cell lines. Mol Pharmacol 64:885–889. doi:10.1124/mol.64.4.885
Kawashima K, Yoshikawa K, Fujii YX, Moriwaki Y, Misawa H (2007) Expression and function of genes encoding cholinergic components in murine immune cells. Life Sci 80:2314–2319. doi:10.1016/j.lfs.2007.02.036
Saeed RW, Varma S, Peng-Nemeroff T, Sherry B, Balakhaneh D, Huston J, Tracey KJ, Al-Abed Y, Metz CN (2005) Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med 201:1113–1123. doi:10.1084/jem.20040463
Zia S, Ndoye A, Lee TX, Webber RJ, Grando SA (2000) Receptor-mediated inhibition of keratinocyte migration by nicotine involves modulations of calcium influx and intracellular concentration. J Pharmacol Exp Ther 293:973–981
Chimienti F, Hogg RC, Plantard L, Lehmann C, Brakch N, Fischer J, Huber M, Bertrand D, Hohl D (2003) Identification of SLURP-1 as an epidermal neuromodulator explains the clinical phenotype of Mal de Meleda. Hum Mol Genet 12:3017–3024. doi:10.1093/hmg/ddg320
Dowling O, Rochelson B, Way K, Al-Abed Y, Metz CN (2007) Nicotine inhibits cytokine production by placenta cells via NFkappaB: potential role in pregnancy-induced hypertension. Mol Med 13:576–583. doi:10.2119/2007-00067.Dowling
Fu XW, Lindstrom J, Spindel ER (2009) Nicotine activates and up-regulates nicotinic acetylcholine receptors in bronchial epithelial cells. Am J Respir Cell Mol Biol 41:93–99. doi:10.1165/rcmb.2008-0352OC
Schedel A, Thornton S, Schloss P, Kluter H, Bugert P (2011) Human platelets express functional alpha7-nicotinic acetylcholine receptors. Arterioscler Thromb Vasc Biol 31:928–934. doi:10.1161/ATVBAHA.110.218297
Cancello R, Zulian A, Maestrini S, Mencarelli M, Della Barba A, Invitti C, Liuzzi A, Di Blasio AM (2012) The nicotinic acetylcholine receptor alpha7 in subcutaneous mature adipocytes: downregulation in human obesity and modulation by diet-induced weight loss. Int J Obes (Lond) 36:1552–1557. doi:10.1038/ijo.2011.275
Waldburger JM, Boyle DL, Pavlov VA, Tracey KJ, Firestein GS (2008) Acetylcholine regulation of synoviocyte cytokine expression by the alpha7 nicotinic receptor. Arthritis Rheum 58:3439–3449. doi:10.1002/art.23987
Fan YY, Yu TS, Wang T, Liu WW, Zhao R, Zhang ST, Ma WX, Zheng JL, Guan DW (2011) Nicotinic acetylcholine receptor alpha7 subunit is time-dependently expressed in distinct cell types during skin wound healing in mice. Histochem Cell Biol 135:375–387. doi:10.1007/s00418-011-0798-y
Bray C, Son JH, Meizel S (2005) Acetylcholine causes an increase of intracellular calcium in human sperm. Mol Hum Reprod 11:881–889. doi:10.1093/molehr/gah245
Del Signore A, Gotti C, Rizzo A, Moretti M, Paggi P (2004) Nicotinic acetylcholine receptor subtypes in the rat sympathetic ganglion: pharmacological characterization, subcellular distribution and effect of pre- and postganglionic nerve crush. J Neuropathol Exp Neurol 63:138–150
Baxter JC, Ramachandra R, Mayne DR, Elmslie KS (2014) Functional expression of alpha7-nicotinic acetylcholine receptors by muscle afferent neurons. J Neurophysiol 112:1549–1558. doi:10.1152/jn.00035.2014
Blumenthal EM, Conroy WG, Romano SJ, Kassner PD, Berg DK (1997) Detection of functional nicotinic receptors blocked by alpha-bungarotoxin on PC12 cells and dependence of their expression on post-translational events. J Neurosci 17:6094–6104
Peng X, Katz M, Gerzanich V, Anand R, Lindstrom J (1994) Human alpha 7 acetylcholine receptor: cloning of the alpha 7 subunit from the SH-SY5Y cell line and determination of pharmacological properties of native receptors and functional alpha 7 homomers expressed in Xenopus oocytes. Mol Pharmacol 45:546–554
Cooper ST, Millar NS (1997) Host cell-specific folding and assembly of the neuronal nicotinic acetylcholine receptor alpha7 subunit. J Neurochem 68:2140–2151
Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang LH, Hudson L, Lin X, Patel N, Johnson SM, Chavan S, Goldstein RS, Czura CJ, Miller EJ, Al-Abed Y, Tracey KJ, Pavlov VA (2008) Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med 14:567–574. doi:10.2119/2008-00079.Parrish
Cui WY, Zhao S, Polanowska-Grabowska R, Wang J, Wei J, Dash B, Chang SL, Saucerman JJ, Gu J, Li MD (2013) Identification and characterization of poly(I:C)-induced molecular responses attenuated by nicotine in mouse macrophages. Mol Pharmacol 83:61–72. doi:10.1124/mol.112.081497
Moriwaki Y, Watanabe Y, Shinagawa T, Kai M, Miyazawa M, Okuda T, Kawashima K, Yabashi A, Waguri S, Misawa H (2009) Primary sensory neuronal expression of SLURP-1, an endogenous nicotinic acetylcholine receptor ligand. Neurosci Res 64:403–412. doi:10.1016/j.neures.2009.04.014
Sugano N, Shimada K, Ito K, Murai S (1998) Nicotine inhibits the production of inflammatory mediators in U937 cells through modulation of nuclear factor-kappaB activation. Biochem Biophys Res Commun 252:25–28
Peng JH, Lucero L, Fryer J, Herl J, Leonard SS, Lukas RJ (1999) Inducible, heterologous expression of human alpha7-nicotinic acetylcholine receptors in a native nicotinic receptor-null human clonal line. Brain Res 825:172–179
Charpantier E, Wiesner A, Huh KH, Ogier R, Hoda JC, Allaman G, Raggenbass M, Feuerbach D, Bertrand D, Fuhrer C (2005) Alpha7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J Neurosci 25:9836–9849. doi:10.1523/JNEUROSCI.3497-05.2005
Feuerbach D, Lingenhohl K, Dobbins P, Mosbacher J, Corbett N, Nozulak J, Hoyer D (2005) Coupling of human nicotinic acetylcholine receptors alpha 7 to calcium channels in GH3 cells. Neuropharmacology 48:215–227. doi:10.1016/j.neuropharm.2004.10.003
Villiger Y, Szanto I, Jaconi S, Blanchet C, Buisson B, Krause KH, Bertrand D, Romand JA (2002) Expression of an alpha7 duplicate nicotinic acetylcholine receptor-related protein in human leukocytes. J Neuroimmunol 126:86–98
Jones IW, Wonnacott S (2005) Why doesn’t nicotinic ACh receptor immunoreactivity knock out? Trends Neurosci 28:343–345. doi:10.1016/j.tins.2005.04.010
Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S (2002) Methyllycaconitine is a potent antagonist of alpha-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther 302:197–204
Gergalova G, Lykhmus O, Komisarenko S, Skok M (2014) alpha7 nicotinic acetylcholine receptors control cytochrome c release from isolated mitochondria through kinase-mediated pathways. Int J Biochem Cell Biol 49:26–31. doi:10.1016/j.biocel.2014.01.001
Lykhmus O, Gergalova G, Koval L, Zhmak M, Komisarenko S, Skok M (2014) Mitochondria express several nicotinic acetylcholine receptor subtypes to control various pathways of apoptosis induction. Int J Biochem Cell Biol 53:246–252. doi:10.1016/j.biocel.2014.05.030
Fucile S (2004) Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 35:1–8
Sharma G, Vijayaraghavan S (2001) Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci USA 98:4148–4153. doi:10.1073/pnas.071540198
Stevens TR, Krueger SR, Fitzsimonds RM, Picciotto MR (2003) Neuroprotection by nicotine in mouse primary cortical cultures involves activation of calcineurin and L-type calcium channel inactivation. J Neurosci 23:10093–10099
Cheng Q, Yakel JL (2015) Activation of alpha7 nicotinic acetylcholine receptors increases intracellular cAMP levels via activation of AC1 in hippocampal neurons. Neuropharmacology 95:405–414. doi:10.1016/j.neuropharm.2015.04.016
Medeiros R, Castello NA, Cheng D, Kitazawa M, Baglietto-Vargas D, Green KN, Esbenshade TA, Bitner RS, Decker MW, LaFerla FM (2014) alpha7 Nicotinic receptor agonist enhances cognition in aged 3xTg-AD mice with robust plaques and tangles. Am J Pathol 184:520–529. doi:10.1016/j.ajpath.2013.10.010
Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A (2001) alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem 276:13541–13546. doi:10.1074/jbc.M008035200
Egea J, Buendia I, Parada E, Navarro E, Leon R, Lopez MG (2015) Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol. doi:10.1016/j.bcp.2015.07.032
Shaw S, Bencherif M, Marrero MB (2002) Janus kinase 2, an early target of alpha 7 nicotinic acetylcholine receptor-mediated neuroprotection against Abeta-(1-42) amyloid. J Biol Chem 277:44920–44924. doi:10.1074/jbc.M204610200
de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE (2005) Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 6:844–851. doi:10.1038/ni1229
Pena G, Cai B, Liu J, van der Zanden EP, Deitch EA, de Jonge WJ, Ulloa L (2010) Unphosphorylated STAT3 modulates alpha 7 nicotinic receptor signaling and cytokine production in sepsis. Eur J Immunol 40:2580–2589. doi:10.1002/eji.201040540
Maldifassi MC, Atienza G, Arnalich F, Lopez-Collazo E, Cedillo JL, Martin-Sanchez C, Bordas A, Renart J, Montiel C (2014) A new IRAK-M-mediated mechanism implicated in the anti-inflammatory effect of nicotine via alpha7 nicotinic receptors in human macrophages. PLoS One 9:e108397. doi:10.1371/journal.pone.0108397
Tracey KJ (2009) Reflex control of immunity. Nat Rev Immunol 9:418–428. doi:10.1038/nri2566
Tyagi E, Agrawal R, Nath C, Shukla R (2010) Inhibitory role of cholinergic system mediated via alpha7 nicotinic acetylcholine receptor in LPS-induced neuro-inflammation. Innate Immun 16:3–13. doi:10.1177/1753425909104680
Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B, Tracey KJ, Ulloa L (2006) Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 203:1623–1628. doi:10.1084/jem.20052362
Kees MG, Pongratz G, Kees F, Scholmerich J, Straub RH (2003) Via beta-adrenoceptors, stimulation of extrasplenic sympathetic nerve fibers inhibits lipopolysaccharide-induced TNF secretion in perfused rat spleen. J Neuroimmunol 145:77–85
Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ (2011) Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334:98–101. doi:10.1126/science.1209985
Vida G, Pena G, Deitch EA, Ulloa L (2011) alpha7-cholinergic receptor mediates vagal induction of splenic norepinephrine. J Immunol 186:4340–4346. doi:10.4049/jimmunol.1003722
Martelli D, McKinley MJ, McAllen RM (2014) The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci 182:65–69. doi:10.1016/j.autneu.2013.12.007
Carrasco-Serrano C, Criado M (2004) Glucocorticoid activation of the neuronal nicotinic acetylcholine receptor alpha7 subunit gene: involvement of transcription factor Egr-1. FEBS Lett 566:247–250. doi:10.1016/j.febslet.2004.04.049
Pandya AA, Yakel JL (2013) Effects of neuronal nicotinic acetylcholine receptor allosteric modulators in animal behavior studies. Biochem Pharmacol 86:1054–1062. doi:10.1016/j.bcp.2013.05.018
Bagdas D, Targowska-Duda KM, Lopez JJ, Perez EG, Arias HR, Damaj MI (2015) The antinociceptive and antiinflammatory properties of 3-furan-2-yl-N-p-tolyl-acrylamide, a positive allosteric modulator of alpha7 nicotinic acetylcholine receptors in mice. Anesth Analg 121:1369–1377. doi:10.1213/ANE.0000000000000902
Kalappa BI, Sun F, Johnson SR, Jin K, Uteshev VV (2013) A positive allosteric modulator of alpha7 nAChRs augments neuroprotective effects of endogenous nicotinic agonists in cerebral ischaemia. Br J Pharmacol 169:1862–1878. doi:10.1111/bph.12247
Sun F, Jin K, Uteshev VV (2013) A type-II positive allosteric modulator of alpha7 nAChRs reduces brain injury and improves neurological function after focal cerebral ischemia in rats. PLoS One 8:e73581. doi:10.1371/journal.pone.0073581
Gatson JW, Simpkins JW, Uteshev VV (2015) High therapeutic potential of positive allosteric modulation of alpha7 nAChRs in a rat model of traumatic brain injury: proof-of-concept. Brain Res Bull 112:35–41. doi:10.1016/j.brainresbull.2015.01.008
Carnevale D, De Simone R, Minghetti L (2007) Microglia-neuron interaction in inflammatory and degenerative diseases: role of cholinergic and noradrenergic systems. CNS Neurol Disord: Drug Targets 6:388–397
Gao Z, Nissen JC, Ji K, Tsirka SE (2014) The experimental autoimmune encephalomyelitis disease course is modulated by nicotine and other cigarette smoke components. PLoS One 9:e107979. doi:10.1371/journal.pone.0107979
Cho CH, Song W, Leitzell K, Teo E, Meleth AD, Quick MW, Lester RA (2005) Rapid upregulation of alpha7 nicotinic acetylcholine receptors by tyrosine dephosphorylation. J Neurosci 25:3712–3723. doi:10.1523/JNEUROSCI.5389-03.2005
Baer K, Burli T, Huh KH, Wiesner A, Erb-Vogtli S, Gockeritz-Dujmovic D, Moransard M, Nishimune A, Rees MI, Henley JM, Fritschy JM, Fuhrer C (2007) PICK1 interacts with alpha7 neuronal nicotinic acetylcholine receptors and controls their clustering. Mol Cell Neurosci 35:339–355. doi:10.1016/j.mcn.2007.03.009
Liu Z, Conroy WG, Stawicki TM, Nai Q, Neff RA, Berg DK (2008) EphB receptors co-distribute with a nicotinic receptor subtype and regulate nicotinic downstream signaling in neurons. Mol Cell Neurosci 38:236–244. doi:10.1016/j.mcn.2008.02.013
Castelan F, Castillo M, Mulet J, Sala S, Sala F, Dominguez Del Toro E, Criado M (2008) Molecular characterization and localization of the RIC-3 protein, an effector of nicotinic acetylcholine receptor expression. J Neurochem 105:617–627. doi:10.1111/j.1471-4159.2007.05169.x
Grando SA (2008) Basic and clinical aspects of non-neuronal acetylcholine: biological and clinical significance of non-canonical ligands of epithelial nicotinic acetylcholine receptors. J Pharmacol Sci 106:174–179
Hoppman-Chaney N, Wain K, Seger PR, Superneau DW, Hodge JC (2013) Identification of single gene deletions at 15q13.3: further evidence that CHRNA7 causes the 15q13.3 microdeletion syndrome phenotype. Clin Genet 83:345–351. doi:10.1111/j.1399-0004.2012.01925.x
Finlay-Schultz J, Canastar A, Short M, El Gazzar M, Coughlan C, Leonard S (2011) Transcriptional repression of the alpha7 nicotinic acetylcholine receptor subunit gene (CHRNA7) by activating protein-2alpha (AP-2alpha). J Biol Chem 286:42123–42132. doi:10.1074/jbc.M111.276014
Yasui DH, Scoles HA, Horike S, Meguro-Horike M, Dunaway KW, Schroeder DI, Lasalle JM (2011) 15q11.2-13.3 chromatin analysis reveals epigenetic regulation of CHRNA7 with deficiencies in Rett and autism brain. Hum Mol Genet 20:4311–4323. doi:10.1093/hmg/ddr357
de Jonge WJ, Ulloa L (2007) The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 151:915–929. doi:10.1038/sj.bjp.0707264
Komal P, Estakhr J, Kamran M, Renda A, Nashmi R (2015) cAMP-dependent protein kinase inhibits alpha7 nicotinic receptor activity in layer 1 cortical interneurons through activation of D1/D5 dopamine receptors. J Physiol. doi:10.1113/JP270469
Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX (2001) The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci 21:7463–7473
Dobelis P, Staley KJ, Cooper DC (2012) Lack of modulation of nicotinic acetylcholine alpha-7 receptor currents by kynurenic acid in adult hippocampal interneurons. PLoS One 7:e41108. doi:10.1371/journal.pone.0041108
Wang Y, Xiao C, Indersmitten T, Freedman R, Leonard S, Lester HA (2014) The duplicated alpha7 subunits assemble and form functional nicotinic receptors with the full-length alpha7. J Biol Chem 289:26451–26463. doi:10.1074/jbc.M114.582858
Costantini TW, Dang X, Coimbra R, Eliceiri BP, Baird A (2015) CHRFAM7A, a human-specific and partially duplicated alpha7-nicotinic acetylcholine receptor gene with the potential to specify a human-specific inflammatory response to injury. J Leukoc Biol 97:247–257. doi:10.1189/jlb.4RU0814-381R
Benfante R, Antonini RA, De Pizzol M, Gotti C, Clementi F, Locati M, Fornasari D (2011) Expression of the alpha7 nAChR subunit duplicate form (CHRFAM7A) is down-regulated in the monocytic cell line THP-1 on treatment with LPS. J Neuroimmunol 230:74–84. doi:10.1016/j.jneuroim.2010.09.008
Woolf NJ, Butcher LL (2011) Cholinergic systems mediate action from movement to higher consciousness. Behav Brain Res 221:488–498. doi:10.1016/j.bbr.2009.12.046
Rowe AR, Mercer L, Casetti V, Sendt KV, Giaroli G, Shergill SS, Tracy DK (2015) Dementia praecox redux: a systematic review of the nicotinic receptor as a target for cognitive symptoms of schizophrenia. J Psychopharmacol 29:197–211. doi:10.1177/0269881114564096
Briggs CA, McKenna DG (1998) Activation and inhibition of the human alpha7 nicotinic acetylcholine receptor by agonists. Neuropharmacology 37:1095–1102
Wessler I, Kirkpatrick CJ (2008) Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol 154:1558–1571. doi:10.1038/bjp.2008.185
Quick MW, Lester RA (2002) Desensitization of neuronal nicotinic receptors. J Neurobiol 53:457–478. doi:10.1002/neu.10109
Papke RL, Kem WR, Soti F, Lopez-Hernandez GY, Horenstein NA (2009) Activation and desensitization of nicotinic alpha7-type acetylcholine receptors by benzylidene anabaseines and nicotine. J Pharmacol Exp Ther 329:791–807. doi:10.1124/jpet.108.150151
Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV (1998) Acetylcholine activates an alpha-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci 18:1187–1195
Wessler I, Reinheimer T, Klapproth H, Schneider FJ, Racke K, Hammer R (1997) Mammalian glial cells in culture synthesize acetylcholine. Naunyn Schmiedebergs Arch Pharmacol 356:694–697
Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX (1997) Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci 9:2734–2742
Bencherif M, Narla ST, Stachowiak MS (2014) Alpha7 neuronal nicotinic receptor: a pluripotent target for diseases of the central nervous system. CNS Neurol Disord Drug Targets 13:836–845
Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A (2008) Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry 65:409–415. doi:10.1001/archpsyc.65.4.409
Kendler KS, Hettema JM, Butera F, Gardner CO, Prescott CA (2003) Life event dimensions of loss, humiliation, entrapment, and danger in the prediction of onsets of major depression and generalized anxiety. Arch Gen Psychiatry 60:789–796. doi:10.1001/archpsyc.60.8.789
Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS, Turner RB (2012) Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci USA 109:5995–5999. doi:10.1073/pnas.1118355109
Copeland WE, Wolke D, Lereya ST, Shanahan L, Worthman C, Costello EJ (2014) Childhood bullying involvement predicts low-grade systemic inflammation into adulthood. Proc Natl Acad Sci USA 111:7570–7575. doi:10.1073/pnas.1323641111
Slavich GM, Cole SW (2013) The emerging field of human social genomics. Clin Psychol Sci 1:331–348
Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, Heim CM (2006) Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry 163:1630–1633. doi:10.1176/appi.ajp.163.9.1630
Miller GE, Chen E, Fok AK, Walker H, Lim A, Nicholls EF, Cole S, Kobor MS (2009) Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc Natl Acad Sci USA 106:14716–14721. doi:10.1073/pnas.0902971106
Weinstein AA, Deuster PA, Francis JL, Bonsall RW, Tracy RP, Kop WJ (2010) Neurohormonal and inflammatory hyper-responsiveness to acute mental stress in depression. Biol Psychol 84:228–234. doi:10.1016/j.biopsycho.2010.01.016
Fang FC (2004) Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol 2:820–832. doi:10.1038/nrmicro1004
Sorci G, Faivre B (2009) Inflammation and oxidative stress in vertebrate host-parasite systems. Philos Trans R Soc Lond B Biol Sci 364:71–83. doi:10.1098/rstb.2008.0151
Neurauter G, Schrocksnadel K, Scholl-Burgi S, Sperner-Unterweger B, Schubert C, Ledochowski M, Fuchs D (2008) Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr Drug Metab 9:622–627
Quik M, Zhang D, McGregor M, Bordia T (2015) Alpha7 nicotinic receptors as therapeutic targets for Parkinson’s disease. Biochem Pharmacol. doi:10.1016/j.bcp.2015.06.014
Bitner RS, Nikkel AL, Markosyan S, Otte S, Puttfarcken P, Gopalakrishnan M (2009) Selective alpha7 nicotinic acetylcholine receptor activation regulates glycogen synthase kinase3beta and decreases tau phosphorylation in vivo. Brain Res 1265:65–74. doi:10.1016/j.brainres.2009.01.069
Garcia-Oscos F, Pena D, Housini M, Cheng D, Lopez D, Cuevas-Olguin R, Saderi N, Salgado Delgado R, Galindo Charles L, Salgado Burgos H, Rose-John S, Flores G, Kilgard MP, Atzori M (2015) Activation of the anti-inflammatory reflex blocks lipopolysaccharide-induced decrease in synaptic inhibition in the temporal cortex of the rat. J Neurosci Res. 93:859–865. doi:10.1002/jnr.23550
Howland RH (2014) Vagus nerve stimulation. Curr Behav Neurosci Rep 1:64–73. doi:10.1007/s40473-014-0010-5
Marangell LB, Rush AJ, George MS, Sackeim HA, Johnson CR, Husain MM, Nahas Z, Lisanby SH (2002) Vagus nerve stimulation (VNS) for major depressive episodes: one year outcomes. Biol Psychiatry 51:280–287
Rush AJ, Sackeim HA, Marangell LB, George MS, Brannan SK, Davis SM, Lavori P, Howland R, Kling MA, Rittberg B, Carpenter L, Ninan P, Moreno F, Schwartz T, Conway C, Burke M, Barry JJ (2005) Effects of 12 months of vagus nerve stimulation in treatment-resistant depression: a naturalistic study. Biol Psychiatry 58:355–363. doi:10.1016/j.biopsych.2005.05.024
Bajbouj M, Merkl A, Schlaepfer TE, Frick C, Zobel A, Maier W, O’Keane V, Corcoran C, Adolfsson R, Trimble M, Rau H, Hoff HJ, Padberg F, Muller-Siecheneder F, Audenaert K, van den Abbeele D, Matthews K, Christmas D, Eljamel S, Heuser I (2010) Two-year outcome of vagus nerve stimulation in treatment-resistant depression. J Clin Psychopharmacol 30:273–281. doi:10.1097/JCP.0b013e3181db8831
Slattery DA, Neumann ID (2010) Chronic icv oxytocin attenuates the pathological high anxiety state of selectively bred Wistar rats. Neuropharmacology 58:56–61. doi:10.1016/j.neuropharm.2009.06.038
McCann MJ, Rogers RC (1990) Oxytocin excites gastric-related neurones in rat dorsal vagal complex. J Physiol 428:95–108
Clodi M, Vila G, Geyeregger R, Riedl M, Stulnig TM, Struck J, Luger TA, Luger A (2008) Oxytocin alleviates the neuroendocrine and cytokine response to bacterial endotoxin in healthy men. Am J Physiol Endocrinol Metab. 295:E686–E691. doi:10.1152/ajpendo.90263.2008
Najjar S, Pearlman DM (2015) Neuroinflammation and white matter pathology in schizophrenia: systematic review. Schizophr Res 161:102–112. doi:10.1016/j.schres.2014.04.041
Meyer U (2013) Developmental neuroinflammation and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 42:20–34. doi:10.1016/j.pnpbp.2011.11.003
Aberg KA, Liu Y, Bukszar J, McClay JL, Khachane AN, Andreassen OA, Blackwood D, Corvin A, Djurovic S, Gurling H, Ophoff R, Pato CN, Pato MT, Riley B, Webb T, Kendler K, O’Donovan M, Craddock N, Kirov G, Owen M, Rujescu D, St Clair D, Werge T, Hultman CM, Delisi LE, Sullivan P, van den Oord EJ (2013) A comprehensive family-based replication study of schizophrenia genes. JAMA Psychiatry 70:573–581. doi:10.1001/jamapsychiatry.2013.288
Marco C, Antonio D, Antonina S, Alessandro S, Concetta C (2015) Genes involved in pruning and inflammation are enriched in a large mega-sample of patients affected by schizophrenia and bipolar disorder and controls. Psychiatry Res 228:945–949. doi:10.1016/j.psychres.2015.06.013
van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, Luurtsema G, Windhorst AD, Cahn W, Lammertsma AA, Kahn RS (2008) Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry 64:820–822. doi:10.1016/j.biopsych.2008.04.025
Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC (2009) Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med 50:1801–1807. doi:10.2967/jnumed.109.066647
Monji A, Kato T, Kanba S (2009) Cytokines and schizophrenia: microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci 63:257–265
Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B (2011) Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 70:663–671. doi:10.1016/j.biopsych.2011.04.013
Sasayama D, Hattori K, Wakabayashi C, Teraishi T, Hori H, Ota M, Yoshida S, Arima K, Higuchi T, Amano N, Kunugi H (2013) Increased cerebrospinal fluid interleukin-6 levels in patients with schizophrenia and those with major depressive disorder. J Psychiatr Res 47:401–406. doi:10.1016/j.jpsychires.2012.12.001
di Penta A, Moreno B, Reix S, Fernandez-Diez B, Villanueva M, Errea O, Escala N, Vandenbroeck K, Comella JX, Villoslada P (2013) Oxidative stress and proinflammatory cytokines contribute to demyelination and axonal damage in a cerebellar culture model of neuroinflammation. PLoS One 8:e54722. doi:10.1371/journal.pone.0054722
Mantyla T, Mantere O, Raij TT, Kieseppa T, Laitinen H, Leiviska J, Torniainen M, Tuominen L, Vaarala O, Suvisaari J (2015) Altered activation of innate immunity associates with white matter volume and diffusion in first-episode psychosis. PLoS One 10:e0125112. doi:10.1371/journal.pone.0125112
Chapman S, Ragg M, McGeechan K (2009) Citation bias in reported smoking prevalence in people with schizophrenia. Aust N Z J Psychiatry 43:277–282. doi:10.1080/00048670802653372
Freedman R, Hall M, Adler LE, Leonard S (1995) Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry 38:22–33. doi:10.1016/0006-3223(94)00252-X
Court J, Spurden D, Lloyd S, McKeith I, Ballard C, Cairns N, Kerwin R, Perry R, Perry E (1999) Neuronal nicotinic receptors in dementia with Lewy bodies and schizophrenia: alpha-bungarotoxin and nicotine binding in the thalamus. J Neurochem 73:1590–1597
Guan ZZ, Zhang X, Blennow K, Nordberg A (1999) Decreased protein level of nicotinic receptor alpha7 subunit in the frontal cortex from schizophrenic brain. NeuroReport 10:1779–1782
Leonard S, Freedman R (2006) Genetics of chromosome 15q13-q14 in schizophrenia. Biol Psychiatry 60:115–122. doi:10.1016/j.biopsych.2006.03.054
Masurel-Paulet A, Andrieux J, Callier P, Cuisset JM, Le Caignec C, Holder M, Thauvin-Robinet C, Doray B, Flori E, Alex-Cordier MP, Beri M, Boute O, Delobel B, Dieux A, Vallee L, Jaillard S, Odent S, Isidor B, Beneteau C, Vigneron J, Bilan F, Gilbert-Dussardier B, Dubourg C, Labalme A, Bidon C, Gautier A, Pernes P, Pinoit JM, Huet F, Mugneret F, Aral B, Jonveaux P, Sanlaville D, Faivre L (2010) Delineation of 15q13.3 microdeletions. Clin Genet 78:149–161. doi:10.1111/j.1399-0004.2010.01374.x
Gillentine MA, Schaaf CP (2015) The human clinical phenotypes of altered CHRNA7 copy number. Biochem Pharmacol. doi:10.1016/j.bcp.2015.06.012
Rakhilin S, Drisdel RC, Sagher D, McGehee DS, Vallejo Y, Green WN (1999) alpha-bungarotoxin receptors contain alpha7 subunits in two different disulfide-bonded conformations. J Cell Biol 146:203–218
Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, Drebing C, Berger R, Venn D, Sirota P, Zerbe G, Olincy A, Ross RG, Adler LE, Freedman R (2002) Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch Gen Psychiatry 59:1085–1096
Sinkus ML, Lee MJ, Gault J, Logel J, Short M, Freedman R, Christian SL, Lyon J, Leonard S (2009) A 2-base pair deletion polymorphism in the partial duplication of the alpha7 nicotinic acetylcholine gene (CHRFAM7A) on chromosome 15q14 is associated with schizophrenia. Brain Res 1291:1–11. doi:10.1016/j.brainres.2009.07.041
Hashimoto K (2015) Targeting of alpha7 nicotinic acetylcholine receptors in the treatment of schizophrenia and the use of auditory sensory gating as a translational biomarker. Curr Pharm Des 21:3797–3806
Beinat C, Banister SD, Herrera M, Law V, Kassiou M (2015) The therapeutic potential of alpha7 nicotinic acetylcholine receptor (alpha7 nAChR) agonists for the treatment of the cognitive deficits associated with schizophrenia. CNS Drugs 29:529–542. doi:10.1007/s40263-015-0260-0
Keefe RS, Meltzer HA, Dgetluck N, Gawryl M, Koenig G, Moebius HJ, Lombardo I, Hilt DC (2015) Randomized, double-blind, placebo-controlled study of encenicline, an alpha7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology. doi:10.1038/npp.2015.176
Noroozian M, Ghasemi S, Hosseini SM, Modabbernia A, Khodaie-Ardakani MR, Mirshafiee O, Farokhnia M, Tajdini M, Rezaei F, Salehi B, Ashrafi M, Yekehtaz H, Tabrizi M, Akhondzadeh S (2013) A placebo-controlled study of tropisetron added to risperidone for the treatment of negative symptoms in chronic and stable schizophrenia. Psychopharmacology 228:595–602. doi:10.1007/s00213-013-3064-2
Umbricht D, Keefe RS, Murray S, Lowe DA, Porter R, Garibaldi G, Santarelli L (2014) A randomized, placebo-controlled study investigating the nicotinic alpha7 agonist, RG3487, for cognitive deficits in schizophrenia. Neuropsychopharmacology 39:1568–1577. doi:10.1038/npp.2014.17
Zhang ZJ, Kang WH, Li Q, Wang XY, Yao SM, Ma AQ (2006) Beneficial effects of ondansetron as an adjunct to haloperidol for chronic, treatment-resistant schizophrenia: a double-blind, randomized, placebo-controlled study. Schizophr Res 88:102–110. doi:10.1016/j.schres.2006.07.010
Lieberman JA, Dunbar G, Segreti AC, Girgis RR, Seoane F, Beaver JS, Duan N, Hosford DA (2013) A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology 38:968–975. doi:10.1038/npp.2012.259
Freedman R, Olincy A, Buchanan RW, Harris JG, Gold JM, Johnson L, Allensworth D, Guzman-Bonilla A, Clement B, Ball MP, Kutnick J, Pender V, Martin LF, Stevens KE, Wagner BD, Zerbe GO, Soti F, Kem WR (2008) Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am J Psychiatry 165:1040–1047. doi:10.1176/appi.ajp.2008.07071135
Michelmore S, Croskery K, Nozulak J, Hoyer D, Longato R, Weber A, Bouhelal R, Feuerbach D (2002) Study of the calcium dynamics of the human alpha4beta2, alpha3beta4 and alpha1beta1gammadelta nicotinic acetylcholine receptors. Naunyn Schmiedebergs Arch Pharmacol 366:235–245. doi:10.1007/s00210-002-0589-z
Boggs DL, Carlson J, Cortes-Briones J, Krystal JH, D’Souza DC (2014) Going up in smoke? A review of nAChRs-based treatment strategies for improving cognition in schizophrenia. Curr Pharm Des 20:5077–5092
Ancin I, Barabash A, Vazquez-Alvarez B, Santos JL, Sanchez-Morla E, Martinez JL, Aparicio A, Pelaez JC, Diaz JA (2010) Evidence for association of the non-duplicated region of CHRNA7 gene with bipolar disorder but not with Schizophrenia. Psychiatr Genet 20:289–297. doi:10.1097/YPG.0b013e32833a9b7a
Lisi DM (2002) Lotronex withdrawal. Arch Intern Med 162:101
Spiller RC (2011) Targeting the 5-HT(3) receptor in the treatment of irritable bowel syndrome. Curr Opin Pharmacol 11:68–74. doi:10.1016/j.coph.2011.02.005
Zhang L, Zhao J (2014) Profile of minocycline and its potential in the treatment of schizophrenia. Neuropsychiatr Dis Treat 10:1103–1111. doi:10.2147/NDT.S64236
Benazzi F (2007) Bipolar disorder–focus on bipolar II disorder and mixed depression. Lancet 369:935–945. doi:10.1016/S0140-6736(07)60453-X
Leboyer M, Soreca I, Scott J, Frye M, Henry C, Tamouza R, Kupfer DJ (2012) Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord 141:1–10. doi:10.1016/j.jad.2011.12.049
Maletic V, Raison C (2014) Integrated neurobiology of bipolar disorder. Front Psychiatry 5:98. doi:10.3389/fpsyt.2014.00098
Kim HK, Chen W, Andreazza AC (2015) The potential role of the NLRP3 inflammasome as a link between mitochondrial complex I dysfunction and inflammation in bipolar disorder. Neural Plast 2015:408136. doi:10.1155/2015/408136
Etain B, Henry C, Bellivier F, Mathieu F, Leboyer M (2008) Beyond genetics: childhood affective trauma in bipolar disorder. Bipolar Disord 10:867–876. doi:10.1111/j.1399-5618.2008.00635.x
Dickerson F, Stallings C, Origoni A, Boronow J, Yolken R (2007) Elevated serum levels of C-reactive protein are associated with mania symptoms in outpatients with bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 31:952–955. doi:10.1016/j.pnpbp.2007.02.018
Goldstein BI, Kemp DE, Soczynska JK, McIntyre RS (2009) Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J Clin Psychiatry 70:1078–1090. doi:10.4088/JCP.08r04505
Munkholm K, Brauner JV, Kessing LV, Vinberg M (2013) Cytokines in bipolar disorder vs. healthy control subjects: a systematic review and meta-analysis. J Psychiatr Res 47:1119–1133. doi:10.1016/j.jpsychires.2013.05.018
Modabbernia A, Taslimi S, Brietzke E, Ashrafi M (2013) Cytokine alterations in bipolar disorder: a meta-analysis of 30 studies. Biol Psychiatry 74:15–25. doi:10.1016/j.biopsych.2013.01.007
Smoller JW (2013) Disorders and borders: psychiatric genetics and nosology. Am J Med Genet B Neuropsychiatr Genet 162B:559–578. doi:10.1002/ajmg.b.32174
Rao JS, Harry GJ, Rapoport SI, Kim HW (2010) Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry 15:384–392. doi:10.1038/mp.2009.47
Jakobsson J, Bjerke M, Sahebi S, Isgren A, Ekman CJ, Sellgren C, Olsson B, Zetterberg H, Blennow K, Palsson E, Landen M (2015) Monocyte and microglial activation in patients with mood-stabilized bipolar disorder. J Psychiatry Neurosci 40:250–258
Haarman BC, Riemersma-van der Lek RF, de Groot JC, Ruhe HG, Klein HC, Zandstra TE, Burger H, Schoevers RA, de Vries EF, Drexhage HA, Nolen WA, Doorduin J (2014) Neuroinflammation in bipolar disorder—a [(11)C]-(R)-PK11195 positron emission tomography study. Brain Behav Immun. 40:219–225. doi:10.1016/j.bbi.2014.03.016
Hong CJ, Lai IC, Liou LL, Tsai SJ (2004) Association study of the human partially duplicated alpha7 nicotinic acetylcholine receptor genetic variant with bipolar disorder. Neurosci Lett 355:69–72
Besson M, David V, Baudonnat M, Cazala P, Guilloux JP, Reperant C, Cloez-Tayarani I, Changeux JP, Gardier AM, Granon S (2012) Alpha7-nicotinic receptors modulate nicotine-induced reinforcement and extracellular dopamine outflow in the mesolimbic system in mice. Psychopharmacology 220:1–14. doi:10.1007/s00213-011-2422-1
Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG (2005) An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122:261–273. doi:10.1016/j.cell.2005.05.012
Li YC, Gao WJ (2011) GSK-3beta activity and hyperdopamine-dependent behaviors. Neurosci Biobehav Rev 35:645–654. doi:10.1016/j.neubiorev.2010.08.001
Zhu LQ, Wang SH, Liu D, Yin YY, Tian Q, Wang XC, Wang Q, Chen JG, Wang JZ (2007) Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J Neurosci 27:12211–12220. doi:10.1523/JNEUROSCI.3321-07.2007
Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL (2007) LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53:703–717. doi:10.1016/j.neuron.2007.01.029
Peineau S, Bradley C, Taghibiglou C, Doherty A, Bortolotto ZA, Wang YT, Collingridge GL (2008) The role of GSK-3 in synaptic plasticity. Br J Pharmacol 153(Suppl 1):S428–S437. doi:10.1038/bjp.2008.2
Frank MJ, Fossella JA (2011) Neurogenetics and pharmacology of learning, motivation, and cognition. Neuropsychopharmacology 36:133–152. doi:10.1038/npp.2010.96
Lin H, Hsu FC, Baumann BH, Coulter DA, Lynch DR (2014) Cortical synaptic NMDA receptor deficits in alpha7 nicotinic acetylcholine receptor gene deletion models: implications for neuropsychiatric diseases. Neurobiol Dis 63:129–140. doi:10.1016/j.nbd.2013.11.021
Nolt MJ, Lin Y, Hruska M, Murphy J, Sheffler-Colins SI, Kayser MS, Passer J, Bennett MV, Zukin RS, Dalva MB (2011) EphB controls NMDA receptor function and synaptic targeting in a subunit-specific manner. J Neurosci 31:5353–5364. doi:10.1523/JNEUROSCI.0282-11.2011
Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, Wong WS, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K (2012) Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488:471–475. doi:10.1038/nature11396
Le Pichon JB, Yu S, Kibiryeva N, Graf WD, Bittel DC (2013) Genome-wide gene expression in a patient with 15q13.3 homozygous microdeletion syndrome. Eur J Hum Genet 21:1093–1099. doi:10.1038/ejhg.2013.1
Ghaleiha A, Ghyasvand M, Mohammadi MR, Farokhnia M, Yadegari N, Tabrizi M, Hajiaghaee R, Yekehtaz H, Akhondzadeh S (2013) Galantamine efficacy and tolerability as an augmentative therapy in autistic children: a randomized, double-blind, placebo-controlled trial. J Psychopharmacol 28:677–685. doi:10.1177/0269881113508830
Angelidou A, Alysandratos KD, Asadi S, Zhang B, Francis K, Vasiadi M, Kalogeromitros D, Theoharides TC (2011) Brief report: “allergic symptoms” in children with autism spectrum disorders. More than meets the eye? J Autism Dev Disord 41:1579–1585. doi:10.1007/s10803-010-1171-z
Theoharides TC, Angelidou A, Alysandratos KD, Zhang B, Asadi S, Francis K, Toniato E, Kalogeromitros D (2012) Mast cell activation and autism. Biochim Biophys Acta 1822:34–41. doi:10.1016/j.bbadis.2010.12.017
Romagnani S (2002) Cytokines and chemoattractants in allergic inflammation. Mol Immunol 38:881–885
Georas SN, Guo J, De Fanis U, Casolaro V (2005) T-helper cell type-2 regulation in allergic disease. Eur Respir J 26:1119–1137. doi:10.1183/09031936.05.00006005
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122:787–795. doi:10.1172/JCI59643
Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J (2015) Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol 11:56–64. doi:10.1038/nrneurol.2014.207
Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, West AB, Arking DE (2014) Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun 5:5748. doi:10.1038/ncomms6748
Siegert RJ, Abernethy DA (2005) Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry 76:469–475. doi:10.1136/jnnp.2004.054635
Stys PK, Zamponi GW, van Minnen J, Geurts JJ (2012) Will the real multiple sclerosis please stand up? Nat Rev Neurosci 13:507–514. doi:10.1038/nrn3275
Hemmer B, Nessler S, Zhou D, Kieseier B, Hartung HP (2006) Immunopathogenesis and immunotherapy of multiple sclerosis. Nat Clin Pract Neurol 2:201–211. doi:10.1038/ncpneuro0154
Nizri E, Irony-Tur-Sinai M, Lory O, Orr-Urtreger A, Lavi E, Brenner T (2009) Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J Immunol. 183:6681–6688. doi:10.4049/jimmunol.0902212
Hilker R, Thomas AV, Klein JC, Weisenbach S, Kalbe E, Burghaus L, Jacobs AH, Herholz K, Heiss WD (2005) Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology 65:1716–1722. doi:10.1212/01.wnl.0000191154.78131.f6
Shimada H, Hirano S, Shinotoh H, Aotsuka A, Sato K, Tanaka N, Ota T, Asahina M, Fukushi K, Kuwabara S, Hattori T, Suhara T, Irie T (2009) Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 73:273–278. doi:10.1212/WNL.0b013e3181ab2b58
Bohnen NI, Kaufer DI, Ivanco LS, Lopresti B, Koeppe RA, Davis JG, Mathis CA, Moore RY, DeKosky ST (2003) Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol 60:1745–1748. doi:10.1001/archneur.60.12.1745
Emre M, Aarsland D, Albanese A, Byrne EJ, Deuschl G, De Deyn PP, Durif F, Kulisevsky J, van Laar T, Lees A, Poewe W, Robillard A, Rosa MM, Wolters E, Quarg P, Tekin S, Lane R (2004) Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 351:2509–2518. doi:10.1056/NEJMoa041470
Stuckenholz V, Bacher M, Balzer-Geldsetzer M, Alvarez-Fischer D, Oertel WH, Dodel RC, Noelker C (2013) The alpha7 nAChR agonist PNU-282987 reduces inflammation and MPTP-induced nigral dopaminergic cell loss in mice. J Parkinsons Dis 3:161–172. doi:10.3233/JPD-120157
Suzuki S, Kawamata J, Matsushita T, Matsumura A, Hisahara S, Takata K, Kitamura Y, Kem W, Shimohama S (2013) 3-[(2,4-Dimethoxy)benzylidene]-anabaseine dihydrochloride protects against 6-hydroxydopamine-induced parkinsonian neurodegeneration through alpha7 nicotinic acetylcholine receptor stimulation in rats. J Neurosci Res 91:462–471. doi:10.1002/jnr.23160
Liu Y, Zeng X, Hui Y, Zhu C, Wu J, Taylor DH, Ji J, Fan W, Huang Z, Hu J (2015) Activation of alpha7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress-induced apoptosis: implications for Parkinson’s disease. Neuropharmacology 91:87–96. doi:10.1016/j.neuropharm.2014.11.028
Bordia T, Grady SR, McIntosh JM, Quik M (2007) Nigrostriatal damage preferentially decreases a subpopulation of alpha6beta2* nAChRs in mouse, monkey, and Parkinson’s disease striatum. Mol Pharmacol 72:52–61. doi:10.1124/mol.107.035998
Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 8:382–397. doi:10.1016/S1474-4422(09)70062-6
Aarsland D, Pahlhagen S, Ballard CG, Ehrt U, Svenningsson P (2012) Depression in Parkinson disease—epidemiology, mechanisms and management. Nat Rev Neurol 8:35–47. doi:10.1038/nrneurol.2011.189
Jacob EL, Gatto NM, Thompson A, Bordelon Y, Ritz B (2010) Occurrence of depression and anxiety prior to Parkinson’s disease. Parkinsonism Relat Disord 16:576–581. doi:10.1016/j.parkreldis.2010.06.014
Lindqvist D, Kaufman E, Brundin L, Hall S, Surova Y, Hansson O (2012) Non-motor symptoms in patients with Parkinson’s disease—correlations with inflammatory cytokines in serum. PLoS One 7:e47387. doi:10.1371/journal.pone.0047387
Beumer W, Gibney SM, Drexhage RC, Pont-Lezica L, Doorduin J, Klein HC, Steiner J, Connor TJ, Harkin A, Versnel MA, Drexhage HA (2012) The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J Leukoc Biol 92:959–975. doi:10.1189/jlb.0212100
Kulak JM, Schneider JS (2004) Differences in alpha7 nicotinic acetylcholine receptor binding in motor symptomatic and asymptomatic MPTP-treated monkeys. Brain Res 999:193–202. doi:10.1016/j.brainres.2003.10.062
Feuerbach D, Pezous N, Weiss M, Shakeri-Nejad K, Lingenhoehl K, Hoyer D, Hurth K, Bilbe G, Pryce CR, McAllister K, Chaperon F, Kucher K, Johns D, Blaettler T, Lopez Lopez C (2015) AQW051, a novel, potent and selective alpha7 nicotinic ACh receptor partial agonist: pharmacological characterization and phase I evaluation. Br J Pharmacol 172:1292–1304. doi:10.1111/bph.13001
Perry VH, Teeling J (2013) Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol 35:601–612. doi:10.1007/s00281-013-0382-8
Shimizu E, Kawahara K, Kajizono M, Sawada M, Nakayama H (2008) IL-4-induced selective clearance of oligomeric beta-amyloid peptide(1–42) by rat primary type 2 microglia. J Immunol 181:6503–6513
Harry GJ (2013) Microglia during development and aging. Pharmacol Ther 139:313–326. doi:10.1016/j.pharmthera.2013.04.013
Hernandez CM, Kayed R, Zheng H, Sweatt JD, Dineley KT (2010) Loss of alpha7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer’s disease. J Neurosci 30:2442–2453. doi:10.1523/JNEUROSCI.5038-09.2010
Zhong G, Wang Y, Zhang Y, Guo JJ, Zhao Y (2015) Smoking is associated with an increased risk of dementia: a meta-analysis of prospective cohort studies with investigation of potential effect modifiers. PLoS One 10:e0118333. doi:10.1371/journal.pone.0118333
Fratiglioni L, Wang HX (2000) Smoking and Parkinson’s and Alzheimer’s disease: review of the epidemiological studies. Behav Brain Res 113:117–120
Parri HR, Hernandez CM, Dineley KT (2011) Research update: alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochem Pharmacol 82:931–942. doi:10.1016/j.bcp.2011.06.039
Liu Q, Huang Y, Xue F, Simard A, DeChon J, Li G, Zhang J, Lucero L, Wang M, Sierks M, Hu G, Chang Y, Lukas RJ, Wu J (2009) A novel nicotinic acetylcholine receptor subtype in basal forebrain cholinergic neurons with high sensitivity to amyloid peptides. J Neurosci 29:918–929. doi:10.1523/JNEUROSCI.3952-08.2009
Thomsen MS, Zwart R, Ursu D, Jensen MM, Pinborg LH, Gilmour G, Wu J, Sher E, Mikkelsen JD (2015) alpha7 and beta2 nicotinic acetylcholine receptor subunits form heteromeric receptor complexes that are expressed in the human cortex and display distinct pharmacological properties. PLoS One 10:e0130572. doi:10.1371/journal.pone.0130572
Foster DJ, Jones CK, Conn PJ (2012) Emerging approaches for treatment of schizophrenia: modulation of cholinergic signaling. Discov Med 14:413–420
Geerts H (2012) alpha7 Nicotinic receptor modulators for cognitive deficits in schizophrenia and Alzheimer’s disease. Expert Opin Investig Drugs 21:59–65. doi:10.1517/13543784.2012.633510
Valles AS, Borroni MV, Barrantes FJ (2014) Targeting brain alpha7 nicotinic acetylcholine receptors in Alzheimer’s disease: rationale and current status. CNS Drugs 28:975–987. doi:10.1007/s40263-014-0201-3
Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, Ellis J, Zerbe GO, Leonard S, Stevens KE, Stevens JO, Martin L, Adler LE, Soti F, Kem WR, Freedman R (2006) Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry 63:630–638. doi:10.1001/archpsyc.63.6.630
Feuerbach D, Lingenhoehl K, Olpe HR, Vassout A, Gentsch C, Chaperon F, Nozulak J, Enz A, Bilbe G, McAllister K, Hoyer D (2009) The selective nicotinic acetylcholine receptor alpha7 agonist JN403 is active in animal models of cognition, sensory gating, epilepsy and pain. Neuropharmacology 56:254–263. doi:10.1016/j.neuropharm.2008.08.025
Stevens KE, Cornejo B, Adams CE, Zheng L, Yonchek J, Hoffman KL, Christians U, Kem WR (2010) Continuous administration of a selective alpha7 nicotinic partial agonist, DMXBA, improves sensory inhibition without causing tachyphylaxis or receptor upregulation in DBA/2 mice. Brain Res 1352:140–146. doi:10.1016/j.brainres.2010.06.063
Tregellas JR, Olincy A, Johnson L, Tanabe J, Shatti S, Martin LF, Singel D, Du YP, Soti F, Kem WR, Freedman R (2010) Functional magnetic resonance imaging of effects of a nicotinic agonist in schizophrenia. Neuropsychopharmacology 35:938–942. doi:10.1038/npp.2009.196
Papke RL, Meyer E, Nutter T, Uteshev VV (2000) alpha7 receptor-selective agonists and modes of alpha7 receptor activation. Eur J Pharmacol 393:179–195
Tietje KR, Anderson DJ, Bitner RS, Blomme EA, Brackemeyer PJ, Briggs CA, Browman KE, Bury D, Curzon P, Drescher KU, Frost JM, Fryer RM, Fox GB, Gronlien JH, Hakerud M, Gubbins EJ, Halm S, Harris R, Helfrich RJ, Kohlhaas KL, Law D, Malysz J, Marsh KC, Martin RL, Meyer MD, Molesky AL, Nikkel AL, Otte S, Pan L, Puttfarcken PS, Radek RJ, Robb HM, Spies E, Thorin-Hagene K, Waring JF, Ween H, Xu H, Gopalakrishnan M, Bunnelle WH (2008) Preclinical characterization of A-582941: a novel alpha7 neuronal nicotinic receptor agonist with broad spectrum cognition-enhancing properties. CNS Neurosci Ther 14:65–82. doi:10.1111/j.1527-3458.2008.00037.x
Arias HR, Gu RX, Feuerbach D, Wei DQ (2010) Different interaction between the agonist JN403 and the competitive antagonist methyllycaconitine with the human alpha7 nicotinic acetylcholine receptor. Biochemistry 49:4169–4180. doi:10.1021/bi901999v
Prickaerts J, van Goethem NP, Chesworth R, Shapiro G, Boess FG, Methfessel C, Reneerkens OA, Flood DG, Hilt D, Gawryl M, Bertrand S, Bertrand D, Konig G (2012) EVP-6124, a novel and selective alpha7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of alpha7 nicotinic acetylcholine receptors. Neuropharmacology 62:1099–1110. doi:10.1016/j.neuropharm.2011.10.024
Cachelin AB, Rust G (1994) Unusual pharmacology of (+)-tubocurarine with rat neuronal nicotinic acetylcholine receptors containing beta 4 subunits. Mol Pharmacol 46:1168–1174
Smyth AM, Lawrie SM (2013) The neuroimmunology of schizophrenia. Clin Psychopharmacol Neurosci 11:107–117. doi:10.9758/cpn.2013.11.3.107
Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, Bernstein HG, Bogerts B (2008) Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res 42:151–157. doi:10.1016/j.jpsychires.2006.10.013
Janelidze S, Mattei D, Westrin A, Traskman-Bendz L, Brundin L (2011) Cytokine levels in the blood may distinguish suicide attempters from depressed patients. Brain Behav Immun 25:335–339. doi:10.1016/j.bbi.2010.10.010
Erhardt S, Lim CK, Linderholm KR, Janelidze S, Lindqvist D, Samuelsson M, Lundberg K, Postolache TT, Traskman-Bendz L, Guillemin GJ, Brundin L (2013) Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 38:743–752. doi:10.1038/npp.2012.248
Schnieder TP, Trencevska I, Rosoklija G, Stankov A, Mann JJ, Smiley J, Dwork AJ (2014) Microglia of prefrontal white matter in suicide. J Neuropathol Exp Neurol 73:880–890. doi:10.1097/NEN.0000000000000107
Brundin L, Erhardt S, Bryleva EY, Achtyes ED, Postolache TT (2015) The role of inflammation in suicidal behaviour. Acta Psychiatr Scand 132:192–203. doi:10.1111/acps.12458
Freland L, Beaulieu JM (2012) Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front Mol Neurosci 5:14. doi:10.3389/fnmol.2012.00014
Beurel E, Michalek SM, Jope RS (2010) Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol 31:24–31. doi:10.1016/j.it.2009.09.007
Baldessarini RJ, Tondo L, Hennen J (2003) Lithium treatment and suicide risk in major affective disorders: update and new findings. J Clin Psychiatry 64(Suppl 5):44–52
Beurel E, Jope RS (2014) Inflammation and lithium: clues to mechanisms contributing to suicide-linked traits. Transl Psychiatry 4:e488. doi:10.1038/tp.2014.129
De Sarno P, Axtell RC, Raman C, Roth KA, Alessi DR, Jope RS (2008) Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J Immunol 181:338–345
Engel T, Hernandez F, Avila J, Lucas JJ (2006) Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci 26:5083–5090. doi:10.1523/JNEUROSCI.0604-06.2006
Fiorentini A, Rosi MC, Grossi C, Luccarini I, Casamenti F (2010) Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mutant mice. PLoS One 5:e14382. doi:10.1371/journal.pone.0014382
Zhang X, Heng X, Li T, Li L, Yang D, Zhang X, Du Y, Doody RS, Le W (2011) Long-term treatment with lithium alleviates memory deficits and reduces amyloid-beta production in an aged Alzheimer’s disease transgenic mouse model. J Alzheimers Dis 24:739–749. doi:10.3233/JAD-2011-101875
Youdim MB, Arraf Z (2004) Prevention of MPTP (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) dopaminergic neurotoxicity in mice by chronic lithium: involvements of Bcl-2 and Bax. Neuropharmacology 46:1130–1140. doi:10.1016/j.neuropharm.2004.02.005
Lieu CA, Dewey CM, Chinta SJ, Rane A, Rajagopalan S, Batir S, Kim YH, Andersen JK (2014) Lithium prevents parkinsonian behavioral and striatal phenotypes in an aged parkin mutant transgenic mouse model. Brain Res 1591:111–117. doi:10.1016/j.brainres.2014.10.032
Lazzara CA, Riley RR, Rane A, Andersen JK, Kim YH (2015) The combination of lithium and l-Dopa/Carbidopa reduces MPTP-induced abnormal involuntary movements (AIMs) via calpain-1 inhibition in a mouse model: relevance for Parkinsons disease therapy. Brain Res 1622:127–136. doi:10.1016/j.brainres.2015.06.018
Di Paolo T, Gregoire L, Feuerbach D, Elbast W, Weiss M, Gomez-Mancilla B (2014) AQW051, a novel and selective nicotinic acetylcholine receptor alpha7 partial agonist, reduces l-Dopa-induced dyskinesias and extends the duration of l-Dopa effects in parkinsonian monkeys. Parkinsonism Relat Disord 20:1119–1123. doi:10.1016/j.parkreldis.2014.05.007
Zhang D, McGregor M, Decker MW, Quik M (2014) The alpha7 nicotinic receptor agonist ABT-107 decreases l-Dopa-induced dyskinesias in parkinsonian monkeys. J Pharmacol Exp Ther 351:25–32. doi:10.1124/jpet.114.216283
Acknowledgments
The authors would like to thank Dr. D. Shimshek for critically reading an earlier version of the manuscript and Dr. C. Parker for his support with the final version of the review.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that this review was prepared without external support. Hans O. Kalkman has no conflicts of interest. Dominik Feuerbach is an employee of Novartis.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kalkman, H.O., Feuerbach, D. Modulatory effects of α7 nAChRs on the immune system and its relevance for CNS disorders. Cell. Mol. Life Sci. 73, 2511–2530 (2016). https://doi.org/10.1007/s00018-016-2175-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2175-4