
Abstract
Amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) represent the two major forms of motoneuron disease. In both forms of disease, spinal and bulbar motoneurons become dysfunctional and degenerate. In ALS, cortical motoneurons are also affected, which contributes to the clinical phenotype. The gene defects for most familial forms of ALS and SMA have been discovered and they point to a broad spectrum of disease mechanisms, including defects in RNA processing, pathological protein aggregation, altered apoptotic signaling, and disturbed energy metabolism. Despite the fact that lack of neurotrophic factors or their corresponding receptors are not found as genetic cause of motoneuron disease, signaling pathways initiated by neurotrophic factors for motoneuron survival, axon growth, presynaptic development, and synaptic function are disturbed in ALS and SMA. Better understanding of how neurotrophic factors and downstream signaling pathways interfere with these disease mechanisms could help to develop new therapies for motoneuron disease and other neurodegenerative disorders.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others
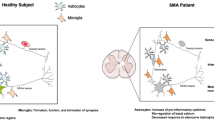
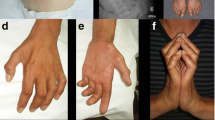

References
Ackermann B et al (2013) Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet 22(7):1328–1347
Al-Chalabi A et al (2012) The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol 124:339–352
Al-Sarraj S et al (2011) p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 122:691–702
Andersen PM, Al-Chalabi A (2011) Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol 7:603–615
Arakawa Y, Sendtner M, Thoenen H (1990) Survival effect of ciliary neurotrophic factor (CNTF) on chick embryonic motoneurons in culture: comparison with other neurotrophic factors and cytokines. J Neurosci 10:3507–3515
Ash PE et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77(4):639–646
Ashley CT Jr, Wilkinson KD, Reines D, Warren ST (1993) FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262:563–566
Ayala YM, Misteli T, Baralle FE (2008) TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci U S A 105:3785–3789
Azzouz M et al (2004) VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 429:413–417, 1995 Sep 28; 377(6547):340–344
Baloh RH et al (1998) Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3-RET receptor complex. Neuron 21:1291–1302
Boillee S et al (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392
Bommel H et al (2002) Missense mutation in the tubulin-specific chaperone E (Tbce) gene in the mouse mutant progressive motor neuronopathy, a model of human motoneuron disease. J Cell Biol 159:563–569
Buratti E, Baralle FE (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276:36337–36343
Buratti E, Baralle FE (2010) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30:277–288
Buratti E, Baralle FE (2008) Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci 13:867–878
Buratti E, Baralle FE (2010a) The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol 7(4):420–429
Buratti E, Baralle FE (2010b) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30: 277–288
Buratti E et al (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20:1774–1784
Buratti E et al (2005) TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 280:37572–37584
Burghes AH, Beattie CE (2009) Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10:597–609
Carmeliet P, Storkebaum E (2002) Vascular and neuronal effects of VEGF in the nervous system: implications for neurological disorders. Semin Cell Dev Biol 13:39–53
Caroni P (1997) Intrinsic neuronal determinants that promote axonal sprouting and elongation. Bioessays 19:767–775
Cartegni L, Krainer AR (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 30:377–384
Chan YB et al (2003) Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet 12:1367–1376
Chao MV (2003) Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 4:299–309
Chen YZ et al (2004) DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74(6):1128–1135
Cho S, Dreyfuss G (2010) A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev 24:438–442
Chow CY et al (2009) Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet 84(1):85–88
Clement AM et al (2003) Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302:113–117
Colombrita C et al (2009) TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem 111:1051–1061
Crawford TO, Pardo CA (1996) The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis 3:97–110
Damiano M et al (2006) Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem 96:1349–1361
De Vos KJ et al (2007) Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet 16:2720–2728
DeJesus-Hernandez M et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256
Deng HX et al (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477(7363):211–215
Dewey CM et al (2011) TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol 31:1098–1108
Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC (2008) Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 3:637–648
Dormann D et al (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29:2841–2857
Drepper C, Sendtner M (2011) A new postal code for dendritic mRNA transport in neurons. EMBO Rep 12:614–616
Drepper C, Herrmann T, Wessig C, Beck M, Sendtner M (2011) C-terminal FUS/TLS mutations in familial and sporadic ALS in Germany. Neurobiol Aging 32(3):548.e1–4
Durham HD, Roy J, Dong L, Figlewicz DA (1997) Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol 56:523–530
Elden AC et al (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466(7310):1069–1075
Elson GC et al (2000) CLF associates with CLC to form a functional heteromeric ligand for the CNTF receptor complex. Nat Neurosci 3:867–872
Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ (2011) Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 7:616–630
Fiesel FC et al (2010) Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J 29:209–221
Fiesel FC, Schurr C, Weber SS, Kahle PJ (2011) TDP-43 knockdown impairs neurite outgrowth dependent on its target histone deacetylase 6. Mol Neurodegener 6:64
Frade JM, RodriguezTebar A, Barde YA (1996) Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383:166–168
Fratta P et al (2012) C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep 2:1016
Freibaum BD, Chitta RK, High AA, Taylor JP (2010) Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 9:1104–1120
Friedlander RM, Brown RH, Gagliardini V, Wang J, Yuan J (1997) Inhibition of ICE slows ALS in mice. Nature 388:31
Gavrilina TO et al (2008) Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum Mol Genet 17:1063–1075
Giess R et al (2002) Early onset of severe familial amyotrophic lateral sclerosis with a SOD-1 mutation: potential impact of CNTF as a candidate modifier gene. Am J Hum Genet 70:1277–1286
Glinka M et al (2010) The heterogeneous nuclear ribonucleoprotein-R is necessary for axonal beta-actin mRNA translocation in spinal motor neurons. Hum Mol Genet 19:1951–1966
Gogliotti RG et al (2012) Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J Neurosci 32:3818–3829
Gravel C, Götz R, Lorrain A, Sendtner M (1997) Adenoviral gene transfer of ciliary neurotrophic factor and brain-derived neurotrophic factor leads to longterm survival of axotomized motoneurons. Nat Med 3:765–770
Greenway MJ et al (2006) ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 38(4):411–413
Grieshammer U et al (1998) Muscle-specific cell ablation conditional upon Cre-mediated DNA recombination in transgenic mice leads to massive spinal and cranial motoneuron loss Neuronal cell death. Neuron 20:633–647
Grosskreutz J, Van Den Bosch L, Keller BU (2010) Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47:165–174
Gubitz AK, Feng W, Dreyfuss G (2004) The SMN complex. Exp Cell Res 296:51–56
Gurney ME et al (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775
Hadano S et al (2001) A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 29(2):166–173
Hamburger V (1934) The effects of wing bud extirpation on the development of the central nervous system in chick embryos. J Exp Zool 68:449–494
Hamburger V (1975) Cell death in the development of the lateral column of the chick embryo. J Comp Neurol 160:535–546
Hand CK et al (2002) A novel locus for familial amyotrophic lateral sclerosis, on chromosome 18q. Am J Hum Genet 70(1):251–256
Haramati S et al (2010) miRNA malfunction causes spinal motor neuron disease. Proc Natl Acad Sci U S A 107:13111–13116
Harraz MM et al (2008) SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest 118:659–670
Hausmanowa-Petrusewicz I (1978) In: Spinal muscular atrophy: infantile and juvenile type. National Library of Medicine & The National Science Foundation, Washington DC
Henderson CE et al (1994) GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science 266:1062–1064
Higashi S et al (2007) Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 1184:284–294
Holtmann B et al (2005) Triple knock-out of CNTF, LIF, and CT-1 defines cooperative and distinct roles of these neurotrophic factors for motoneuron maintenance and function. J Neurosci 25:1778–1787
Hu F et al (2010) Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68:654–667
Hughes RA, Sendtner M, Goldfarb M, Lindholm D, Thoenen H (1993a) Evidence that fibroblast growth factor 5 is a major muscle derived survival factor for cultured spinal motoneurons. Neuron 10:369–377
Hughes RA, Sendtner M, Thoenen H (1993b) Members of several gene families influence survival of rat motoneurons in vitro and in vivo. J Neurosci Res 36(6):663–671
Imlach WL et al (2012) SMN is required for sensory-motor circuit function in Drosophila. Cell 151:427–439
Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N (2011) Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol 69:152–162
Jablonka S, Beck M, Lechner BD, Mayer C, Sendtner M (2007) Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol 179:139–149
Johnson JO et al (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68(5):857–864
Kabashi E et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40(5):572–574
Kashima T, Manley JL (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet 34:460–463
Kerkhoff H, Jennekens FGI, Troost D, Veldman H (1991) Nerve growth factor receptor immunostaining in the spinal cord and peripheral nerves in amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 81:649–656
Kiebler MA, Bassell GJ (2006) Neuronal RNA granules: movers and makers. Neuron 51:685–690
Kiernan MC et al (2011) Amyotrophic lateral sclerosis. Lancet 377:942–955
Klein RD et al (1997) A GPI-linked protein that interacts with Ret to form a candidate neurturin receptor. Nature 387:717–721
Kong L et al (2009) Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci 29:842–851
Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S (1997) Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science 277:559–562
Krecic AM, Swanson MS (1999) hnRNP complexes: composition, structure, and function. Curr Opin Cell Biol 11:363–371
Kuroda M et al (2000) Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. EMBO J 19:453–462
Kwiatkowski TJ Jr et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Leibrock J et al (1989) Molecular cloning and expression of brain-derived neurotrophic factor. Nature 341:149–152
Ling SC et al (2010) ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci U S A 107:13318–13323
Liu X, Ernfors P, Wu H, Jaenisch R (1995) Sensory but not motor neuron deficits in mice lacking NT4 and BDNF. Nature 375:238–241
Lotti F et al (2012) An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151:440–454
Luty AA et al (2010) Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann Neurol 68(5):639–649
Mackenzie IRA, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9:995–1007
Maekawa S et al (2009) TDP-43 is consistently co-localized with ubiquitinated inclusions in sporadic and Guam amyotrophic lateral sclerosis but not in familial amyotrophic lateral sclerosis with and without SOD1 mutations. Neuropathology 29:672–683
Maruyama H et al (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465(7295):223–226
McDonald KK et al (2011) TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet 20:1400–1410
McGovern VL, Gavrilina TO, Beattie CE, Burghes AH (2008) Embryonic motor axon development in the severe SMA mouse. Hum Mol Genet 17:2900–2909
McWhorter ML, Monani UR, Burghes AH, Beattie CE (2003) Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol 162:919–931
Mentis GZ et al (2011) Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69:453–467
Meyer M, Matsuoka I, Wetmore C, Olson L, Thoenen H (1992) Enhanced synthesis of brain-derived neurotrophic factor in the lesioned peripheral nerve: different mechanisms are responsible for the regulation of BDNF and NGF mRNA. J Cell Biol 119:45–54
Milbrandt J et al (1998) Persephin, a novel neurotrophic factor related to GDNF and neurturin. Neuron 20:245–253
Moisse K et al (2009a) Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res 1249:202–211
Moisse K et al (2009b) Cytosolic TDP-43 expression following axotomy is associated with caspase 3 activation in NFL-/- mice: support for a role for TDP-43 in the physiological response to neuronal injury. Brain Res 1296:176–186
Monani UR et al (2000) The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy [In Process Citation]. Hum Mol Genet 9:333–339
Mori K et al (2013a) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125(3):413–423
Mori K et al (2013b) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339(6125):1335–1338
Morlando M et al (2012) FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J 31:4502–4510
Mourelatos Z, Abel L, Yong J, Kataoka N, Dreyfuss G (2001) SMN interacts with a novel family of hnRNP and spliceosomal proteins. EMBO J 20:5443–5452
Nagai M et al (2007) Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10:615–622
Neumann M et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Ning K et al (2010) PTEN depletion rescues axonal growth defect and improves survival in SMN-deficient motor neurons. Hum Mol Genet 19:3159–3168
Nishihira Y et al (2009) Sporadic amyotrophic lateral sclerosis of long duration is associated with relatively mild TDP-43 pathology. Acta Neuropathol 117:45–53
Nishimura AL et al (2004) A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75(5):822–831
Novak KD, Prevette D, Wang S, Gould TW, Oppenheim RW (2000) Hepatocyte growth factor/scatter factor is a neurotrophic survival factor for lumbar but not for other somatic motoneurons in the chick embryo. J Neurosci 20:326–337
Oppenheim RW (1985) Naturally occuring cell death during neural development. Trends Neurosci 8:487–493
Oppenheim RW et al (2000) Glial cell line-derived neurotrophic factor and developing mammalian motoneurons: regulation of programmed cell death among motoneuron subtypes. J Neurosci 20:5001–5011
Oppenheim RW et al (2001) Cardiotrophin-1, a muscle-derived cytokine, is required for the survival of subpopulations of developing motoneurons. J Neurosci 21:1283–1291
Oprea GE et al (2008) Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320:524–527
Orlacchio A et al (2010) SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 133(Pt 2):591–598
Parkinson N et al (2006) ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67(6):1074–1077
Pascale A, Govoni S (2012) The complex world of post-transcriptional mechanisms: is their deregulation a common link for diseases? Focus on ELAV-like RNA-binding proteins. Cell Mol Life Sci 69:501–517
Pasinelli P et al (2004) Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 43:19–30
Pellizzoni L (2007) Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep 8:340–345
Pennica D et al (1996) Cardiotrophin-1, a cytokine present in embryonic muscle, supports long-term survival of spinal motoneurons. Neuron 17:63–74
Pesiridis GS, Lee VM, Trojanowski JQ (2009) Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 18:R156–R162
Pesiridis GS, Tripathy K, Tanik S, Trojanowski JQ, Lee VM (2011) A “two-hit” hypothesis for inclusion formation by carboxyl-terminal fragments of TDP-43 protein linked to RNA depletion and impaired microtubule-dependent transport. J Biol Chem 286:18845–18855
Plachta N et al (2007) Identification of a lectin causing the degeneration of neuronal processes using engineered embryonic stem cells. Nat Neurosci 10:712–719
Poesen K et al (2008) Novel role for vascular endothelial growth factor (VEGF) receptor-1 and its ligand VEGF-B in motor neuron degeneration. J Neurosci 28:10451–10459
Polymenidou M et al (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14(4):459–468
Pun S, Santos AF, Saxena S, Xu L, Caroni P (2006) Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9:408–419
Rayaprolu S et al (2012) Angiogenin variation and Parkinson disease. Ann Neurol 71(5):725–727; author reply 727–728
Reaume AG et al (1996) Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13:43–47
Renton AE et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268
Riethmacher D et al (1997) Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 389:725–730
Rosen DR et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Rossoll W et al (2002) Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum Mol Genet 11:93–105
Rossoll W et al (2003) Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol 163:801–812
Ruiz R, Casanas JJ, Torres-Benito L, Cano R, Tabares L (2010) Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J Neurosci 30:849–857
Rutherford NJ et al (2008) Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 4:e1000193
Sagot Y, Tan SA, Hammang JP, Aebischer P, Kato AC (1996a) GDNF slows loss of motoneurons but not axonal degeneration or premature death of pmn/pmn mice. J Neurosci 16:2335–2341
Sagot Y et al (1996b) Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci 11:7727–7733
Sapp PC et al (2003) Identification of two novel loci for dominantly inherited familial amyotrophic lateral sclerosis. Am J Hum Genet 73(2):397–403
Schrank B et al (1997) Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A 94:9920–9925
Schwab C, Arai T, Hasegawa M, Yu S, McGeer PL (2008) Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J Neuropathol Exp Neurol 67:1159–1165
Seeburger JL, Tarras S, Natter H, Springer JE (1993) Spinal cord motoneurons express p75 NGFR and p145 trkB mRNA in amyotrophic lateral sclerosis. Brain Res 621:111–115
Selvaraj BT, Frank N, Bender FL, Asan E, Sendtner M (2012) Local axonal function of STAT3 rescues axon degeneration in the pmn model of motoneuron disease. J Cell Biol 199:437–451
Sendtner M (2001) Molecular mechanisms in spinal muscular atrophy: models and perspectives. Curr Opin Neurol 14:629–634
Sendtner M (2010) Therapy development in spinal muscular atrophy. Nat Neurosci 13:795–799
Sendtner M, Kreutzberg GW, Thoenen H (1990) Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature 345:440–441
Sendtner M, Holtmann B, Kolbeck R, Thoenen H, Barde YA (1992a) Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature 360:757–758, 1995 Sep 28; 377(6547):340–344
Sendtner M et al (1992b) Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy. Nature 358:502–504
Sephton CF et al (2011) Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem 286:1204–1215
Simon CM, Jablonka S, Ruiz R, Tabares L, Sendtner M (2010) Ciliary neurotrophic factor-induced sprouting preserves motor function in a mouse model of mild spinal muscular atrophy. Hum Mol Genet 19:973–986
Simpson EP, Henry YK, Henkel JS, Smith RG, Appel SH (2004) Increased lipid peroxidation in sera of ALS patients: a potential biomarker of disease burden. Neurology 62:1758–1765
Singh KK et al (2008) Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nat Neurosci 11:649–658
Smith RG, Henry YK, Mattson MP, Appel SH (1998) Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann Neurol 44:696–699
Sreedharan J et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319(5870):1668–1672
Stockli KA et al (1989) Molecular cloning, expression and regional distribution of rat ciliary neurotrophic factor. Nature 342:920–923
Strong MJ et al (2007) TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci 35:320–327
Subramanian M et al (2011) G-quadruplex RNA structure as a signal for neurite mRNA targeting. EMBO Rep 12:697–704
Thoenen H, Sendtner M (2002) Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat Neurosci 5(Suppl):1046–1050
Tollervey JR et al (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14:452–458
Torres-Benito L, Ruiz R, Tabares L (2012) Synaptic defects in spinal muscular atrophy animal models. Dev Neurobiol 72:126–133
Urushitani M et al (2006) Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci 9:108–118
Van Damme P et al (2007) Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci U S A 104:14825–14830
Vance C et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211
Volkening K, Leystra-Lantz C, Yang W, Jaffee H, Strong MJ (2009) Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res 1305:168–182
Wang IF, Reddy NM, Shen CK (2002) Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci U S A 99:13583–13588
Wegorzewska I, Baloh RH (2011) TDP-43-based animal models of neurodegeneration: new insights into ALS pathology and pathophysiology. Neurodegener Dis 8:262–274
Weidner KM et al (1991) Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc Natl Acad Sci U S A 88:7001–7005
Wirth B (2000) An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat 15:228–237
Wong PC et al (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14:1105–1116
Wood-Allum C, Shaw PJ (2010) Motor neurone disease: a practical update on diagnosis and management. Clin Med 10:252–258
Wu CH et al (2012) Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488(7412):499–503
Yamamoto Y et al (1997) Hepatocyte growth factor (HGF/SF) is a muscle-derived survival factor for a subpopulation of embryonic motoneurons. Development 124:2903–2913
Yamazaki T et al (2012) FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep 2:799–806
Zinszner H, Albalat R, Ron D (1994) A novel effector domain from the RNA-binding protein TLS or EWS is required for oncogenic transformation by CHOP. Genes Dev 8:2513–2526
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg 2014
About this chapter
Cite this chapter
Sendtner, M. (2014). Motoneuron Disease. In: Lewin, G., Carter, B. (eds) Neurotrophic Factors. Handbook of Experimental Pharmacology, vol 220. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-45106-5_15
Download citation
DOI: https://doi.org/10.1007/978-3-642-45106-5_15
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-45105-8
Online ISBN: 978-3-642-45106-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)