
Opinion statement
Giant cell arteritis (GCA) is a systemic inflammatory vasculopathy mediated by pathogenic responses of a diverse array of innate and adaptive immune cells as well as vascular cells. Current therapy relies on broadly immunosuppressant glucocorticoids, which rapidly control systemic inflammation, but fail to eliminate vascular inflammation. Despite steady growth of the immunosuppressive armamentarium overall, it has been difficult to identify clinically meaningful and steroid-sparing effects of targeted biologic therapies in GCA. Here, we propose that this reflects the multiplicity of pathogenic elements that converge to induce and maintain GCA: (a) GCA is a disease of the elderly, posing additional risks when aggressively disabling immunity. (b) GCA is a chronic disease and persists as smoldering vasculitis. (c) The burden of disease is difficult to measure, complicating adjustments of immunosuppressive therapy. In the absence of objective activity markers, physicians and patients lean toward overutilization of glucocorticoids. (d) Key pathogenic drivers are T cells and macrophages, which assemble in granulomas and are highly diverse in their functional product portfolio. The diversity of vasculitogenic effector functions confounds simple therapeutic targeting. (e) Ultimate vascular complications are dependent on vessel wall cells, requiring therapeutic strategies beyond immunosuppression. Immediate goals in improving the therapeutic management of GCA should focus on optimizing glucocorticoid dosing to avoid overutilization and on adapting therapy to the needs of the immunosenescent host. Building a spectrum of possible therapeutic targets will enable carefully designed multipronged combination therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammation, a life-saving immune response, leads to tissue damage and possible organ failure when unopposed and chronic. Inflammatory destruction of medium- and large-sized arteries is particularly dangerous, as injury to the aorta and its major branches poses a threat to survival. Such arteries are the target of the autoinflammatory and autoimmune syndrome giant cell arteritis (GCA), which causes aortitis and vasculitic occlusion of the second to fifth aortic branch vessels [1–3, 4••, 5••, 6, 7]. Clinically, the most dreaded complication of GCA is ischemic damage to the optic nerve or acute injury to the aortic wall. In most patients, GCA progresses slowly, demanding sensitive and precise tools to measure disease load and necessitating a spectrum of therapeutic interventions to tailor treatment to the needs of each patient [1–3, 4••, 5••, 6].
GCA is a chronic disease of the elderly, and treatment needs to be aligned with the needs of the patient. Acute complications are anterior ischemic optic neuropathy (AION), stroke, and ischemic damage along the visual axis. Aortic dissection and rapidly progressive aneurysm formation require emergency intervention. Chronic manifestations present as recurrent polymyalgia rheumatica and slowly progressive changes in the subclavian arteries and the aorta. Treatment goals in acute and chronic disease are different. It is currently unknown whether disease remission is achievable and how to balance risks and benefits in 65–85-year-old individuals [1–3, 4••, 5••, 6].
In contrast to many other immune-mediated diseases, the therapeutic spectrum in GCA remains narrow. This may reflect the distinctiveness of pathogenic processes that produce vasculitis. Glucocorticoids have been the therapeutic gold standard, and it will be difficult to replace them, given their superb therapeutic efficacy and the need for prompt immunosuppression in newly diagnosed patients. Glucocorticoids are given in high doses during induction therapy, and the vast majority of patients have an excellent response [8, 9]. Challenges arise from the side effects that come with chronic application of glucocorticoids and from the difficulties of proper dose adjustments. Given the potentially devastating complications of GCA, both physicians and patients tend to lean toward glucocorticoid overdosing.
Broadening of the therapeutic armamentarium in GCA has to rely on understanding relevant pathogenic events and on targeting pinnacle disease pathways. Recent progress in comprehending which parts of the immune system are involved promises to widen the tool box and expand the treatment strategies for acute induction therapy as well as chronic maintenance therapy. GCA is a prototypic granulomatous disease, identifying T cells and macrophages as the major drivers of pathology [1–3, 4••, 5••, 6]. Among T cells, CD4 T cells dominate the cellular infiltrates in the arterial wall layers and intermingle with highly activated macrophages (MΦ), often called histiocytes. In many, but not all patients, such activated MΦ give rise to multinucleated giant cells (GC). Classically, such multinucleated GC are aligned along the fragmented lamina elastic interna; placement in other parts of the wall is unusual. Additional cells involved in pathogenic processes are of vascular origin, especially myofibroblasts and endothelial cells. A key driver of granulomatous vasculitis is dendritic cells (DC), located in the adventitia, generally at the adventitia-media junction in close proximity to the lamina elastic externa. Such vascular DC hold checkpoint positions and enable CD4 T cells and MΦ to assemble in granulomatous lesions in an unusual tissue niche. The blood vessel wall responds with a maladaptive response that eventually leads to luminal occlusion, disturbed blood flow, and tissue ischemia.
Cellular players in giant cell arteritis
Identifying and characterizing the cellular elements in GCA promise to provide clues toward novel diagnostic and therapeutic approaches. While there is no doubt that granulomatous lesions are composed of immune cells, it is the vessel wall that directs the immune system toward a restricted tissue niche and vascular cells mount an injury response that results in the clinical complications of GCA. Here, we will summarize what is currently known about the cellular players contributing to arteritis.
Innate immunity in giant cell arteritis
Protective and pathogenic immune responses begin with activation of innate immune cells. Neutrophils and eosinophils are rarely encountered in the mural infiltrates, and occasionally, a few mast cells are seen in tissue sections of inflamed temporal arteries [10, 11]. NK cells have, so far, not been implicated [12]. But, two types of innate immune cells have central importance in driving disease and, as such, are critical targets for anti-vasculitic therapy, dendritic cells (DC), and MΦ.
Dendritic cells
DC, specifically wall-embedded vascular DC (vasDC), function as sentinel cells in initiating and sustaining vasculitis [13, 14]. Notably, vasDC are present in normal, noninflamed human arteries. Their placement in the proximal adventitia strongly supports the concept that immune surveillance is an important part of arterial function. Even more importantly, the distribution and molecular signature of vasDC are vessel specific, meaning that each medium and large human artery has a unique pattern of vasDC expression [15]. Immune sensing capability defines the vascular tree as part of the innate immune system itself.
In vasculitic lesions, adventitial DC are activated, CD83+, and produce T cell-attracting chemokines, such as CCL19 and CCL21 [13, 14]. Normal human arteries, populated by resting vasDC, are resistant to vasculitis, suggesting gate keeper function for this immune sentinel. The nature of the DC activator is unknown, but in experimental systems, a “danger signal” provided by bacterial products is sufficient to break the DC-imposed tolerance. Interestingly, different danger signals bias the downstream immune responses toward distinct vasculitic patterns [16].
Together, these data support the notion that GCA begins with a danger signal in the adventitia, sensed by vasDC, which triggers a functional switch of the sentinels, resulting in the recruitment of T cells and MΦ that stabilize the local inflammatory response by building granulomas. As the inflammatory reactions move deeper into the wall, DC increase in number and accompany the tissue-invasive lymphocytes. The origin of such amplifying DC is currently unknown.
The pinnacle position of vasDC identifies them as key elements in the disease process and encourages therapeutic targeting of this cell population to hold early steps in vasculitis. Given the vessel-specific distribution of vasDC, better understanding of their regulation may provide valuable insights into the tissue tropism of GCA.
Macrophages
GCA is intimately connected to macrophage (MΦ) biology. MΦ are a condition sine qua non to form granulomas. Also, multinucleated GC, recognized as a typical phenomenon of the disease in the term GCA, are offspring of MΦ. Finally, the proinflammatory cytokine IL-6, which induces the exuberant acute phase response that is elegantly captured in the highly elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) [9, 17], is a MΦ product.
The precursor cells of MΦ, monocytes, originate in the bone marrow from a common myeloid precursor, circulate in the peripheral blood, and eventually enter the tissue, where they differentiate into MΦ. Interestingly, circulating monocytes in patients with GCA are no longer in a resting state, but 60–80 % are able to produce the inflammatory cytokines IL-1β and IL-6 [18]. These data suggest a fundamental abnormality in monocyte biology and redirect the focus on “initial triggers” away from the tissue site. Once part of the granulomatous infiltrates in the vessel wall, MΦ differentiate into heterogeneous populations which are characterized by core products, such as TGF-β, iNOS, or collagenase [19]. A close correlation between positioning in the arterial wall and the product portfolio has given rise to the concept that microenvironmental signals direct the functional lineage commitment of the cells.
Proteomic studies in the serum and plasma of patients with GCA have drawn attention to additional MΦ products that are highly elevated. Vascular endothelial growth factor (VEGF), an angiogenic molecule, has been associated with ischemic complications manifesting as visual loss [20]. Earlier studies have assigned VEGF production to multinucleated GC and MΦ localized in the vicinity of newly formed microvessels that support the outgrowth of the intimal neotissue blocking blood flow. Also, GCA MΦ are highly efficient in producing the interferon gamma (IFN-γ)–dependent chemokines CXCL9, 10, and 11 [20]. A recent study has confirmed that GCA MΦ are responsive to these chemokines, placing the T cell product IFN-γ into a key position in organizing the cellular recruitment into the arterial wall [21]. Immunohistochemical examination of tissue-residing MΦ has revealed that MΦ activation patterns in GCA and primary angiitis of the central nervous system (PACNS) are fundamentally different [22]. GCA MΦ express more MRP14, whereas PACNS MΦ tend to express MRP8, suggesting that GCA and PACNS represent distinct entities within the spectrum of granulomatous vasculitides [22].
Finally, vascular cells are dependent on MΦ-derived cytokines and growth factors to mobilize, migrate, and deposit extracellular matrix, generating the hyperplastic intimal layer that occludes the vascular lumen. Here again, multinucleated GC and media-homing MΦ are the cellular source of platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) [23], driving the maladaptive response to injury of the artery that is under attack by the immune system.
Overall, innate immune cells orchestrate the dysfunctional immune response leading to vasculitis, guide the disease into the mural layers, support vascular cell populations in the all remodeling process, and participate in the systemic inflammatory response by producing proinflammatory cytokines. Almost certainly, such innate immune cells are the main target of glucocorticoids and suppression of DC and MΦ function is responsible for the prompt therapeutic response during induction therapy of acute GCA. The need for high doses of glucocorticoids calls for therapeutic alternatives and emphasizes a fundamental and persistent abnormality in innate immune regulation underlying GCA.
Adaptive immunity in giant cell arteritis
B cells
Clinical observations have not supported a direct role for B cells in the pathogenesis of GCA, separating this vasculitic entity from ANCA-associated vasculitis. In contrast to AAV, no autoantibody has been associated with GCA and patients usually lack lymphadenopathy and hypergammaglobulinemia. In general, B cells are a minor cell population in granulomatous infiltrates, and in immunohistochemical studies, B cells are scarcely detected in temporal artery biopsy specimens [24]. Recent studies have provided evidence that the inflammatory syndrome in untreated GCA is associated with altered distribution of circulating B cells. Specifically, untreated patients have decreased numbers of circulating B cells and the cell numbers normalized after treatment [25, 26]. Also, B cells from GCA patients with active disease had an enhanced capacity for IL-6 production and B cell-activating factor was strongly associated with disease activity [25, 26]. These studies suggest that B cells may contribute to the inflammatory syndrome via IL-6 production, but further studies are required to investigate whether immunoglobulins have a place in pathogenesis.
T cells
A dominant cell population in the pathognomonic granulomas is CD4 T cells. In line with accumulation of such cells in peripheral tissue lesions, almost all CD4 T cells have switched to a memory phenotype. The close interactions between T cells and DC in the arterial wall infiltrates suggest that T cell priming could occur in the periphery, but formal proof is lacking. Whether the lymphoid microarchitectures build in the adventitia and media of an inflamed vessel have all structural elements to allow for T cell priming awaits experimental confirmation.
CD8 T cells are infrequent in GCA lesions. Work published more than 20 years ago raised the question whether GCA is associated with CD8 T cell lymphopenia, suggesting as one possibility that CD8 T cell provides protective functions against vasculitic T cell responses [27].
The process of granuloma formation requires T cell help. It is currently unclear whether T cells dedicated to different functional lineages could possibly support the initiation and the persistence of granulomatous infiltrates. Previous work has indicated that at least two distinct T cell lineages are present in inflamed temporal arteries [28–30] and that both of these lineages may play a distinct role in the disease process. Recent studies suggest not only that Th1 and Th17 cells have vasculitogenic potential, but also that other subtypes of CD4 T cells participate in the infiltrates. These findings may enable novel pathogenic concepts, such as multiple different types of antigens functioning as drivers of vascular inflammation.
Here, we will summarize how each helper T cell type contributes to GCA.
Th1 cells
Th1 cells are known to emerge in IL-12–rich microenvironments and differentiate into effector cells with a defined cytokine production profile. Typically, Th1 cells secrete IFN-γ, IL-2, and GM-CSF. IL-2 is a T cell growth factor, functioning as a feed-forward loop to stabilize Th1 immunity. Among the Th1-derived products, IFN-γ is the most important, with a wide range of regulatory power. Specifically, IFN-γ induces MΦ activation and thus holds control over the most important partner in granuloma formation. IFN-γ–activated MΦ release chemokines (e.g., CXCL9, CXCL10, CXCL11) secrete metalloproteases (MMP, e.g., MMP-2, MMP-9) and provide reactive oxygen species through the respiratory burst [31–33]. IFN-γ turns MΦ into producers for angiogenic factors (e.g., VEGF) and growth factors (e.g., PDGF and FGF) [23]. Notably, IFN-γ cooperates with Toll-like receptor ligands to initiate a MΦ activation program associated with tissue inflammation. In inflamed temporal arteries, IFN-γ may coactivate MΦ that also receive stimulatory signals from danger signals in the tissue microenvironment.
An important aspect of GCA pathogenesis is the formation of multinucleated giant cells (GC). Although often considered an IL-4–dependent process, MΦ fusion and multinucleation are equally responsive to IFN-γ [34–38]. In GCA, multinucleated GC serve as a cellular source of numerous cytokines, thus functioning as an important amplification mechanism [39].
Interestingly, IFN-γ–producing Th1 cells are relatively insensitive to glucocorticoid therapy and persist in steroid-treated patients [29, 40]. Evidence has been provided that IL-12 production has a similar tendency to continue despite steroid therapy [29]. This raises the concept that IL-12 and predominance of Th1 cells in GCA patients may derive from a nonphagocytic cell population.
Persistent IFN-γ overproduction is a critical mechanism in the chronicity of disease. Thus, a simple approach to treating chronic GCA could lie in curbing IFN-γ production. However, this suggestion needs to be carefully evaluated. IFN-γ is a cornerstone in immunoprotection, particularly against malignancy and viral infection [41–44]. IFN-γ–dependent immunoprotection is particularly important in the elderly, thus in the patient populations at risk for GCA. Recent data have alerted us to the presence of varicella zoster antigen in inflamed temporal arteries [45, 46]. While the pathogenic role of such local viral antigen remains to be determined, it raises concerns about the competence of anti-viral immunity in GCA patients. Targeting IFN-γ would render patients immunocompromised, possibly more than is acceptable.
Th17 cells
IL-6, a well-established biomarker of disease activity in GCA, sways T cells to differentiate into Th17 cells. Frequencies of Th17 cells are elevated in untreated patients, and Th17 cells are among the lesional T cells. The product portfolio of Th17 cells includes IL-17, IL-21, IL-22, IL-8, and IL-26. Th17 cells cannot produce IL-2, thus fail to self-expand. Accordingly, they account for a relatively small number of T cells; e.g., in healthy controls, 0.2 % of T cells are Th17 cells, and in untreated patterns, this frequency increased to 1.5–2.0 % [29]. IL-17 receptors are expressed on several disease-relevant target cells, including MΦ, EC, and fibroblasts. It is still unclear which pathogenic processes in the disease lesions are directed by IL-17.
Th17 cells are a typical component of early, untreated disease. They are highly sensitive to glucocorticoids and usually normalize rapidly under treatment.
Low doses of steroids seem capable to control Th17 frequencies. It may be relatively easy to therapeutically target this T cell component and develop novel therapies, but their absence in chronic vasculitis questions how successful this strategy would be in management of chronic disease.
IL-21–producing CD4 T cells
A recent study has implicated the T cell product IL-21 in the inflammatory cocktail present in vascular lesions [30]. Like the other T cell lineages, the increase in IL-21–producing T cells was easily detectable in the peripheral blood [30]. IL-21 is a pleiotropic cytokine produced by several T helper cell subtypes, specifically Th17 cells, Th9 cells, and follicular helper T (Tfh) cells [47]. IL-21 is not only a critical regulator of Th17 development; it also induces cytotoxicity of CD8 T cells and NK cells [48]. In germinal centers, IL-21–producing Tfh cells are the master regulators of B cell differentiation, representing an essential element of antibody responses. Essentially, IL-21 participates in multiple effector mechanism in protective and pathogenic immunity [48]. Fortunately, IL-21, similar to IL-17, is highly sensitive to glucocorticoid treatment [30]. More precise targeting of IL-21 would require the identification of the producing cell type and a better understanding of which effector pathways are IL-21–dependent.
Other T cell subtypes in vasculitis
With a growing array of T cells being implicated in the chronic inflammation associated with autoimmune disease, it is to be expected that more and more T cell lineages will be associated with GCA. A novel subset of helper CD4 T cells producing the marker cytokine IL-9, Th9 cells, have recently been recognized to be important in the pathogenesis of ulcerative colitis [49]. Th9 development is regulated by the lineage-determining transcription factor PU.1. IL-9 has been observed in tissue sections of inflamed temporal arteries [50]. IL-9 was correlated with the intensity of the systemic inflammatory response, measured by ESR and CRP [50]. IL-9 can be produced by an array of inflammatory cells, mostly mast cells, NKT cells, and T cells. IL-9–producing mast cells have fostered the concept that IL-9 is important in allergic responses. Currently, the cellular origin of IL-9 in GCA lesions remains undetermined and further studies are needed to clarify its role in the disease process.
Th22 cells represent another novel T cell subset, equipped with the potential to promote chronic, tissue destructive inflammation [51]. Their marker cytokine is IL-22, a member of the IL-10 family of cytokines. Th22 cells are often considered to be siblings of Th17 cells [51]. Several lines of evidence suggest that IL-22 may be a player in starting and maintaining tissue inflammation [51, 52]. Its precise role in GCA remains to be explored.
Vascular cells in giant cell arteritis
Endothelial cells
Endothelial cells (EC) occupy the immediate interphase between the vessel wall and the immune system. They also are a critical component of the wall-remodeling process, since neovessels provide oxygen for the growing intimal layer. It is currently unknown how many different types of EC are represented in medium and large human arteries. Besides, the endothelial layer covering the main lumen, vasa vasorum, in the adventitia builds a special support system. It is suspected that neovessels supplying the thickening intimal layer originate from the vasa vasorum network and that vasa vasorum provides immune cells access to the tissue niche of the arterial wall. EC heterogeneity in healthy and inflamed temporal arteries and in healthy and inflamed aortas is suspected, but experimental data are lacking.
In other vascular diseases, specifically atherosclerosis, dysfunction of EC is considered pathognomic. In GCA patients, EC function is similarly abnormal [53]. EC in the inflamed arteries express high levels of endothelin, suggesting their activation [54]. Anti-EC antibodies are sometimes detected in the sera of GCA patients, and those antibodies may induce EC injury [55]. A recent study shows that serum amyloid A protein (SAA), one of the many acute phase reactants elevated in GCA patients, induces proliferation of EC, leading to myofibroblast outgrowth and angiogenesis [56]. It would be important to understand why SAA could have such a role in highly selected arteries, whereas other vascular territories are completely spared.
Smooth muscle cells
The most important cell type in the wall remodeling process is the vascular smooth muscle cell (VSMC). Current paradigm holds that vessel wall injury triggers a phenotypic switch, turning contractile VSMC into synthetic myofibroblasts. Such dedifferentiated cells acquire migratory and invasive functions, leave the adventitia and media, and move across the destroyed lamina elastic interna to form the matrix-rich hyperplastic intima. VSMC in GCA arteries is known to actively participate in the inflammatory response. They upregulate the enzyme aldose reductase to protect themselves and neighboring cells from oxidative damage [57]. Also, they actively interfere with immune cell recruitment. By expressing NOTCH ligands, they are able to interact with NOTCH receptors on CD4 T cells, thus selectively interacting with vasculitogenic T cells [40]. This NOTCH-NOTCH ligand pathway has been implicated in the amplification of vasculitis, and blockade of this signaling pathway can effectively suppress vascular inflammation [40]. A recent study has pointed out yet another critical function of VSMC. By producing neurotrophins, wall-residing VSMC can influence the neuronal regulation of blood vessels [58]. Neurotrophins control the survival, the development, and the function of neurons, which could possibly provide both proinflammatory and anti-inflammatory signals. Further studies are needed to define the precise origin of the cells that form the neointima and to characterize the molecular pathways that induce myofibroblast mobility and matrix production. A better understanding of VSMC biology holds great promise to develop entirely new therapeutic approaches in GCA.
Conclusions
The pathogenic concept of GCA has evolved. Multinucleated GC and granuloma formation have encouraged speculations about infectious triggers. Also, elastic components of the wall were suspected to elicit fusion of highly stimulated MΦ, unable to digest the inert parts of the elastic lamina. Clinical management assumed that the vasculitis is a self-limited syndrome that requires about 2 years of immunosuppression. This simplified approach has been replaced with the more complex concept that immune stimulators other than indigestible elastic fibers or persistent microbes may give rise to persistent granulomatous vasculitis.
As in most inflammatory diseases, a broad array of inflammatory cytokines participates in GCA. The question remains whether one or two of such proinflammatory mediators hold pinnacle positions and are key disease drivers. Blocking TNF-α, a highly successful intervention in rheumatoid arthritis and inflammatory bowel disease, has no steroid-sparing effects in GCA [59].
Separating it from other inflammatory conditions, GCA is associated with an intense acute phase response. Highly elevated ESR and CRP levels suggest a role for IL-6, a potent upstream inducer of CRP. One of the predictable therapeutic effects of glucocorticoids is the prompt suppression of IL-6 production. This aspect of glucocorticoids can likely be replaced with more targeted therapeutic approaches. IL-6 may also be involved in inducing Th17 cell differentiation, relevant in some stages of disease.
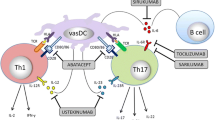
A major lesson from pathogenic studies has been the unexpected diversity of functional T cell subsets in the vascular lesions (Fig. 1). T cells producing IFN-γ, IL-2, IL-17, IL-9, and IL-21 have been described, crossing the boundaries between classical lineage assignments. Of all T cell products, IFN-γ not only may be the most resistant to therapy, but also represents a key protection and survival factor, particularly in the naturally immunocompromised elderly. The diversity of differentiated T cells in the vasculitic infiltrates is strongly suggestive for a multi-antigen stimulation and discourages the concept that a single instigator causes GCA. It is equally important in the development of novel therapeutic strategies. A cocktail of immunomodulators will be required to manage the abnormal adaptive immunity in GCA.

Pathogenic pathways in GCA. Healthy medium and large arteries are composed of three layers: the endothelial cell (EC) barrier in the intima, sheets of vascular smooth muscle cells (VSMC) in the media, and the adventitial support layer with vascular dendritic cells (vasDC) and vasa vasorum networks. Inflammatory cascades of early GCA start with a breakdown of the immune privilege in the adventitia, triggered by activation and immobility of vasDC. Resulting chemokine production recruits various lineages of T cells and MΦ. T cells committed to at least four functional lineages have been identified by their marker cytokines: IFN-γ, IL-17, IL-9, and IL-21. A common principle of recruited T cells and MΦ is their arrangement in granulomas, stabilizing lymphoid microstructure that optimizes inflammation. The artery, represented by VSMC, EC, and fibroblasts, responds with a maladaptive repair program, leading to medial thinning and lumen-occlusive hyperplastic intima.
Table 1 outlines the cellular players in GCA and how current and future therapies can target their pathogenic role. Several clinical trials are currently exploring the use of cytokine blockers and of costimulatory inhibitors. Some currently available T cell immunosuppressants are used empirically in patients requiring long-term therapy, but their use has not been investigated in appropriately designed clinical trials. These include azathioprine, mycophenolate, cyclosporine, tacrolimus, and others. On the horizon are novel treatment approaches that may be able to reach more than a single vasculitogenic cell population [60–64]. As the clinical complications of GCA are related to the vascular wall remodeling, EC and VSMC emerge as intriguing targets. Of particular interest are cell-based therapies, which may be able to harness the immune system’s natural anti-inflammatory potential to bring to a halt a damage-inducing immune response and induce disease remission.
The superior therapeutic efficacy of glucocorticoids almost certainly lies in their pleiotropic effects, targeting innate as well as adaptive immune cells. This desirable aspect of glucocorticoids will be difficult to replace with a single or even a few immunomodulatory medications. Nature had millions of years to develop such a versatile immunosuppressant as glucocorticoids. It will take some time to design a cocktail of man-made immunomodulators that have equal efficacy.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. Lancet. 2008;372(9634):234–45.
Salvarani C, Pipitone N, Versari A, Hunder GG. Clinical features of polymyalgia rheumatica and giant cell arteritis. Nat Rev Rheumatol. 2012;8(9):509–21.
Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med. 2003;349(2):160–9.
Weyand CM, Goronzy JJ. Clinical practice. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med. 2014;371(1):50–7. This review article provides clinical information on the evaluation, management, and problems of GCA.
Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. 2013;9(12):731–40. This review article covers detailed information on the pathophysiology of GCA.
Weyand CM, Liao YJ, Goronzy JJ. The immunopathology of giant cell arteritis: diagnostic and therapeutic implications. J Neuroophthalmol. 2012;32(3):259–65.
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
Mazlumzadeh M, Hunder GG, Easley KA, Calamia KT, Matteson EL, Griffing WL, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum. 2006;54(10):3310–8.
Weyand CM, Fulbright JW, Hunder GG, Evans JM, Goronzy JJ. Treatment of giant cell arteritis: interleukin-6 as a biologic marker of disease activity. Arthritis Rheum. 2000;43(5):1041–8.
Banks PM, Cohen MD, Ginsburg WW, Hunder GG. Immunohistologic and cytochemical studies of temporal arteritis. Arthritis Rheum. 1983;26(10):1201–7.
Cavazza A, Muratore F, Boiardi L, Restuccia G, Pipitone N, Pazzola G, et al. Inflamed temporal artery: histologic findings in 354 biopsies, with clinical correlations. Am J Surg Pathol. 2014;38(10):1360–70.
Cid MC, Campo E, Ercilla G, Palacin A, Vilaseca J, Villalta J, et al. Immunohistochemical analysis of lymphoid and macrophage cell subsets and their immunologic activation markers in temporal arteritis. Influence of corticosteroid treatment. Arthritis Rheum. 1989;32(7):884–93.
Han JW, Shimada K, Ma-Krupa W, Johnson TL, Nerem RM, Goronzy JJ, et al. Vessel wall-embedded dendritic cells induce T-cell autoreactivity and initiate vascular inflammation. Circ Res. 2008;102(5):546–53.
Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med. 2004;199(2):173–83.
Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation. 2008;118(12):1276–84.
Deng J, Ma-Krupa W, Gewirtz AT, Younge BR, Goronzy JJ, Weyand CM. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ Res. 2009;104(4):488–95.
Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum. 1993;36(9):1286–94.
Weyand CM, Wagner AD, Bjornsson J, Goronzy JJ. Correlation of the topographical arrangement and the functional pattern of tissue-infiltrating macrophages in giant cell arteritis. J Clin Invest. 1996;98(7):1642–9.
Wagner AD, Goronzy JJ, Weyand CM. Functional profile of tissue-infiltrating and circulating CD68+ cells in giant cell arteritis. Evidence for two components of the disease. J Clin Invest. 1994;94(3):1134–40.
Baldini M, Maugeri N, Ramirez GA, Giacomassi C, Castiglioni A, Prieto-Gonzalez S, et al. Selective up-regulation of the soluble pattern-recognition receptor pentraxin 3 and of vascular endothelial growth factor in giant cell arteritis: relevance for recent optic nerve ischemia. Arthritis Rheum. 2012;64(3):854–65.
Corbera-Bellalta M, Planas-Rigol E, Lozano E, Terrades-Garcia N, Alba MA, Prieto-Gonzalez S, et al. Blocking interferon gamma reduces expression of chemokines CXCL9, CXCL10 and CXCL11 and decreases macrophage infiltration in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis. 2015. doi:10.1136/annrheumdis-2015-208371.
Mihm B, Bergmann M, Bruck W, Probst-Cousin S. The activation pattern of macrophages in giant cell (temporal) arteritis and primary angiitis of the central nervous system. Neuropathology. 2014;34(3):236–42.
Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ. Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum. 1998;41(4):623–33.
Martinez-Taboada V, Brack A, Hunder GG, Goronzy JJ, Weyand CM. The inflammatory infiltrate in giant cell arteritis selects against B lymphocytes. J Rheumatol. 1996;23(6):1011–4.
van der Geest KS, Abdulahad WH, Chalan P, Rutgers A, Horst G, Huitema MG, et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol. 2014;66(7):1927–38.
van der Geest KS, Abdulahad WH, Rutgers A, Horst G, Bijzet J, Arends S, et al. Serum markers associated with disease activity in giant cell arteritis and polymyalgia rheumatica. Rheumatology (Oxford). 2015;54(8):1397–402.
Martinez-Taboada VM, Goronzy JJ, Weyand CM. Clonally expanded CD8 T cells in patients with polymyalgia rheumatica and giant cell arteritis. Clin Immunol Immunopathol. 1996;79(3):263–70.
Weyand CM, Younge BR, Goronzy JJ. IFN-gamma and IL-17: the two faces of T-cell pathology in giant cell arteritis. Curr Opin Rheumatol. 2011;23(1):43–9.
Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. 2010;121(7):906–15.
Terrier B, Geri G, Chaara W, Allenbach Y, Rosenzwajg M, Costedoat-Chalumeau N, et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum. 2012;64(6):2001–11.
Rittner HL, Kaiser M, Brack A, Szweda LI, Goronzy JJ, Weyand CM. Tissue-destructive macrophages in giant cell arteritis. Circ Res. 1999;84(9):1050–8.
Hilhorst M, Shirai T, Berry G, Goronzy JJ, Weyand CM. T cell-macrophage interactions and granuloma formation in vasculitis. Front Immunol. 2014;5:432.
Shirai T, Hilhorst M, Harrison DG, Goronzy JJ, Weyand CM. Macrophages in vascular inflammation—from atherosclerosis to vasculitis. Autoimmunity. 2015;48(3):139–51.
Weyand CM, Tetzlaff N, Bjornsson J, Brack A, Younge B, Goronzy JJ. Disease patterns and tissue cytokine profiles in giant cell arteritis. Arthritis Rheum. 1997;40(1):19–26.
Most J, Neumayer HP, Dierich MP. Cytokine-induced generation of multinucleated giant cells in vitro requires interferon-gamma and expression of LFA-1. Eur J Immunol. 1990;20(8):1661–7.
Fais S, Burgio VL, Silvestri M, Capobianchi MR, Pacchiarotti A, Pallone F. Multinucleated giant cells generation induced by interferon-gamma. Changes in the expression and distribution of the intercellular adhesion molecule-1 during macrophages fusion and multinucleated giant cell formation. Lab Invest. 1994;71(5):737–44.
Helming L, Gordon S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009;19(10):514–22.
Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83.
Kaiser M, Younge B, Bjornsson J, Goronzy JJ, Weyand CM. Formation of new vasa vasorum in vasculitis. Production of angiogenic cytokines by multinucleated giant cells. Am J Pathol. 1999;155(3):765–74.
Piggott K, Deng J, Warrington K, Younge B, Kubo JT, Desai M, et al. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation. 2011;123(3):309–18.
Farrar MA, Schreiber RD. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol. 1993;11:571–611.
Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–91.
Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259(5102):1739–42.
Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107–11.
Nagel MA, Khmeleva N, Boyer PJ, Choe A, Bert R, Gilden D. Varicella zoster virus in the temporal artery of a patient with giant cell arteritis. J Neurol Sci. 2013;335(1–2):228–30.
Lavi E, Gilden D, Nagel M, White T, Grose C. Prevalence and distribution of VZV in temporal arteries of patients with giant cell arteritis. Neurology. 2015;85(21):1914–5.
Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol. 2015;15(5):295–307.
Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol. 2008;26:57–79.
Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol. 2014;15(7):676–86.
Ciccia F, Rizzo A, Guggino G, Cavazza A, Alessandro R, Maugeri R, et al. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatology (Oxford). 2015;54(9):1596–604.
Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. 2015;33:747–85.
Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 2015;74(1):5–17.
Gonzalez-Juanatey C, Llorca J, Garcia-Porrua C, Sanchez-Andrade A, Martin J, Gonzalez-Gay MA. Steroid therapy improves endothelial function in patients with biopsy-proven giant cell arteritis. J Rheumatol. 2006;33(1):74–8.
Dimitrijevic I, Andersson C, Rissler P, Edvinsson L. Increased tissue endothelin-1 and endothelin-B receptor expression in temporal arteries from patients with giant cell arteritis. Ophthalmology. 2010;117(3):628–36.
Regent A, Dib H, Ly KH, Agard C, Tamby MC, Tamas N, et al. Identification of target antigens of anti-endothelial cell and anti-vascular smooth muscle cell antibodies in patients with giant cell arteritis: a proteomic approach. Arthritis Res Ther. 2011;13(3):R107.
O’Neill L, Rooney P, Molloy D, Connolly M, McCormick J, McCarthy G, et al. Regulation of inflammation and angiogenesis in giant cell arteritis by acute-phase serum amyloid A. Arthritis Rheumatol. 2015;67(9):2447–56.
Rittner HL, Hafner V, Klimiuk PA, Szweda LI, Goronzy JJ, Weyand CM. Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J Clin Invest. 1999;103(7):1007–13.
Ly KH, Regent A, Molina E, Saada S, Sindou P, Le-Jeunne C, et al. Neurotrophins are expressed in giant cell arteritis lesions and may contribute to vascular remodeling. Arthritis Res Ther. 2014;16(6):487.
Hoffman GS, Cid MC, Rendt-Zagar KE, Merkel PA, Weyand CM, Stone JH, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146(9):621–30.
Goveia J, Stapor P, Carmeliet P. Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol Med. 2014;6(9):1105–20.
Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8(9):726–36.
Berair R, Hollins F, Brightling C. Airway smooth muscle hypercontractility in asthma. J Allergy (Cairo). 2013;2013:185971.
Kim JY, Kang JS, Kim HM, Ryu HS, Kim HS, Lee HK, et al. Inhibition of bone marrow-derived dendritic cell maturation by glabridin. Int Immunopharmacol. 2010;10(10):1185–93.
Camoretti-Mercado B. Targeting the airway smooth muscle for asthma treatment. Transl Res. 2009;154(4):165–74.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
RW declares that he has no conflict of interest. JJG declares that he has no conflict of interest. GB declares that he has no conflict of interest. YJL declares that she has no conflict of interest. CMW declares that she has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Funding
This work was supported by grants from the National Institutes of Health (R01 AR042527, R01 AI044142, HL 117913, R01 AI108906, and P01 HL058000 to CMW and R01 AI108891, R01 AG045779, and I01 BX001669 to JJG) and the Govenar Discovery Fund (CMW). YJL was supported by North American Neuro-Ophthalmology Society Pilot Grant.
Additional information
This article is part of the Topical Collection on Vasculitis
Rights and permissions
About this article

Cite this article
Watanabe, R., Goronzy, J.J., Berry, G. et al. Giant Cell Arteritis: From Pathogenesis to Therapeutic Management. Curr Treat Options in Rheum 2, 126–137 (2016). https://doi.org/10.1007/s40674-016-0043-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40674-016-0043-x