
Abstract
Purpose
This consensus review article considers the question of whether glucocorticoid (GC) therapy is still relevant in the treatment of rheumatic diseases, with a particular focus on rheumatoid arthritis (RA), and whether its side effects can be adequately managed. Recent basic and clinical research on the molecular, cellular and clinical effects of GCs have considerably advanced our knowledge in this field. An overview of the subject seems appropriate.
Methods
This review is the result of a multidisciplinary expert working group, organised by European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis. The recent literature was surveyed and the salient evidence synthetized.
Results
The pathophysiological basis of RA (and other inflammatory rheumatic diseases) now strongly implicates the adaptive immune system in addition to innate mechanisms. The molecular effect of GCs and differential GC sensitivity is better understood, although exploiting this knowledge is still in its infancy. The newer treatment strategies of early and aggressive control of RA have gr eatly improved clinical outcomes, but improvements are still possible. Newer targeted anti-inflammatory drugs have made an important impact, yet they too are associated with numerous side effects.
Discussion
Short durations of moderate doses of GCs are generally well tolerated and have a positive benefit/risk ratio. Patients should be assessed for fracture risk and bone preserving agents and be prescribed calcium and vitamin D supplementation.
Conclusions
Within a strategy of a disease modifying approach to inflammatory disease, combination therapy including a GC is effective approach.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The decade of the human genome (2000–2010) [1] brought in a number of significant advances to the understanding of pathophysiology of rheumatoid arthritis (RA); not only the genetic components of the disease, but also the involvement of activating environmental factors and the subsequent immune response. That decade also bore witness to a number of clinical advances in the treatment of RA, forged out of the collaborative efforts of American College of Rheumatology (ACR) and the European League against Rheumatism (EULAR) to standardise the clinical approach and from the fresh insights from innovative trials that gave rise to the ideas of the “window of opportunity” in early RA, and the “tight control”, or treat-to-target, which is necessary to optimise the remission rate [2–4]. Glucocorticoid (GC) therapy, which is increasingly associated with greater risks of adverse outcomes, particularly osteoporotic fractures, has been omitted from some treatment guidelines, but some experts have remained convinced of its value in rapidly controlling the inflammation in the joints. In parallel with these developments, that decade saw the launch of several biological disease-modifying agents (biological DMARDs) which have made an important impact in managing immunoinflammatory diseases.
The clinical management of rheumatic diseases is a very active research field and effects of GC therapy continue to provoke much debate. What are the risks and how does GC therapy measure up to newer treatment strategies with biological DMARDs and can GCs be incorporated in these strategies? Can we consider the risks associated with GCs, in both muscles and bone, as well as in other organ systems as manageable? Could the stratification of patients according to their risk of developing side effects from using GC’s optimise benefit?
Such were the questions at a recent expert working group of the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO) convened to discuss the current evidence for and against the use of GC’s in rheumatic diseases. With a focus on RA, this review attempts to put into perspective the progression of our knowledge of the adverse effects of GCs (within the limits of our appreciation of cause and effect) with the advances in our understanding of the inflammatory response.
New insights into the pathophysiological basis of RA
The pathogenesis of RA has been well described by Klareskog and colleagues in an excellent review, so only a few salient points will be described here. Because of the multiplicity of interacting factors that give rise to this clinical condition, they describe RA as a “complex genetic disease” [5].
It is now clear that neutrophil-dominated inflammation (an innate immune response) plays an important role in RA, as is also the case for inflammatory pulmonary diseases such as asthma, chronic obstructive pulmonary disease (COPD), bronchiolitis and cystic fibrosis [6]. This similarity seems pertinent since early manifestations of RA are frequently in the lung [7]. Indeed, smoking and other forms of pulmonary insult increase the risk of RA, especially in persons with specific genetic modifications at the human leukocyte antigen (HLA) gene, thus giving rise to altered patterns of citrullination of mucosal proteins and other barrier tissues and subsequently antibodies to those entities [5, 8].
The adaptive immune system is also strongly implicated and is now considered to be a key driver of the disease process. Of particular importance is the involvement of the Th17 T-cells (Fig. 1), which become activated to secrete interleukin-17 (IL-17) and migrate along with neutrophils into the synovium. Once there, they induce the local mesenchymal cells to produce proinflammatory cytokines such as interleukin-1 (IL-1), interleukin-6 (IL-6), tumour necrosis factor-alpha (TNFα), a variety of growth factors, and matrix metalloproteinases, with resulting damage to the joint and bone (see [9]). The neutrophils survive for an extended time in the synovium because of the upregulation of a transcription factor that inhibits a regulatory apoptotic pathway [10, 11]. Much effort is now focussed on trying to re-activate apoptosis or otherwise down-regulate inflammatory T cell activity.

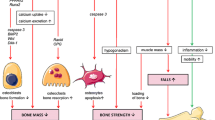
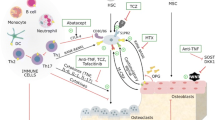
Cellular interactions within the joint in rheumatoid arthritis. In the symptomatic phase of the disease, cells from the innate immune system located in the synovium, including mast cells, neutrophils and macrophages, respond to stimulation by ACPA-immune complexes and/or rheumatoid factor and/or other triggering factors, by the release of cytokines and chemokines. The chemokines attract activated cells of the adaptive immune system (T- and B-cells) which invade the synovium and start to secrete inflammatory cytokines (line arrows indicate the movement of cell types; square arrows link the sources and targets). A positive feedback loop involving cytokines, prostaglandins, reactive oxygen intermediates and trophic factors ensues (only a few of the implicated signalling molecules are illustrated), marshalling immune cells, synovial fibroblasts, chondrocytes, and osteoclasts. This, together with the release of matrix proteases and the molecular products of damage, drives the chronic phase of rheumatoid arthritis pathogenesis. Note that endogenous glucocorticoid signalling is locally enhanced via the upregulation of 11β-HSD1 production in most cells. The main inflammatory signals in the synovium include tumor necrosis factor α (TNF-α), interleukin-1 (IL-1), interleukin-6 (IL-6) and interferon-α (IFN-α). Important stimulators of immune cell activation/proliferation include interleukin-15 (IL-15) and granulocyte colony-stimulating factor (G-CSF), while the stimulators of mesenchymal cell proliferation/differentiation are mainly platelet-derived growth factor (PDGF), fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF). Other secreted entities are the matrix metalloproteinases (MMPs), transforming growth factor β (TGF-β), dickkopf-1 (DKK-1) and the receptor activator of nuclear factor κB ligand (RANKL). In the adjacent bone, the differentiation of osteoclasts is stimulated by the increased production of RANKL and IL-32. RANKL also stimulates the activity and therefore bone resorption of these cells. The expression of DKK-1, an inhibitor of wnt signalling is also believed to contribute to bone remodelling. References: McInnes et al. [8]; Klareskog et al. [5]; Weinstein [160]; Clark [171]
The adaptive immune system is also invoked to produce antibodies against rheumatoid factor (RF) and/or against cyclic citrullinated peptides [anti-CCP; also known as antibodies to citrullinated protein antigens (ACPA)]. The plasma levels of these antibodies give important clues as to the activity of the disease [12, 13]. Higher levels of these antibodies are associated with an increased risk of joint damage and cardiovascular diseases [14]. Whilst the effect of these antibodies in the disease process is not fully understood, recent work has shown that ACPA can stimulate osteoclast differentiation and that in pre-RA individuals, those who are ACPA-positive, there is more frequent evidence of bone loss and reductions in cortical thickness as compared with ACPA-negative individuals [15]. The production of these antibodies is associated with more generalised inflammation, as indicated by higher levels of C-reactive protein (CRP) and higher erythrocyte sedimentation rates [16]. Evidence supports that the presence of ACPA as a better diagnostic test than anti-RF for the disease in asymptomatic individuals [5], however, 20 % of patients with RA (even when well established), are negative for one or other (or both) of these antibodies [13]. The development of ACPA is largely dependent on environmental factors and lifestyle, rather than genetics, while genetic factors have a relatively important effect in determining which ACPA-positive individuals will go on to develop RA [17].
The impact of RA on organ systems
Epidemiological research in the 1970s and 1980s showed that morbidity and mortality in patients with RA were much higher than in the general population [18]. The possible reasons for this seem to be related to increased cardiovascular (CV) risk, but the contribution of RA to the overall risk profile has proved difficult to determine. Indeed this patient population tends to show high CV risk factor profile, where smoking, hypertension, obesity and diabetes are frequent. This situation created a strong possibility of confounding in the epidemiological studies that was not always adequately controlled and still gives rise to uncertainty. We will therefore provide a brief description of this risk profile of RA patients before discussing the possible contribution of the disease in organ failure.
Smoking is the strongest environmental risk factor for RA and the prevalence of cigarette smoking tends to be higher in RA. In a 2011 meta-analysis of 15 case–control studies (2956 patients and 3713 controls), Boyer et al. found that the prevalence of smoking in RA patients was 56 % higher than in the case controls [odds ratio (OR) 1.56; 95 % confidence interval (CI) 1.34–1.80] [19]. Cigarette smoking has also been associated with increased severity of RA particularly in men with seropositive disease [20]. Hypertension may be more prevalent in this RA patients, with some researchers reporting rates of 50 % [21]. However, in the Boyer et al. meta-analysis, no clear increase was demonstrated. A tendency towards hypertension is plausible in this population, since several studies have reported an increase in arterial wall stiffness in RA patients (see [22]). The relationship between corpulence and RA risk has been investigated in several studies, but the correlation between BMI and RA remains uncertain [23]. What seems to be more relevant is relative body composition, where increased body fat and reduced muscle mass leads to a condition known as rheumatoid cachexia [23]. Increased central adiposity is common in RA, which in itself is a risk factor for insulin resistance and endothelial dysfunction and could also play a role in RA. An increased prevalence of insulin resistance has been reported in individuals with RA, associates particularly with those having high-grade inflammation [22]. While the association between RA and diabetes mellitus is widely debated, the Boyer et al. meta-analysis did find that the prevalence of diabetes mellitus was significantly increased in RA as compared to controls with an odds ratio of 1.74 (95 % CI 1.22–2.50) (P = 0.003) [19]. The COMORA study has highlighted other important comorbidities in RA (that are highly variable between countries) including depression and asthma [24].
Adverse effects of RA on cardiovascular outcomes
RA patients have a higher CV risk than in the general population [25–27], a risk which is comparable to that of patients with type-2 diabetes [28]. According to one prospective cohort study the age- and sex-adjusted hazard ratio for CV disease was 1.94 versus the general population [28] and in a 2012 meta-analysis of 41,490 patients in observational studies, the patients with RA had a 48 % increased risk of CV disease [i.e. a pooled RR (relative risk) of 1.48, 95 % CI 1.36–1.62] with the risks of myocardial infarction and cerebrovascular accidents increased by 68 and 41 % [29].
It has been expected that the improvements in RA treatment since the 1990’s would be reflected by a decrease on annualised CV risk. Initially this hypothesis seemed to be supported by data from the North American ARAMIS registry [30], but other results were conflicting [31]. Additional important confounding factors that were perhaps not fully considered at the time were the drug-related deaths in association with the use of rofecoxib, a specific COX-2 inhibitor, and rosiglitazone, an anti-diabetic drug in the thiazolidinedione class of drugs. Both of these were approved in 1999 (USA), but rofecoxib was withdrawn in 2004 and rosiglitazone, which remains on the US market, was withdrawn from the European market in 2010. Both drugs have been associated with higher rates of MI [32, 33]. It might also be reminded that the non-steroidal anti-inflammatories, as a drug class, are also not without risk. In a recent network meta-analysis of 31 trials (116,429 patients) [34], ibuprofen and diclofenac (versus placebo) were associated with increased risk of stroke; and etoricoxib and diclofenac were associated with increased risk of CV death. Naproxen appeared to be the least harmful.
A key factor in the increased CV risk, particularly atherosclerosis, is the alterations in circulating lipid profiles seen with RA and other systemic inflammatory conditions [35, 36]. Indeed, a significant proportion of patients diagnosed with RA (and without cardiovascular disease) are reported to have lipid profiles that would mandate statin therapy [37, 38]. Interestingly, some of the RA susceptibility genes seem to be involved in the regulation of lipid metabolism [39]. The risk of incident CV events in RA, however, is not strongly correlated with traditional CV risk factors [22, 40], but more closely associated with inflammation, the extra-articular manifestations of RA [41] and the presence of anti-CCP antibodies [14]. In vitro experiments have shown that IL-17 and TNFα provoke endothelial cells to produce pro-coagulant and pro-thrombotic factors that could change the underlying CV risk [42] and that these effect can be inhibited by simvastatin [43]. A statin therapy in RA patients has also been shown to reduce inflammatory biomarkers and improve the disease activity score (DAS-28) [44].
Indeed, chronic systemic inflammation and changes in lipid metabolism are tightly linked, influencing each other, as well as immune responses. Metabolic “fingerprints” of lipid profiles are distinctive of underlying inflammation and in patients with RA it would seem that this fingerprint is different depending on whether the disease is early or established [45]. Effective RA therapy (defined as at least a 20 % improvement in symptoms), has been associated with “amelioration” in the lipid profile (increased HDL-cholesterol and apolipoprotein A1 levels, but unchanged LDL-cholesterol levels) which could improve CV risk [46]. The relationship between chronic inflammation and CV disease may become clearer with the termination of two clinical studies currently underway: the Cardiovascular Inflammation Reduction Trial testing MTX 15–20 mg in coronary patients (estimated completion date: December 2018) and the Novartis trial of canakinumab, a monoclonal antibody directed against IL-1β, in post-MI patients (estimated completion date: July 2016).
Also associated with RA is an increased risk of stroke, particularly ischemic stroke, which is correlated with RA severity, cardiovascular risk factors and comorbidity [47].
Adverse effects of RA on bone health
The destruction of cartilage and bone are major effects of RA [5, 8]. The activation of matrix metalloproteases is the main driver of the degradation of cartilage, although other processes contribute. In the bone, the increased expression of the “receptor activator of nuclear factor κB ligand” (RANKL) and the expression of osteoprotegerin, results in enhanced promotion of osteoclast differentiation and subsequent bone loss (see [9] for references). The erosion tends to occur at sites subjected to mechanical stress or loading, thus the periarticular, cortical sites are more vulnerable. Also, as stated above, recent evidence suggests that ACPA can directly stimulate this process via enhanced osteoclast differentiation [15].
Adverse effects of RA on the immune system
Patients with RA have a greater risk of infection than matched controls [48]. This may be due to a combination of factors including the immunomodulatory effects of RA, smoking, increasingly sedentary lifestyle and the effect of drugs used in RA treatment. However, the chronic systemic inflammation in RA also seems to induce a defect in the immune response via an effect on T helper cells [49]. The short-term administration of low-doses of GC has a beneficial effect on this defect (Miossec, personal communication).
GC therapeutic strategies in RA
In the early 1990s, the first-line treatment for RA was non-steroidal anti-inflammatory drugs (NSAIDs). GC therapy was mostly as a second-line with varying strategies of step-down, step-up or pulse dosing. The clinical efficacy of GCs was considered as limited since the beneficial effects ended rapidly after treatment withdrawal. Other second-line therapies in RA, such as methotrexate (MTX) and sulfasalazine (SSZ), were beginning to be recognised as having good effectiveness profiles [50], especially when administered in combination [51].
In 1997, the COBRA trial results revealed the possibilities of a different strategy [52]: the treatment newly diagnosed (early) RA with a combination of SSZ and MTX, plus high doses of prednisolone in a step-down regimen. In this randomised study with a blinded outcome assessment (disease activity score and radiographic damage score), the GC arm showed a higher rate of improvement at week 28 than with SSZ alone (72 versus 49 % of patients improved according to ACR criteria. This study and a number of important clinical studies that followed in the period 1999–2013 (one could mention: Fin-RACo [53], BARFOT [54], TICORA [3], BeSt [55], CAMERA-II [56], DREAM [57] IMPROVED [58] and COBRA-light [59]) helped build on the idea that there was a “window of opportunity” in early RA and that tight control the inflammation or “treating-to-target” (i.e. remission or minimal disease activity) resulted in better long-term outcomes.
Several systematic reviews have concluded that low dose (≤7.5 mg/day) GC can provide a useful “bridging-therapy” below the therapeutic effects of the slower acting DMARDs start to manifest [60–63]. Conclusion accepted by EULAR in their 2010 recommendations [64] and further confirmed in 2014 [65]. The ACR has been rather more reticent on the use of GCs and in its 2012 update of recommendations, focussed on conventional DMARDs and biological therapies and did not address the use of GCs [66].
The risks of GC therapies
The most commonly self-reported adverse events by patients who are prescribed with longer-term GC use are weight gain (about 70 % of individuals), skin bruising (~55 %), sleeping problems (~45 %) and mood problems (~45 %) and all show a positive relationship with GC exposure [67]. It might be added that a constructive patient-practitioner dialogue at the start of any GC therapy is very important [68]. The adverse reactions associated with GC therapy have been described in more detail in recent reviews [69, 70]. From a clinical aspect, the more serious adverse reactions are outlined below.
Adverse effects of GCs on bone health and fracture: epidemiology
In the early 1990s, it was clear that GCs could negatively impact bone health, but the magnitude of effect was unclear. Confirmation of a bone demineralising effect of GC therapy accrued gradually throughout the 1990s with the publication of a review with meta-analysis [71] and with the very large cohort analyses of the UK’s GPRD database by van Staa et al. [72, 73]. The latter group demonstrated that patients exposed to GCs had higher relative risk of fracture (versus age-matched non-exposed individuals), with relative risk ratios varying from 1.1 for forearm fracture, through 1.6 for hip, to 2.6 for vertebral fracture. Again, one of the major difficulties encountered in observational studies, is controlling for confounding factors, since the decision to prescribe a GC (and the dose selection) is greatly influenced by the activity of the underlying disease and the age of the patient. The rate of bone loss will vary also according to these factors, as well as others (patient sex, baseline BMD, previous fracture history) [74].
The effects of GC on fracture risk are not only dose-related effect, but dependent on an integration of daily dose and duration of exposure. The cumulative dose per patient was found to be pertinent metric in a case-controlled study by Vestergaard et al. on a Danish registry [75]. This study a slight increase in hip fracture risk for patients who received 130–499 mg (OR 1.17), a higher risk for patients at 500–1499 mg (OR 1.36) and the greatest risk for patients at levels of 1500 mg or higher (OR 1.65). Van Staa and colleagues, in contrast, in their post hoc analysis of two prospective studies testing bisphosphonate efficacy in postmenopausal GC users [76], reported that daily GC dose (and not cumulative) was the stronger predictor of vertebral fracture risk compared with nonusers. This issue is not resolved, but with short treatment durations, the daily dose seems to be the key risk factor, but with longer durations (>3 months) and without any high dose administration, it would seem that cumulative dose is more important. This “exposure integral” further complicates the identification of a threshold above which adverse effects become more likely.
In a meta-analysis published in 2004 of the findings from seven prospective cohorts (42,500 individuals) Kanis and colleagues [77] also came to the conclusion that GC exposure increases the risk of any fracture, osteoporotic fracture and hip fracture. This increase in risk was only partly explained by a decrease in BMD, suggesting that bone remodelling induced by GC exposure weakens bone more than is indicated by the change in BMD [77]. Such remodelling could be fairly localised and not easily observed in standard DXA scans. It has, for instance, been argued that a lateral subregional approach to areal BMD measurement can show relevant BMD loss in GC exposed individuals which is masked in the standard posterior-anterior projection [78]. Trabecular thinning appears to be an important consequence of GC therapy and contributory to the increased bone fragility [79–81]. The recently developed trabecular bone score may find a role in the assessment of fracture risk in GC treatment [82].
GCs effects on bone: pathophysiology
The pathophysiological effects of GC on bone metabolism appear relatively rapidly after initiation of treatment with GC. In a pioneering study in 1995, Lems et al. [83] showed that the levels of serum osteocalcin (markers of bone formation) in healthy male volunteers decreased significantly with exposure to 10 mg prednisone per day for 1 week. A finding that was later confirmed in a larger study of healthy postmenopausal women [84]. Both studies observed that, whereas markers of bone formation were reduced during treatment, the markers of bone resorption were relatively unchanged. Both furthermore showed that the levels of bone formation markers rebounded back to pre-treatment levels after the end of treatment.
Serious bone problems remain uncommon, but association of higher GC exposure with osteonecrosis was described in an analysis of 71 patients with severe acute respiratory syndrome (SARS) who were treated with high doses of GCs [85]. Ten percent of the cohort developed radiographic evidence of osteonecrosis and they had received total GC doses >2.5 g prednisolone equivalents and treatment duration exceeding 18 days. The assessment of bone biomarkers, early during treatment and at repeated intervals, did not indicate any predictive pattern in those patients who went on to develop osteonecrosis. In orthopaedic clinics, it is estimated that about 15 % of the hip replacements are due to osteonecrosis and about half of those are associated with GC use (Einhorn, personal communication). Possible risk factors for osteonecrosis are considered to be related to the vasculature and blood clotting (there is an increased risk in lupus), but also lipid intermediaries and the presence of anti-phospholipid antibodies. It is of considerable interest in this respect, the recent discovery of specialised endothelial cells within bone which directly stimulate osteoprogenitor cells [86, 87]. The possibility is also raised therein of a novel method to stimulate the regeneration of osteoblasts.
GCs effects on bone: counteracting bone loss
The risk of GC-induced osteoporosis became a major concern in the late 1990s, especially in persons older than 70 years, [88, 89], and physicians were encouraged to prescribe bone preserving agents. Widely prescribed were the bisphosphonates [90, 91], as well as calcium and vitamin D supplementation [92]. The combination of bisphosphonates with GC and a DMARD can protect against bone loss and may even increase BMD in the lumbar region [93]. The evidence of long-term safety of bisphosphonates in GC-users is sparse [94] and it may be that patients should be switched to a different bone-preserving agent after 2 years since their continued use may be associated with low bone turnover and atypical fractures [95]. The data from the longitudinal GLOW study (in postmenopausal women) [96] would suggest that management of bone health for GC-users is however far from optimal. BMD testing is more frequent in GC users than non-GC users and their awareness of osteoporosis risk is better, but their treatments, even calcium and vitamin D supplementation remain inadequate; especially in Europe.
Adverse effects of GCs on the cardiovascular outcomes
Since systemic inflammatory diseases increase CV risk, the evaluation of the added effect of GC treatment is complicated. In general, GC therapy has been charged with tipping the balance further to the side of harm [97, 98], which is understandable given that long-term GC therapy use has been associated with hyperlipidaemia [99], hyperglycaemia [100], increased body mass, blood pressure and cholesterol [101]; all known CV risk factors. The evidence however has mostly been indirect. Wolfe and colleagues noted that the strongest explanatory factor for the increased MI risk in patients with RA was the activity of the disease itself and the patients with overt RA were more likely to be treated with a GC. Since there was evidence to link GC use with the increased risks for diabetes and hypertension, confounding likely accounted for the observed association between GC use and the overall risk of MI [102]. It remains possible however that some interaction exists between RF-positivity and exposure to glucocorticoids in the risk of CV events [103]. Longer-term GC use in the medium dose range in patients with RA has been linked with hypertension [104] and it may also increase the risk of venous thromboembolism [105].
Adverse effects of GCs on the immune system
High endogenous GC secretion (hypersecretion), as well as high exogenous GC doses have been associated with lung infections and opportunistic fungal infections [106]. In a nested case–controlled analysis using data from a Canadian administrative database, a 2011 publication [107] reported that there was a dose response in the prevalence of serious infection in older patients with RA according to GC use (cumulative dose weighted by the closeness in time to the index date, i.e. diagnosis of infection). The odds ratio for current GC exposure (at 5 mg prednisolone/day) was 1.5 and the risk was found to increase with the duration of treatment (comparative groups: 3, 6 months and 3 years). The immune function recovered following GC withdrawal, but relatively slowly. Thus discontinuation of a 2-year course of 10 mg prednisolone halved the risk as compared to on-going use, 6 months later, but the cumulative exposure still appeared to have an impact on current risk [108]. A retrospective analysis published in 2006 [109] reported that RA patients who received low-dose GC therapy for more than 10 years had increased mortality compared to those who did not receive GCs and that this increased mortality was mainly due to infections and complications related to systemic amyloidosis.
Once again, determining the relationship between GC dose and additional risk of opportunist infections is not an easy task. The observational studies are often compromised by confounding by disease severity, intermittent patterns of GC use and administration route; controlled interventional trials by heterogeneous reporting and publication bias [110]. A recent systematic review of serious infection with biological treatments for RA concluded that an increase in risk exist with respect to traditional DMARDs but put the absolute risk of infection with a standard dose regimen at 6 per 1000 [111].
Effects of GCs on glycaemia
The hypothalamic-pituitary-adrenal (HPA) axis is closely implicated in the regulation of adipose tissue and metabolic pathways [112, 113] and studies have shown that glucocorticoids induce hyperglycaemia and may cause diabetes or aggravate pre-existing diabetes [114, 115]. The mechanisms underlying the effect remain poorly understood, but appear to include increases in gluconeogenesis and hepatic glucose output, as well as inducing insulin resistance [116]. In a cross-sectional analysis of patients with established RA and without type 2 diabetes mellitus, tests showed that there was no difference between chronic GC users and GC-naïve patients in terms of glucose tolerance, insulin sensitivity and β-cell function [117]. As already described, RA patients tend to have decreased insulin sensitivity and impaired β-cell function when compared to control subjects of comparable age with normal glucose tolerance. Low doses of prednisolone have been shown to acutely affect carbohydrate metabolism [117] and GC-induced hyperglycemia is common in individuals with or without diabetes [114]. However, within the context of RA, the long-term effects of low-dose GC are not clear and longitudinal studies are required in this area. Short-term high dose GC treatment (30 or 60 mg/day), seems to have a variable effect on glucose metabolism with some patients showing improvements and others deterioration [118]. In a cross-sectional analysis of the NHANES III population of individuals aged 60 years or more (n = 5302), the prevalence of diabetes in individuals with RA was 17 % and the estimated increase in the risk of DM associated with RA (adjusted odds ratio of 1.3) had a 95 % CI that spanned 1 and therefore did not reach statistical significance [119].
Effects of GCs on weight gain
The question of weight gain with GC treatment was examined in a sub-study of CAMERA-II, which compared a combination therapy for RA (prednisone 10 mg/day + MTX) versus MTX alone [120]. The study team found a moderate weight increase in the GC group (mean +2.9 versus +1.3 kg), but no independent association of a change in BMI. A risk of developing metabolic syndrome, on the other hand, does not appear to be associated with GC therapy [121]. While some research has suggested that the synthetic prednisolone derivative, deflazacort, has a more moderate effect on glycaemia than prednisone, this seems to be related to its lower (~80 %) therapeutic potency [122, 123]. Further work in this field is probably needed to identify the individual risk factors associated with clinically relevant metabolic effects of low dose GC therapy [124].
Effects of GCs on vision
Prolonged use of glucocorticoids is a significant risk factor for the development of cataract [125]. The mechanisms underlying the effect remains unknown, but may be linked to gene transcription events in lens epithelial cells [126]. Other risk factors for these typically posterior subcapsular cataracts include age, male gender, diabetes, increased weight change and thyroid hormone use [127].
Effects of GCs on depression
Cortisol has a complex role in depression and higher levels are a risk factor for depression [128]. The circadian pattern of GC release also seems to be important and the constant levels of an exogenous GC seem to disturb some individuals. Depression is highly prevalent in patients with RA with rates of major depressive disorder, according to a recent meta-analysis [129] of 17 % (95 % CI 10–24 %), which is similar to the 15 % prevalence reported in the COMORA study [24]. According to some self-reported scales however the presence of depressive symptoms may affect 35–40 % of individuals [129]. The presence of depression has been found to reduce the effect of prednisolone treatment by as much as 50 % (on DAS-28 and HAQ scores) [130].
Optimising the GC response
The timing of GC administration
Modified release preparations of prednisone, which allow the higher levels of GCs to be attained during the night and therefore simulate the endogenous pattern of cortisol release, could provide superior results than traditional formulations. The loss of the endogenous cortisol rhythm is particularly associated with sleep disturbance, depression, and metabolic abnormalities [128, 133–135]. So far the studies with these compounds have been short-term (3–4 months) with low doses (5 mg/day), but the results seem to be encouraging.
The choice of administration route
By far the most clinical research into the efficacy and safety of GC therapy in rheumatic diseases has been on orally administered doses. Other routes are associated with local adverse side effects of a more restricted nature:
-
Intraarticular administration of GCs on systemic bone metabolism are considered relatively benign, or at least have less adverse effects than systemic oral GC use [136], however high quality randomised trials that actively assess joint damage or fracture risk over time are very rare. One randomised study which did use this strategy was TICORA [3] and no adverse events relating to these injections were described. Some physicians are somewhat less concerned by cartilage damage, since that consider this treatment effective only to postpone an inevitable joint replacement, but the results of TICORA and more recently OPERA [137] would suggest that this approach deserves greater attention. OPERA used a treat-to-target strategy of methotrexate (20 mg/week) and triamcinolone hexacetonide (20 mg/mL, 0.5–2 mL/joint), with or without an additional biologic DMARD. At 6-monthly intervals over 1 year, a maximum four swollen joints per patient were injected with; equivalent to (according to the authors) about 1 mg prednisolone a day during the 12-months’ follow-up. The blinded MRI outcome assessment (synovitis, osteitis and tenosynovitis scores, bone erosion and joint-space narrowing) revealed a significant decrease in disease activity and the absence of further bone erosion or structural damage. The addition of a biologic DMARD (adalimumab) gave better results on the tenosynovitis and osteitis scores than without.
-
Topical GCs are known to have various degrees of systemic exposure but rarely have consequential effects on bone [138, 139].
The choice of GC formulation
There are distinct differences between GC formulations in the residence time in the joint, which can vary from ~2.8 days for methylprednisolone and betamethasone, ~4 days for triamcinolone acetonide, 6 days for triamcinolone hexacetonide and 25 days for rimexolone [140, 141]. Triamcinolone hexacetonide was the preferred choice in a recent review [142] based on clinical response. For the treatment of adhesive capsulitis a 20 mg dose of triamcinolone hexacetonide was recently been demonstrated to be non-inferior to a 40 mg dose, so it is possible that similar dose comparisons might prove valuable in RA [143]. Further long-term safety studies are also warranted since fluorinated glucocorticoid preparations, such as triamcinolone, may be more strongly associated with myopathy [144].
The expectation has been held for some time that chemical modification could provide GCs with significantly better benefit-risk ratios [145–147] but so far these remain elusive. Thus, the nitrosteroids and the selective glucocorticoid-receptor agonists (SEGRAs) or dissociating glucocorticoids have, as yet, failed to “sharpen the old spear.”
The bioavailability of GC is regulated by several different mechanisms. A large portion of the circulating GCs is bound to corticosteroid-binding globulin and serum albumin. GCs are also metabolised in the circulation and may be reactivated in certain tissues by the enzyme 11-beta hydroxysteroid dehydrogenase (11β-HSD, type 1; expressed in brain, bone, synovium, liver and adipose tissue), which catalyses the conversion of (inactive) cortisone to cortisol thus modulating the local bioavailability of active GC.
GC resistance
GC resistance is a fairly well documented phenomenon in respiratory diseases such as asthma, but comparatively less so in other chronic inflammatory conditions [148]. The result of this “resistance” is that relatively high doses are necessary before an anti-inflammatory effect is seen [148, 149] and to some degree this physiological dysregulation is probably responsible for a high basal immune activation state. The prevalence of GC resistance is difficult to measure since several different definitions exist, but it is estimated that about 30 % of patients with RA have a poor clinical response to glucocorticoids [112, 150]. Various treatment are being explored to reverse acquired GC resistance [151].
Genetic polymorphisms of the GC receptor is one reason for GC resistance and this may play a role in RA [152, 153]. Acquired GC resistance may also be a factor in GC resistance and the oxidative stress caused by smoking is also associated with a reduction in the anti-inflammatory effects of GCs in respiratory disease [148]. Chronic stress is known to have a marked (central) effect on the hypothalamic-pituitary-gonadal axis—a result well known to behaviorists [154]—and such a mechanism has not been entirely ruled out in RA [155, 156]. Indeed it has been argued that to understand the development of chronic inflammatory diseases, a complete workup of the hypothalamic-pituitary-gonadal axis as well as the hypothalamic-pituitary-adrenal axis is required. Imbalances within these complex systems and their interactions with the immune system could be crucial in the development of such diseases and their response to treatment [157].
Current treatments for RA
Use of GCs
The subject of glucocorticoid-induced osteoporosis was addressed in two consensus statements on [158, 159], which seemed to agree that two important steps in GC discussion are a daily dose ≤7.5/>7.5 mg/day prednisone and a treatment duration <3/≥3 months. Differences persist however in the thresholds above which anti-osteoporosis drugs should be described [160]. A framework for treatment management decisions for GC-induced osteoporosis in patients whose therapy last for 3 months or longer has recently been proposed [161]. EULAR has published recommendations for monitoring for potential side-effects of systemic GC therapy in rheumatic diseases [162] and for the management of medium to high GC doses (i.e. >7.5 and ≤ 100 mg prednisone equivalent daily) [163], which could help to lay the foundations of a stratification procedure for the use of GC in patients with RA.
GC-withdrawal
GC-withdrawal has been fairly recently recognised as serious problem where there is long-lasting suppression in the functioning of the HPA axis [131]. The symptoms seem to be quite varied and include vomiting, abdominal pain, weakness, malaise, fever, hypotension, hypoglycaemia, and hypernatremia. The discontinuation of long-term GC therapy has also been associated with an increased risk of both depression and delirium/confusion [132]. More research and awareness are needed on this issue, particularly in elderly, and to understand how this is related to the recovery of trabecular bone density.
Special considerations for GC therapy in older patients
Fracture risk should be estimated (FRAX score or other tool) and, depending on risk score and access to densitometry, a BMD measurement at the level of the lumbar vertebrae and femoral neck [164]. In post-menopausal women and at-risk men, the biomarkers of bone metabolism may be helpful for determining therapeutic strategy with the prescription of bone sparing treatments [165] and for this it should be noted that blood sampling is best performed in the morning in adequately hydrated patients in a fasting condition. Appropriate regimens of supplementation with calcium and vitamin D are usually warranted [92] although it is cautioned that upper thresholds are not exceeded (2000 mg/day calcium (diet + supplement); 800–1000 IU/day vitamin D). It should also be noted that vitamin D deficiency has been associated with various autoimmune diseases including RA and lupus [166].
CV risk is known to increase with age [167] and, as we have seen systemic immune disease further increases this risk. It is important therefore to submit patients to a thorough CV risk assessment prior to initiating therapy with GC’s. Any hyperglycaemia, hypertension or dyslipidaemia should be managed as much as possible before prescribing a GC. Traditional CV risk factors and RA-associated risk differ, but recent work shows how an existing risk score might be adjusted in the context of RA [168], integrating disease activity, disability, daily prednisone use (yes/no), and disease duration (less than 10 or 10 years or more).
Additional baseline assessments should also be made of ankle oedema and risk factors for glaucoma [169].
Conclusions
The decision to add a GC, within a strategy of combination therapy, to bring early RA under control still seems to divide rheumatologists. Doubts remain as to the best pre-therapeutic work-up of patients and to their subsequent monitoring; also from the patients themselves, who may have preconceived negative opinions [170]. On the other hand, it should also be borne in mind that GCs are naturally produced endocrine hormones. So, while it may be imprudent to speak of a “safe” dose of exogenous GC, it is likely that at the lower end of the therapeutic GC exposition, there is a favourable benefit-risk ratio. The problem is to find the ideal dose, by disease state, since it is not the same for all individuals. There is an urgent need therefore to be able to test patients for GC sensitivity.
This clinical research topic is very large this review barely does justice to the knowledge base. The scope was given as GC therapy in rheumatic diseases and regrettably little other than RA was discussed because of space. We are getting close to the goal of being able to individualise therapeutic options and select optimal dose regimens, but still more refinement is needed in the tools available. At the present the following points seem pertinent:
-
Early treatment and treat-to-target are important strategies in RA, and probably other systemic inflammatory diseases, resulting in improved patient outcomes.
-
In RA, the option of a combination treatment with a fast-acting GC and a DMARD should be considered in most cases. Oral GC therapy, either at a stable low dose (≤7.5 mg/day) or an initial medium dose (~30 mg/day) that is rapidly tapered down, in combination with a traditional DMARD, (usually MTX) frequently results in rapid suppression of inflammation and can lead to clinical remission. It should be noted that monotherapy with GC is not advocated in RA.
-
High oral GC doses (>30–100 mg prednisone equivalents/day) are associated with more adverse effects with a risk to bone loss in particular. When these are used over even short (≤3 months) durations there is a cumulative effect that considerably increases the probably of serious side effects (such as aseptic necrosis). In MTX-resistant RA, recourse to a biologic DMARD seems to be more effective than GC pulse therapy.
-
All GC preparations, given sufficient systemic bioavailability, will have measurable effects on the HPA axis and on bone metabolism, but these are dependent on dose, time of administration and duration of exposure. Low doses and relatively short durations of moderate doses are generally well tolerated and have a positive benefit/risk ratio. Depending on the indication intraarticular GC may be a suitable option.
-
The fracture risk in patients and possibility BMD should be assessed and bone preserving agents prescribed accordingly. At a minimum, patient-specific calcium and vitamin D supplementation are advisable.
-
GC’s are effective adjuvants in the management in some other inflammatory rheumatic diseases such as lupus, vasculitis and giant-cell arteritis. These often require much higher GC exposure and the optimal therapeutic regimens are far from clear.
-
More research is needed on the assessment of individual GC sensitivity, also on GC withdrawal and further optimisation of combination therapies.
References
Baltimore D (2001) Our genome unveiled. Nature 409(6822):814–816
Boers M (2003) Understanding the window of opportunity concept in early rheumatoid arthritis. Arthritis Rheum 48(7):1771–1774
Grigor C, Capell H, Stirling A et al (2004) Effect of a treatment strategy of tight control for rheumatoid arthritis (the TICORA study): a single-blind randomised controlled trial. Lancet 364(9430):263–269
Bakker MF, Jacobs JW, Verstappen SM et al (2007) Tight control in the treatment of rheumatoid arthritis: efficacy and feasibility. Ann Rheum Dis. doi:10.1136/ard.2007.078360
Klareskog L, Catrina AI, Paget S (2009) Rheumatoid arthritis. Lancet. doi:10.1016/S0140-6736(09)60008-8
Saffar AS, Ashdown H, Gounni AS (2011) The molecular mechanisms of glucocorticoids-mediated neutrophil survival. Curr Drug Targets 12(4):556–562
Ytterberg AJ, Joshua V, Reynisdottir G et al (2014) Shared immunological targets in the lungs and joints of patients with rheumatoid arthritis: identification and validation. Ann Rheum Dis. doi:10.1136/annrheumdis-2013-204912
McInnes IB, Schett G (2011) The pathogenesis of rheumatoid arthritis. N Engl J Med. doi:10.1056/NEJMra1004965
Miossec P, Korn T, Kuchroo VK (2009) Interleukin-17 and type 17 helper T cells. N Engl J Med. doi:10.1056/NEJMra0707449
Jonsson H, Allen P, Peng SL (2005) Inflammatory arthritis requires Foxo3a to prevent Fas ligand-induced neutrophil apoptosis. Nat Med 11(6):666–671
Turrel-Davin F, Tournadre A, Pachot A et al (2010) FoxO3a involved in neutrophil and T cell survival is overexpressed in rheumatoid blood and synovial tissue. Ann Rheum Dis. doi:10.1136/ard.2009
Westwood OM, Nelson PN, Hay FC (2006) Rheumatoid factors: what’s new? Rheumatology (Oxford) 45(4):379–385
Zendman AJ, van Venrooij WJ, Pruijn GJ (2006) Use and significance of anti-CCP autoantibodies in rheumatoid arthritis. Rheumatology (Oxford) 45(1):20–25
Lopez-Longo FJ, Oliver-Minarro D, de la Torre I et al (2009) Association between anti-cyclic citrullinated peptide antibodies and ischemic heart disease in patients with rheumatoid arthritis. Arthritis Rheum. doi:10.1002/art.24390
Kleyer A, Finzel S, Rech J et al (2013) Bone loss before the clinical onset of rheumatoid arthritis in subjects with anticitrullinated protein antibodies. Ann Rheum Dis. doi:10.1136/annrheumdis-2012-202958
Crowson CS, Rahman MU, Matteson EL (2009) Which measure of inflammation to use? A comparison of erythrocyte sedimentation rate and C-reactive protein measurements from randomized clinical trials of golimumab in rheumatoid arthritis. J Rheumatol. doi:10.3899/jrheum.081188
Hensvold AH, Magnusson PK, Joshua V et al (2015) Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: an epidemiological investigation in twins. Ann Rheum Dis. doi:10.1136/annrheumdis-2013-203947
Wolfe F, Mitchell DM, Sibley JT et al (1994) The mortality of rheumatoid arthritis. Arthritis Rheum 37(4):481–494
Boyer JF, Gourraud PA, Cantagrel A et al (2011) Traditional cardiovascular risk factors in rheumatoid arthritis: a meta-analysis. Joint Bone Spine. 78(2):179–183
Wolfe F (2000) The effect of smoking on clinical, laboratory, and radiographic status in rheumatoid arthritis. J Rheumatol 27(3):630–637
Dessein PH, Joffe BI (2006) Insulin resistance and impaired beta cell function in rheumatoid arthritis. Arthritis Rheum 54(9):2765–2775
Liao KP, Solomon DH (2013) Traditional cardiovascular risk factors, inflammation and cardiovascular risk in rheumatoid arthritis. Rheumatology (Oxford). doi:10.1093/rheumatology/kes243
Stavropoulos-Kalinoglou A, Metsios GS, Koutedakis Y et al (2011) Obesity in rheumatoid arthritis. Rheumatology (Oxford) 50(3):450–462
Dougados M, Soubrier M, Antunez A et al (2014) Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: results of an international, cross-sectional study (COMORA). Ann Rheum Dis 73(1):62–68
Wallberg-Jonsson S, Ohman ML, Dahlqvist SR (1997) Cardiovascular morbidity and mortality in patients with seropositive rheumatoid arthritis in Northern Sweden. J Rheumatol 24(3):445–451
Watson DJ, Rhodes T, Guess HA (2003) All-cause mortality and vascular events among patients with rheumatoid arthritis, osteoarthritis, or no arthritis in the UK General Practice Research Database. J Rheumatol 30(6):1196–1202
Sihvonen S, Korpela M, Laippala P et al (2004) Death rates and causes of death in patients with rheumatoid arthritis: a population-based study. Scand J Rheumatol 33(4):221–227
Peters MJ, van Halm VP, Voskuyl AE et al (2009) Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. doi:10.1002/art.24836
Avina-Zubieta JA, Thomas J, Sadatsafavi M et al (2012) Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. doi:10.1136/annrheumdis-2011-20072
Krishnan E, Lingala VB, Singh G (2004) Declines in mortality from acute myocardial infarction in successive incidence and birth cohorts of patients with rheumatoid arthritis. Circulation 110(13):1774–1779
Meune C, Touze E, Trinquart L et al (2009) Trends in cardiovascular mortality in patients with rheumatoid arthritis over 50 years: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford). doi:10.1093/rheumatology/kep252
Juni P, Nartey L, Reichenbach S et al (2004) Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet 364(9450):2021–2029
Nissen SE, Wolski K (2007) Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 356(24):2457–2471
Trelle S, Reichenbach S, Wandel S et al (2011) Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. doi:10.1136/bmj.c7086
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 115(5):1111–1119
Bensinger SJ, Tontonoz P (2008) Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. doi:10.1038/nature07202
Toms TE, Panoulas VF, Douglas KM et al (2010) Statin use in rheumatoid arthritis in relation to actual cardiovascular risk: evidence for substantial undertreatment of lipid-associated cardiovascular risk? Ann Rheum Dis. doi:10.1136/ard.2009.115717
Curtis JR, John A, Baser O (2012) Dyslipidemia and changes in lipid profiles associated with rheumatoid arthritis and initiation of anti-tumor necrosis factor therapy. Arthritis Care Res (Hoboken). doi:10.1002/acr.21693
Toms TE, Panoulas VF, Smith JP et al (2011) Rheumatoid arthritis susceptibility genes associate with lipid levels in patients with rheumatoid arthritis. Ann Rheum Dis. doi:10.1136/ard.2010.144634
del Rincon ID, Williams K, Stern MP et al (2001) High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum 44(12):2737–2745
Turesson C, McClelland RL, Christianson TJ et al (2007) Severe extra-articular disease manifestations are associated with an increased risk of first ever cardiovascular events in patients with rheumatoid arthritis. Ann Rheum Dis 66(1):70–75
Hot A, Lenief V, Miossec P (2012) Combination of IL-17 and TNFalpha induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Ann Rheum Dis. doi:10.1136/annrheumdis-2011-200468
Hot A, Lavocat F, Lenief V et al (2013) Simvastatin inhibits the pro-inflammatory and pro-thrombotic effects of IL-17 and TNF-alpha on endothelial cells. Ann Rheum Dis. doi:10.1136/annrheumdis-2012-201887
McCarey DW, McInnes IB, Madhok R et al (2004) Trial of atorvastatin in rheumatoid arthritis (TARA): double-blind, randomised placebo-controlled trial. Lancet 363(9426):2015–2021
Young SP, Kapoor SR, Viant MR et al (2013) The impact of inflammation on metabolomic profiles in patients with arthritis. Arthritis Rheum. doi:10.1002/art.38021
Park YB, Choi HK, Kim MY et al (2002) Effects of antirheumatic therapy on serum lipid levels in patients with rheumatoid arthritis: a prospective study. Am J Med 113(3):188–193
Nadareishvili Z, Michaud K, Hallenbeck JM et al (2008) Cardiovascular, rheumatologic, and pharmacologic predictors of stroke in patients with rheumatoid arthritis: a nested, case–control study. Arthritis Rheum. doi:10.1002/art.23935
Doran MF, Crowson CS, Pond GR et al (2002) Frequency of infection in patients with rheumatoid arthritis compared with controls: a population-based study. Arthritis Rheum 46(9):2287–2293
Toh ML, Kawashima M, Hot A et al (2010) Role of IL-17 in the Th1 systemic defects in rheumatoid arthritis through selective IL-12Rbeta2 inhibition. Ann Rheum Dis. doi:10.1136/ard.2009.111757
Felson DT, Anderson JJ, Meenan RF (1990) The comparative efficacy and toxicity of second-line drugs in rheumatoid arthritis. Results of two meta analyses. Arthritis Rheum. 33(10):1449–1461
O’Dell JR, Haire CE, Erikson N et al (1996) Treatment of rheumatoid arthritis with methotrexate alone, sulfasalazine and hydroxychloroquine, or a combination of all three medications. N Engl J Med 334(20):1287–1291
Boers M, Verhoeven AC, Markusse HM et al (1997) Randomised comparison of combined step-down prednisolone, methotrexate and sulphasalazine with sulphasalazine alone in early rheumatoid arthritis. Lancet 350(9074):309–318
Mottonen T, Hannonen P, Leirisalo-Repo M et al (1999) Comparison of combination therapy with single-drug therapy in early rheumatoid arthritis: a randomised trial. FIN-RACo trial group. Lancet 353(9164):1568–1573
Svensson B, Boonen A, Albertsson K et al (2005) Low-dose prednisolone in addition to the initial disease-modifying antirheumatic drug in patients with early active rheumatoid arthritis reduces joint destruction and increases the remission rate: a 2-year randomized trial. Arthritis Rheum 52(11):3360–3370
Goekoop-Ruiterman YP, de Vries-Bouwstra JK, Allaart CF et al (2005) Clinical and radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): a randomized, controlled trial. Arthritis Rheum 52(11):3381–3390
Bakker MF, Jacobs JW, Welsing PM et al (2012) Low-dose prednisone inclusion in a methotrexate-based, tight control strategy for early rheumatoid arthritis: a randomized trial. Ann Intern Med 156(5):329–339
Vermeer M, Kuper HH, Hoekstra M et al (2011) Implementation of a treat-to-target strategy in very early rheumatoid arthritis: results of the Dutch Rheumatoid Arthritis Monitoring remission induction cohort study. Arthritis Rheum. doi:10.1002/art.30494
Heimans L, Wevers-de Boer KV, Visser K et al (2013) A two-step treatment strategy trial in patients with early arthritis aimed at achieving remission: the IMPROVED study. Ann Rheum Dis. doi:10.1136/annrheumdis-2013-203243
den Uyl D, Ter Wee M, Boers M et al (2013) A non-inferiority trial of an attenuated combination strategy (‘COBRA-light’) compared to the original COBRA strategy: clinical results after 26 weeks. Ann Rheum Dis. doi:10.1136/annrheumdis-2012-202818
Kirwan JR, Bijlsma JW, Boers M et al (2007) Effects of glucocorticoids on radiological progression in rheumatoid arthritis. Cochrane Database Syst Rev 1:CD006356
Gorter SL, Bijlsma JW, Cutolo M et al (2010) Current evidence for the management of rheumatoid arthritis with glucocorticoids: a systematic literature review informing the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. doi:10.1136/ard.2009.127332
Knevel R, Schoels M, Huizinga TW et al (2010) Current evidence for a strategic approach to the management of rheumatoid arthritis with disease-modifying antirheumatic drugs: a systematic literature review informing the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. doi:10.1136/ard.2009.126748
Smolen JS, Aletaha D, Bijlsma JW et al (2010) Treating rheumatoid arthritis to target: recommendations of an international task force. Ann Rheum Dis. doi:10.1136/ard.2009.123919
Smolen JS, Landewe R, Breedveld FC et al (2013) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. doi:10.1136/annrheumdis-2013-204573
Gaujoux-Viala C, Nam J, Ramiro S et al (2014) Efficacy of conventional synthetic disease-modifying antirheumatic drugs, glucocorticoids and tofacitinib: a systematic literature review informing the 2013 update of the EULAR recommendations for management of rheumatoid arthritis. Ann Rheum Dis. doi:10.1136/annrheumdis-2013-204588
Singh JA, Furst DE, Bharat A et al (2012) 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken). doi:10.1002/acr.21641
Curtis JR, Westfall AO, Allison J et al (2006) Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum 55(3):420–426
van der Goes MC, Jacobs JW, Boers M et al (2010) Patient and rheumatologist perspectives on glucocorticoids: an exercise to improve the implementation of the European League Against Rheumatism (EULAR) recommendations on the management of systemic glucocorticoid therapy in rheumatic diseases. Ann Rheum Dis. doi:10.1136/ard.2009.114579
Saag KG (2012) Short-term and long-term safety of glucocorticoids in rheumatoid arthritis. Bull NYU Hosp Jt Dis 70(Suppl 1):21–25
Santiago T, da Silva JA (2014) Safety of low- to medium-dose glucocorticoid treatment in rheumatoid arthritis: myths and reality over the years. Ann N Y Acad Sci. doi:10.1111/nyas.12428
Saag KG (1997) Low-dose corticosteroid therapy in rheumatoid arthritis: balancing the evidence. Am J Med 103(6A):31S–39S
van Staa TP, Leufkens HG, Abenhaim L et al (2000) Use of oral corticosteroids and risk of fractures. J Bone Miner Res 15(6):993–1000
van Staa TP, Leufkens HG, Abenhaim L et al (2000) Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 39(12):1383–1389
da Silva JA, Jacobs JW, Kirwan JR et al (2006) Safety of low dose glucocorticoid treatment in rheumatoid arthritis: published evidence and prospective trial data. Ann Rheum Dis 65(3):285–293
Vestergaard P, Olsen ML, Paaske JS et al (2003) Corticosteroid use and risk of hip fracture: a population-based case-control study in Denmark. J Intern Med 254(5):486–493
van Staa TP, Laan RF, Barton IP et al (2003) Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 48(11):3224–3229
Kanis JA, Johansson H, Oden A et al (2004) A meta-analysis of prior corticosteroid use and fracture risk. J Bone Miner Res 19(6):893–899
Manning LI, Briggs AM, van Doornum DS et al (2013) Glucocorticoid-induced bone loss is associated with abnormal intravertebral areal bone mineral density distribution. Int J Endocrinol. doi:10.1155/2013/768579
Dalle Carbonare L, Arlot ME, Chavassieux PM et al (2001) Comparison of trabecular bone microarchitecture and remodeling in glucocorticoid-induced and postmenopausal osteoporosis. J Bone Miner Res 16(1):97–103
Sambrook PN, Hughes DR, Nelson AE et al (2003) Osteocyte viability with glucocorticoid treatment: relation to histomorphometry. Ann Rheum Dis 62(12):1215–1217
Aeberli D, Schett G (2013) Cortical remodeling during menopause, rheumatoid arthritis, glucocorticoid and bisphosphonate therapy. Arthritis Res Ther. doi:10.1186/ar4180
Harvey NC, Gluer CC, Binkley N et al (2015) Trabecular bone score (TBS) as a new complementary approach for osteoporosis evaluation in clinical practice. Bone. doi:10.1016/j.bone.2015.05.016
Lems WF, Jacobs JW, Van Rijn HJ et al (1995) Changes in calcium and bone metabolism during treatment with low dose prednisone in young, healthy, male volunteers. Clin Rheumatol 14(4):420–424
Ton FN, Gunawardene SC, Lee H et al (2005) Effects of low-dose prednisone on bone metabolism. J Bone Miner Res 20(3):464–470
Chan M, Chan PK, Griffith JF et al (2006) Steroid-induced osteonecrosis in severe acute respiratory syndrome: a retrospective analysis of biochemical markers of bone metabolism and corticosteroid therapy. Pathology. 38(3):229–235
Kusumbe AP, Ramasamy SK, Adams RH (2014) Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. doi:10.1038/nature13145
Ramasamy SK, Kusumbe AP, Wang L et al (2014) Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature. doi:10.1038/nature13146
van Staa TP, Leufkens HG, Abenhaim L et al (2000) Use of oral corticosteroids in the UK. QJM 93(2):105–111
van Staa TP, Abenhaim L, Cooper C et al (2001) Public health impact of adverse bone effects of oral corticosteroids. Br J Clin Pharmacol 51(6):601–607
Saag KG, Emkey R, Schnitzer TJ et al (1998) Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 339(5):292–299
Cohen S, Levy RM, Keller M et al (1999) Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 42(11):2309–2318
Devogelaer JP (2006) Glucocorticoid-induced osteoporosis: mechanisms and therapeutic approach. Rheum Dis Clin North Am 32(4):733–757
van der Goes MC, Jacobs JW, Jurgens MS et al (2013) Are changes in bone mineral density different between groups of early rheumatoid arthritis patients treated according to a tight control strategy with or without prednisone if osteoporosis prophylaxis is applied? Osteoporos Int. doi:10.1007/s00198-012-2073-z
Homik J, Cranney A, Shea B et al (2000) Bisphosphonates for steroid induced osteoporosis. Cochrane Database Syst Rev 2:D001347
Lems WF, Saag K (2015) Bisphosphonates and glucocorticoid-induced osteoporosis: cons. Endocrine. doi:10.1007/s12020-015-0639-1
Silverman S, Curtis J, Saag K et al (2015) International management of bone health in glucocorticoid-exposed individuals in the observational GLOW study. Osteoporos Int. doi:10.1007/s00198-014-2883-2
Souverein PC, Berard A, van Staa TP et al (2004) Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case–control study. Heart 90(8):859–865
Ruyssen-Witrand A, Fautrel B, Saraux A et al (2011) Cardiovascular risk induced by low-dose corticosteroids in rheumatoid arthritis: a systematic literature review. Joint Bone Spine. doi:10.1016/j.jbspin.2010.02.040
el-Shaboury AH, Hayes TM (1973) Hyperlipidaemia in asthmatic patients receiving long-term steroid therapy. Br Med J 2(5858):85–86
Panthakalam S, Bhatnagar D, Klimiuk P (2004) The prevalence and management of hyperglycaemia in patients with rheumatoid arthritis on corticosteroid therapy. Scott Med J 49(4):139–141
Fraser R, Ingram MC, Anderson NH et al (1999) Cortisol effects on body mass, blood pressure, and cholesterol in the general population. Hypertension 33(6):1364–1368
Wolfe F, Michaud K (2008) The risk of myocardial infarction and pharmacologic and nonpharmacologic myocardial infarction predictors in rheumatoid arthritis: a cohort and nested case–control analysis. Arthritis Rheum. doi:10.1002/art.23811
Davis JM III, Maradit KH, Crowson CS et al (2007) Glucocorticoids and cardiovascular events in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 56(3):820–830
Panoulas VF, Douglas KM, Stavropoulos-Kalinoglou A et al (2008) Long-term exposure to medium-dose glucocorticoid therapy associates with hypertension in patients with rheumatoid arthritis. Rheumatology (Oxford) 47(1):72–75
Johannesdottir SA, Horvath-Puho E, Dekkers OM et al (2013) Use of glucocorticoids and risk of venous thromboembolism: a nationwide population-based case–control study. JAMA Intern Med. doi:10.1001/jamainternmed.2013.122
Cutolo M, Seriolo B, Pizzorni C et al (2008) Use of glucocorticoids and risk of infections. Autoimmun Rev. doi:10.1016/j.autrev.2008.07.010
Dixon WG, Kezouh A, Bernatsky S et al (2011) The influence of systemic glucocorticoid therapy upon the risk of non-serious infection in older patients with rheumatoid arthritis: a nested case–control study. Ann Rheum Dis. doi:10.1136/ard.2010.144741
Dixon WG, Abrahamowicz M, Beauchamp ME et al (2012) Immediate and delayed impact of oral glucocorticoid therapy on risk of serious infection in older patients with rheumatoid arthritis: a nested case–control analysis. Ann Rheum Dis. doi:10.1136/annrheumdis-2011-200702
Sihvonen S, Korpela M, Mustonen J et al (2006) Mortality in patients with rheumatoid arthritis treated with low-dose oral glucocorticoids. A population-based cohort study. J Rheumatol 33(9):1740–1746
Dixon WG, Suissa S, Hudson M (2011) The association between systemic glucocorticoid therapy and the risk of infection in patients with rheumatoid arthritis: systematic review and meta-analyses. Arthritis Res Ther. doi:10.1186/ar3453
Singh JA, Cameron C, Noorbaloochi S et al (2015) Risk of serious infection in biological treatment of patients with rheumatoid arthritis: a systematic review and meta-analysis. Lancet. doi:10.1016/S0140-6736(14)61704-9
Silverman MN, Sternberg EM (2008) Neuroendocrine-immune interactions in rheumatoid arthritis: mechanisms of glucocorticoid resistance. Neuroimmunomodulation. doi:10.1159/000135620
Kadmiel M, Cidlowski JA (2013) Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. doi:10.1016/j.tips.2013.07.003
Clore JN, Thurby-Hay L (2009) Glucocorticoid-induced hyperglycemia. Endocr Pract. doi:10.4158/EP08331.RAR
Angelopoulos TP, Tentolouris NK, Bertsias GK et al (2014) Steroid-induced diabetes in rheumatologic patients. Clin Exp Rheumatol 32(1):126–130
Iwamoto T, Kagawa Y, Naito Y et al (2004) Steroid-induced diabetes mellitus and related risk factors in patients with neurologic diseases. Pharmacotherapy. 24(4):508–514
Hoes JN, van der Goes MC, van Raalte DH et al (2011) Glucose tolerance, insulin sensitivity and beta-cell function in patients with rheumatoid arthritis treated with or without low-to-medium dose glucocorticoids. Ann Rheum Dis. doi:10.1136/ard.2011.151464
den Uyl D, van Raalte DH, Nurmohamed MT et al (2012) Metabolic effects of high-dose prednisolone treatment in early rheumatoid arthritis: balance between diabetogenic effects and inflammation reduction. Arthritis Rheum. doi:10.1002/art.3337
Simard JF, Mittleman MA (2007) Prevalent rheumatoid arthritis and diabetes among NHANES III participants aged 60 and older. J Rheumatol 34(3):469–473
Jurgens MS, Jacobs JW, Geenen R et al (2013) Increase of body mass index in a tight controlled methotrexate-based strategy with prednisone in early rheumatoid arthritis: side effect of the prednisone or better control of disease activity? Arthritis Care Res (Hoboken). doi:10.1002/acr.21797
Toms TE, Panoulas VF, Douglas KM et al (2008) Lack of association between glucocorticoid use and presence of the metabolic syndrome in patients with rheumatoid arthritis: a cross-sectional study. Arthritis Res Ther. doi:10.1186/ar2578
Babadjanova G, Allolio B, Vollmer M et al (1996) Comparison of the pharmacodynamic effects of deflazacort and prednisolone in healthy subjects. Eur J Clin Pharmacol 51(1):53–57
Saez Barcelona JA, Carmona MM, Navarro LV et al (1999) An open comparison of the diabetogenic effect of deflazacort and prednisone at a dosage ratio of 1.5 mg:1 mg. Eur J Clin Pharmacol 55(2):105–109
Fardet L, Feve B (2014) Systemic glucocorticoid therapy: a review of its metabolic and cardiovascular adverse events. Drugs. doi:10.1007/s40265-014-0282-9
James ER (2007) The etiology of steroid cataract. J Ocul Pharmacol Ther 23(5):403–420
Celojevic D, Carlsson T, Johansson B et al (2012) Cell adhesion molecule expression in human lens epithelial cells after corticosteroid exposure. Open Ophthalmol J. doi:10.2174/1874364101206010042
Chang JR, Koo E, Agron E et al (2011) Risk factors associated with incident cataracts and cataract surgery in the Age-related Eye Disease Study (AREDS): AREDS report number 32. Ophthalmology. doi:10.1016/j.ophtha.2011.03.032
Herbert J (2013) Cortisol and depression: three questions for psychiatry. Psychol Med. doi:10.1017/S0033291712000955
Matcham F, Rayner LF, Steer S et al (2013) The prevalence of depression in rheumatoid arthritis: a systematic review and meta-analysis. Rheumatology (Oxford). doi:10.1093/rheumatology/ket169
Matcham F, Norton S, Scott DL et al (2015) Symptoms of depression and anxiety predict treatment response and long-term physical health outcomes in rheumatoid arthritis: secondary analysis of a randomized controlled trial. Rheumatology (Oxford). doi:10.1093/rheumatology/kev306
Dinsen S, Baslund B, Klose M et al (2013) Why glucocorticoid withdrawal may sometimes be as dangerous as the treatment itself. Eur J Intern Med. doi:10.1016/j.ejim.2013.05.014
Fardet L, Nazareth I, Whitaker HJ et al (2013) Severe neuropsychiatric outcomes following discontinuation of long-term glucocorticoid therapy: a cohort study. J Clin Psychiatry. doi:10.4088/JCP.12m08034
Debono M, Ross RJ (2011) Optimal glucocorticoid therapy. Endocr Dev. doi:10.1159/000321241
Cutolo M, Iaccarino L, Doria A et al (2013) Efficacy of the switch to modified-release prednisone in rheumatoid arthritis patients treated with standard glucocorticoids. Clin Exp Rheumatol 31(4):498–505
Buttgereit F, Mehta D, Kirwan J et al (2013) Low-dose prednisone chronotherapy for rheumatoid arthritis: a randomised clinical trial (CAPRA-2). Ann Rheum Dis. doi:10.1136/annrheumdis-2011-201067
Emkey RD, Lindsay R, Lyssy J et al (1996) The systemic effect of intraarticular administration of corticosteroid on markers of bone formation and bone resorption in patients with rheumatoid arthritis. Arthritis Rheum 39(2):277–282
Axelsen MB, Eshed I, Horslev-Petersen K et al (2015) A treat-to-target strategy with methotrexate and intra-articular triamcinolone with or without adalimumab effectively reduces MRI synovitis, osteitis and tenosynovitis and halts structural damage progression in early rheumatoid arthritis: results from the OPERA randomised controlled trial. Ann Rheum Dis. doi:10.1136/annrheumdis-2013-204537
Osamura H (1982) Penetration of topical corticosteroids through human epidermis. J Dermatol 9(1):45–58
Vestergaard P, Rejnmark L, Mosekilde L (2005) Fracture risk associated with systemic and topical corticosteroids. J Intern Med 257(4):374–384
Derendorf H, Mollmann H, Gruner A et al (1986) Pharmacokinetics and pharmacodynamics of glucocorticoid suspensions after intra-articular administration. Clin Pharmacol Ther 39(3):313–317
Derendorf H, Möllmann H, Barth J (1990) Pharmakokinetik von intraartikulär applizierten Glukokortikoiden. Akt Rheumatol 15:145–153
Dernis E, Ruyssen-Witrand A, Mouterde G et al (2010) Use of glucocorticoids in rheumatoid arthritis—pratical modalities of glucocorticoid therapy: recommendations for clinical practice based on data from the literature and expert opinion. Joint Bone Spine. doi:10.1016/j.jbspin.2009.12.010
Yoon SH, Lee HY, Lee HJ et al (2013) Optimal dose of intra-articular corticosteroids for adhesive capsulitis: a randomized, triple-blind, placebo-controlled trial. Am J Sports Med. doi:10.1177/0363546513480475
Pereira RM, Freire de Carvalho CJ (2011) Glucocorticoid-induced myopathy. Joint Bone Spine. doi:10.1016/j.jbspin.2010.02.025
Buttgereit F, Burmester GR, Lipworth BJ (2005) Optimised glucocorticoid therapy: the sharpening of an old spear. Lancet 365(9461):801–803
van der Laan S, Meijer OC (2008) Pharmacology of glucocorticoids: beyond receptors. Eur J Pharmacol. doi:10.1016/j.ejphar.2008.01.060
De Bosscher K, Haegeman G, Elewaut D (2010) Targeting inflammation using selective glucocorticoid receptor modulators. Curr Opin Pharmacol. doi:10.1016/j.coph.2010.04.007
Barnes PJ, Adcock IM (2009) Glucocorticoid resistance in inflammatory diseases. Lancet. doi:10.1016/S0140-6736(09)60326-3
Biddie SC, Conway-Campbell BL, Lightman SL (2012) Dynamic regulation of glucocorticoid signalling in health and disease. Rheumatology (Oxford). doi:10.1093/rheumatology/ker215
Quax RA, Koper JW, de Jong PH et al (2012) In vitro glucocorticoid sensitivity is associated with clinical glucocorticoid therapy outcome in rheumatoid arthritis. Arthritis Res Ther. doi:10.1186/ar4029
Barnes PJ (2011) Glucocorticosteroids: current and future directions. Br J Pharmacol. doi:10.1111/j.1476-5381.2010.01199.x
van Oosten MJ, Dolhain RJ, Koper JW et al (2010) Polymorphisms in the glucocorticoid receptor gene that modulate glucocorticoid sensitivity are associated with rheumatoid arthritis. Arthritis Res Ther. doi:10.1186/ar3118
Quax RA, Koper JW, Huisman AM et al (2015) Polymorphisms in the glucocorticoid receptor gene and in the glucocorticoid-induced transcript 1 gene are associated with disease activity and response to glucocorticoid bridging therapy in rheumatoid arthritis. Rheumatol Int. doi:10.1007/s00296-015-3235-z
Sorrells SF, Sapolsky RM (2007) An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun 21(3):259–272
Harbuz MS, Jessop DS (1999) Is there a defect in cortisol production in rheumatoid arthritis? Rheumatology (Oxford) 38(4):298–302
Cutolo M, Foppiani L, Minuto F (2002) Hypothalamic-pituitary-adrenal axis impairment in the pathogenesis of rheumatoid arthritis and polymyalgia rheumatica. J Endocrinol Invest 25(10 Suppl):19–23
Straub RH, Bijlsma JW, Masi A et al (2013) Role of neuroendocrine and neuroimmune mechanisms in chronic inflammatory rheumatic diseases–the 10-year update. Semin Arthritis Rheum. doi:10.1016/j.semarthrit.2013.04.008
Devogelaer JP, Goemaere S, Boonen S et al (2006) Evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis: a consensus document of the Belgian Bone Club. Osteoporos Int 17(1):8–19
Grossman JM, Gordon R, Ranganath VK et al (2010) American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken). doi:10.1002/acr.20295
Weinstein RS (2011) Clinical practice. Glucocorticoid-induced bone disease. N Engl J Med. doi:10.1056/NEJMcp1012926
Lekamwasam S, Adachi JD, Agnusdei D et al (2012) A framework for the development of guidelines for the management of glucocorticoid-induced osteoporosis. Osteoporos Int. doi:10.1007/s00198-012-1958-1
Hoes JN, Jacobs JW, Boers M et al (2007) EULAR evidence-based recommendations on the management of systemic glucocorticoid therapy in rheumatic diseases. Ann Rheum Dis 66(12):1560–1567
Duru N, van der Goes MC, Jacobs JW et al (2013) EULAR evidence-based and consensus-based recommendations on the management of medium to high-dose glucocorticoid therapy in rheumatic diseases. Ann Rheum Dis. doi:10.1136/annrheumdis-2013-203249
Kanis JA, McCloskey EV, Johansson H et al (2013) European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int. doi:10.1007/s00198-012-2074-y
Eastell R, Krege JH, Chen P et al (2006) Development of an algorithm for using PINP to monitor treatment of patients with teriparatide. Curr Med Res Opin 22(1):61–66
Arnson Y, Amital H, Shoenfeld Y (2007) Vitamin D and autoimmunity: new aetiological and therapeutic considerations. Ann Rheum Dis 66(9):1137–1142
Sans S, Kesteloot H, Kromhout D (1997) The burden of cardiovascular diseases mortality in Europe. Task Force of the European Society of Cardiology on Cardiovascular Mortality and Morbidity Statistics in Europe. Eur Heart J 18(12):1231–1248
Solomon DH, Greenberg J, Curtis JR et al (2015) Derivation and Internal Validation of an Expanded Cardiovascular Risk Prediction Score for Rheumatoid Arthritis: a Consortium of Rheumatology Researchers of North America Registry Study. Arthritis Rheumatol. doi:10.1002/art.39195
van der Goes MC, Jacobs JW, Boers M et al (2010) Monitoring adverse events of low-dose glucocorticoid therapy: EULAR recommendations for clinical trials and daily practice. Ann Rheum Dis. doi:10.1136/ard.2009.124958
Fardet L (2013) Long term glucocorticoid therapy: so much remains to be done…. Rev Med Intern. doi:10.1016/j.revmed.2013.02.017
Clark AR (2007) Anti-inflammatory functions of glucocorticoid-induced genes. Mol Cell Endocrinol 275:79–97. doi:10.1016/j.mce.2007.04.013
Acknowledgments
ESCEO is a non-profit Belgian organization (receiving funding from government and industry) dedicated to providing clinical scientists and practitioners with the latest information in evidence-based medicine and health economics. Professor Cooper wishes to acknowledge the Medical Research Council of UK for funding. Professor Miossec acknowledges funding from the IHU prometteur OPERA (Grant ANR-10-IBHU-004) and the EU Marie Curie Network EUTRAIN. The authors would like to thank Jeremy Grierson, PhD, medical writer, for his assistance in preparing the drafts of the manuscript from the presentations and discussions of the working group participants, as well as graphic and bibliographic support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that they have no conflict of interest.
Statement of human and animal rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and /or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Studies performed using animal tissues were done in accordance with accepted ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Cooper, C., Bardin, T., Brandi, ML. et al. Balancing benefits and risks of glucocorticoids in rheumatic diseases and other inflammatory joint disorders: new insights from emerging data. An expert consensus paper from the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO). Aging Clin Exp Res 28, 1–16 (2016). https://doi.org/10.1007/s40520-015-0522-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40520-015-0522-1