
Abstract
Background
Desmin-related myopathy (DRM) is an autosomally inherited skeletal and cardiac myopathy, mainly caused by dominant mutations in the desmin gene (DES). We describe new families carrying the p.S13F or p.N342D DES mutations, the cardiac phenotype of all carriers, and the founder effects.
Methods
We collected the clinical details of all carriers of p.S13F or p.N342D. The founder effects were studied using genealogy and haplotype analysis.
Results
We identified three new index patients carrying the p.S13F mutation and two new families carrying the p.N342D mutation. In total, we summarised the clinical details of 39 p.S13F carriers (eight index patients) and of 21 p.N342D carriers (three index patients). The cardiac phenotype of p.S13F carriers is fully penetrant and severe, characterised by cardiac conduction disease and cardiomyopathy, often with right ventricular involvement. Although muscle weakness is a prominent and presenting symptom in p.N342D carriers, their cardiac phenotype is similar to that of p.S13F carriers. The founder effects of p.S13F and p.N342D were demonstrated by genealogy and haplotype analysis.
Conclusion
DRM may occur as an apparently isolated cardiological disorder. The cardiac phenotypes of the DES founder mutations p.S13F and p.N342D are characterised by cardiac conduction disease and cardiomyopathy, often with right ventricular involvement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mutations in the gene encoding desmin (DES), a major intermediate filament protein of skeletal and cardiac muscle, are the main cause of desmin-related myopathy (DRM, OMIM #601419) [1, 2]. The desmin protein provides structural and functional integrity by coordinating mechanical stress transmission, organelle positioning and assembly of sarcomeres, signal transduction, and apoptosis [3]. DRM’s clinical phenotype encompasses myopathies, cardiomyopathies, cardiac conduction disease, and combinations of these disorders. More than 50 different DES mutations have been identified worldwide; most of these are inherited missense mutations, mainly located in the 2B domain of the protein (Fig. 1). Potential genotype-phenotype relationships are now emerging [4]. Desmin-immunoreactive deposits in skeletal and cardiac muscles and granulofilamentous material at the ultrastructural level are considered morphological hallmarks of DRM [5].

Schematic representation of desmin with locations of p.S13F (‘head’ structure) and p. N342D (2B domain). The desmin molecule is organised in three domains: a highly conserved alpha-helical core (with four consecutive helical rod segments (domain 1A, 1B, 2A, and 2B) connected by short non-helical linkers) flanked by globular N- and C- terminal (‘head’ and ‘tail’) structures
Recently, we performed a meta-analysis of 159 DES mutation carriers with 40 different mutations, focusing on their clinical characteristics [4]. Both neurological and cardiological signs were present in 48% of carriers, whereas isolated neurological or isolated cardiological signs were each present in 22%. Up to 50% of carriers had cardiomyopathy, with dilated cardiomyopathy being most prevalent. In 62% cardiac conduction disease and/or arrhythmias were reported, with the most frequent being atrioventricular block. Symptoms started at a mean age of 35 years (range 11–72 years), while a quarter of carriers had died at a mean age of 49 years (range 28–62 years). Only 5 out of 159 carriers (three women aged between 39–54 years and two 5-year-old children) showed no phenotype at the time of evaluation [4].
In the Netherlands, two DES mutations (p.S13F and p.N342D) have been identified in several index patients. We have previously described some of these families [6–8]. For the current study, we were able to extend these data with new families/patients and additional information on the cardiac phenotypes of previously published patients. Our aims were (1) to describe new families carrying the p.S13F or p.N342D DES mutations, (2) to investigate the cardiac phenotype of all known Dutch patients and carriers described in the literature, and (3) to study the founder effects of the two mutations in the Netherlands.
Methods
Mutation analysis
Since 2004 two clinical genetics centres (at Maastricht University Medical Centre and University Medical Center Groningen) perform genetic screening for DES in the Netherlands. Genomic DNA was isolated from blood samples obtained from the index patients and relatives using standardised procedures. Analysis of DES was performed according to the method we have described previously [9]. Written informed consent was obtained from DES mutation carriers according to the hospitals’ medical ethics committees.
Clinical evaluation
We collected data from cardiological evaluation (12-lead ECG, echocardiography, 24-hour Holter monitoring, and exercise testing) from all evaluated known and obligate Dutch carriers (35 with p.S13F and 19 with p.N342D). Some of these data have been described previously [6–8]. We were able to extend these data with new patients/families and extra information on the cardiac phenotype of previously published patients. If only ECG and X-ray of the heart were available we consider cardiac enlargement to be a cardiomyopathic feature if these individuals also had indications of cardiac conduction disorders. We have also summarised the published cardiological data on p.S13F and p.N342D carriers of other origin.
Genealogy
To discover possible distant relationships between index patients, the genealogies were investigated using civil registers and state archives. The pedigrees were reconstructed to approximately AD 1750 (covering 6–8 generations).
Haplotype analysis
Ten microsatellite markers around DES were selected with the National Center for Biotechnology Information (NCBI) Map Viewer and analysed in carriers and control persons (primers and conditions available upon request).
Geographical distribution of carriers
We mapped the geographical distribution of proven (n = 24 and n = 12 respectively) and obligate/likely carriers (n = 2 and n = 13 respectively) of p.S13F or p.N342D in the Netherlands by collecting the postal codes. For each mutation, the data were converted into an overview of the postal code areas. These make up 90 different regions, each containing about 180,000 inhabitants.
Results
Yield of mutation analyses of the DES gene in the Netherlands
Five different pathogenic mutations in DES have been identified in the Netherlands, resulting in the following changes at the protein level: p.S13F, p.N342D, p.R454W, p.T442I, and p.T453I (Table 1). Previously, we published on p.S13F, p.N342D, and p.R454W [6–8]. The two other mutations (p.T442I and p.T453I) have been newly identified in the Netherlands, each in one index patient, but they have been described in the literature (Table 1) [10, 11]. We also found several variants in DES that are unclassified, or which seem to be a modifier or polymorphism, resulting in the following changes at the protein level: p.G27S, p.L136L, p.A213V, p.A285V, p.R315H, p.N342N. In addition, the following intronic and 3’UTR variants were identified: c.578 + 11 G > A, c.735 + 20 C > T and c.*468 C > T.
In this study we focused on the two Dutch founder mutations (p.S13F and p.N342D). Mutation p.S13F (c.38 C > T) is a missense mutation in the head domain (Fig. 1). Mutation p.N342D (c.1024A > G) is a missense mutation in the 2B domain (Fig. 1). Both mutations alter highly conserved residues, change polarity, co-segregate with disease in the families of all identified index patients, and were absent in at least 300 ethnically matched control alleles. Twenty-seven proven and eight obligate p.S13F carriers were identified by family cascade screening of eight index patients. Twelve proven and seven obligate p.N342D carriers were identified by family cascade screening of three index patients.
Cardiac phenotype of p.S13F
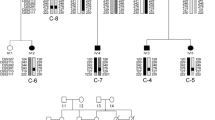
We collected the clinical characteristics from three new index patients (F-III-1, G-III-1, H-III-1; Table 2) with a family history suspected for DRM (Fig. 2). We also gathered additional information (echocardiography and new electrocardiogram) on one previously published patient (E-I-2; Table 2) and cardiological findings on three newly identified carriers in a previously described family (D-II-7, D-III-3, D-III-4; Table 2). For information on the clinical characteristics of previously published individuals and their pedigrees, see Table 1 and Fig. 1 from our previous study [8]. One of the previously published patients (C-VII-10) died at the age of 48, but we have no details about the course of events leading to death. In total, 35 known and obligate p.S13F carriers (including eight index patients) have now been identified and clinically evaluated in the Netherlands (June 2011).

Pedigrees of three recently identified index patients with mutation p.S13F. Square symbols indicate men, circles women. Black solid symbols: clinical diagnosis DRM; half-closed symbols (vertical bar) indicate a possible diagnosis of DRM, dots indicate mutation carriers. Diagonal lines through symbols indicate the patient has died, arrow indicates index patient in each family. Abbreviations: (c)AVB = (complete) atrioventricular block, D = death, DCM = dilated cardiomyopathy, E: S13F = carrier of p.S13F mutation, E: - = not a carrier of p.S13F mutation, ICD = implantable cardioverter defibrillator, LBTB = left bundle branch block, LGMD = limb-girdle muscular dystrophy, LV = left ventricle, PM = pacemaker, PVC premature ventricular contraction, y = years
We have summarised our Dutch data together with the data published on four Singapore-Chinese p.S13F carriers [12] (Table 2). Many patients show a severe cardiac phenotype, including sudden cardiac death or progressive heart failure: 14 of 39 known and obligate carriers died, underwent transplantation, or experienced appropriate implantable cardioverter defibrillator (ICD) interventions at a mean age of 48.4 years (range 27–63 years). Data on initial presentation were available on 31/39 carriers, all of whom had a cardiac phenotype: 21/31 individuals (68%) presented initially with a cardiac phenotype, five individuals (16%) presented with cardiological symptoms preceding neuromuscular symptoms, while the other five individuals (16%) presented with neuromuscular symptoms before or simultaneously with cardiac pathology. Thus, more than 80% presented with isolated cardiological symptoms. We had information on the cardiomyopathic phenotype available for 28 carriers: 17/28 (61%) had developed a cardiomyopathic phenotype: nine dilated cardiomyopathy; five left ventricular hypertrophy, diastolic dysfunction, or a restrictive cardiomyopathy-like phenotype; one had an unspecified form of cardiomyopathy; and two had arrhythmogenic right ventricular cardiomyopathy (ARVC). In addition to these ARVC patients, right ventricular involvement was noticeable due to right ventricular failure in six other patients, and ventricular tachycardias originating from the right ventricle in two patients. Furthermore, as many as 13 patients showed right bundle branch block (RBBB) which also suggests involvement of the right ventricle, although this remains speculative. Regarding the electrocardiographic data from 31 carriers: 21/31 (68%) demonstrated RBBB, left bundle branch block, or atrioventricular block at the time of initial presentation (Fig. 3), suggesting that this is an early manifestation of the disease.
Genealogy and haplotype analysis of families with p.S13F
Common ancestors for three index patients (from families A, B, and C) were previously revealed by their genealogy [8]. Additional genealogical investigations revealed several links between all eight index patients. Haplotype analysis demonstrated an identical haplotype for six microsatellite markers in carriers of the available (7/8) Dutch families (see online supplement). Analysis revealed a different haplotype in a Singapore-Chinese p.S13F carrier (individual Pica II-3) [12] (see online supplement).
Cardiac phenotype of p.N342D
In 1975 two large Dutch families with “inflammatory myopathy in scapulo-ilio-peroneal atrophy with cardiopathy” were reported [13]. We identified a p.N342D mutation in these families, thereby proving that these families had DRM. Figure 4 shows updated pedigrees for these families. In total, 19 known and obligate p.N342D and 10 probable carriers (including three index patients) have now been identified and clinically evaluated in the Netherlands. See Table 3 for the clinical characteristics. The data summary below is based only on the known and obligate carriers. For the pedigree of family A, we refer to Fig. 1 from a previous publication [7].

A 12-lead electrocardiogram with RBBB (a) and a rhythm strip with high-grade atrioventricular block (b) from patients with p.S13F
We summarised our data together with that published on two p.N342D carriers of Irish-German decent [1, 14]. Data on presentation were available from 17 carriers: they all presented with neuromuscular symptoms (16 weakness of muscles, one painful calves), only one individual was reported to have cardiac complaints (palpitations) simultaneously with neuromuscular symptoms. The mean age at presentation was 29.6 (range 16–42 years). We had information on cardiomyopathic phenotype for 11 carriers: 6/11 (55%) had a cardiomyopathic phenotype: three cardiac enlargement (not further specified), one left ventricular dysfunction, one ARVC, and one left ventricular hypertrophy. We had electrocardiographic data available for 18 carriers: 13/18 showed abnormalities on ECG, 10 (63%) of them had cardiac conduction disease. RBBB was identified in four patients.
Genealogy and haplotype analysis of families with p.N342D
Genealogy revealed a common ancestral couple for families O and A, but no link between families O and U. Haplotype analysis around DES revealed an identical haplotype for eight microsatellite markers in carriers from all three Dutch families (see online supplement).
Geographical distribution of carriers of p.S13F or p.N342D
Figure 5 shows the geographical distribution of known, obligate and likely carriers of p.S13F (n = 26) or p.N342D (n = 25) in the Netherlands (Fig. 5a, b), with the number of such carriers per region. Each region contains on average 180,000 inhabitants. The geographical distribution of these mutation carriers reflects the founder effect of both mutations, although a difference in physicians’ knowledge of DRM and in their referral for DES analysis might also have affected this distribution (Fig. 5).

Pedigrees of families O and U with mutation p.N342D. Updated pedigrees of the two families (O and U) previously published by Jennekens et al. [13] in which we identified mutation p.N342D. Square symbols indicate men, circles women, diamonds unknown sex. Black filled symbols: clinical diagnosis DRM. Diagonal lines through symbols indicate deceased. The number in a symbol indicates the number of individuals with this symbol

Geographical distribution of carriers of p.S13F (A) and p.N342D (B). The geographical distribution of known, obligate and likely carriers of p.S13F (n = 26) and p.N342D (n = 25), respectively, in the Netherlands, showing the number of carriers per home region (in brackets: the number of regions). Each region contains on average 180,000 inhabitants
Discussion
We summarise the cardiac phenotypes of all carriers of two Dutch DES founder mutations (p.S13F and p.N342D), including three new index patients with p.S13F and two new large families with p.N342D. This is the largest series to date of carriers of single DES mutations.
The cardiac phenotype of p.S13F seems to be fully penetrant and is characterised by cardiomyopathy and cardiac conduction disorders and/or arrhythmias, including right ventricular involvement and atrioventricular block at a young age. Most patients present with cardiological symptoms and/or signs, compatible with conduction disease/arrhythmias or cardiomyopathy/heart failure. Severe expression of the cardiac phenotype, including sudden cardiac death or progressive heart failure, is seen frequently. Neuromuscular symptoms are not always present. In our series even the youngest patient (H-IV-2 with cardiac evaluation at age 9) already showed some cardiac conduction abnormalities. But clinicians have to be aware that the penetrance is age dependent, thus carriers can also develop their first cardiac abnormalities at a later age.
The cardiac phenotype of p.N342D is in accordance with the phenotype of p.S13F. However, the neuromuscular phenotype is more prominent in carriers with p.N342D than in those with p.S13F, because all p.N342D carriers presented with a neuromuscular phenotype. Unfortunately, we did not have full information on the severity of heart failure or the exact causes of death of some p.N342D carriers. The type of rhythm and conduction disorders and the young ages at death in many individuals suggest that the cardiac phenotype is also severe. But in some cases respiratory problems seem to have been the cause of death. Five p.N342D carriers (5/18, aged between 21-30 years) did not show cardiological abnormalities at the time of evaluation, but they may have developed a cardiac phenotype at a later age. Thus full (age dependent) penetrance of the cardiac phenotype of p.N342D cannot be excluded.
One possible explanation for the variability of phenotypes associated with these two DES mutations is the location of the mutation. Mutations in the 2B domain such as p.N342D are predominantly involved when patients have an isolated neuromuscular phenotype, whereas head and tail domain mutations are predominant in patients who have an isolated cardiological phenotype [4]. Although the neuromuscular phenotype in carriers of the 2B domain mutation p.N342D is more prominent compared with carriers of the head mutation p.S13F, these mutations both cause cardiological as well as neuromuscular signs.
From the literature, there are indications that ICD therapy is better than pacemaker therapy for DES mutation carriers in need of a pacemaker [4, 15]. Our study underscores this: individual H-II-4 experienced several collapses despite having a pacemaker and died suddenly without signs of heart failure; individual G-III-1 collapsed despite a pacemaker and received an ICD (no appropriate shocks in 2 years of follow-up), and individual D-II-3 died suddenly 5 months after he had received a pacemaker for a complete atrioventricular block after cardiomyoplasty. Autopsy did not show abnormalities related to cardiomyoplasty so that ventricular fibrillation was suggested as the cause of death. However, future studies are necessary to evaluate whether ICD therapy is better than pacemaker therapy for DES mutation carriers in need of a pacemaker.
Historically, it is possible that the founder mutation was inherited from a person travelling with the Dutch East Indies Company to Singapore or China in the 17th or 18th century. But our analysis revealed a different haplotype in a p.S13F carrier of the Singapore-Chinese family to that found in the Dutch families. It is therefore unlikely that the mutation in the Singapore-Chinese family is the same founder mutation. So this is a recurrent mutation and it is possible that more carriers of this particular mutation will be identified worldwide.
We are convinced that there are many unidentified carriers of p.S13F and p.N342D founder mutations in the Netherlands. Our estimation of the number of unidentified p.S13F carriers is several hundreds, taking into account that the genealogical links between the identified families are usually more than five generations ago and that death occurs in the majority of cases after reproductive age. We hope this study will lead to better recognition of the phenotype and identification of carriers.
Recognition of DRM and identifying the underlying mutation, followed by family cascade screening, is important. DRM should be considered in patients with a combination of skeletal myopathy and any type of cardiomyopathy (especially with right ventricular dysfunction) or cardiac conduction disease (especially atrioventricular block), but also in patients with isolated cardiomyopathy or skeletal myopathy. Family H shows that DRM can even present as an apparently isolated, familial cardiac conduction disorder. We recommend paying attention to the family history and referring patients to a cardiogenetics outpatient clinic. Early diagnosis of DRM provides the opportunity to prevent complications and reduce morbidity and mortality. We propose regular cardiological monitoring of all DES mutation carriers starting at the age of 10 years and we recommend ICD therapy be considered instead of pacemaker therapy in those carriers who need a pacemaker.
References
Dalakas MC, Park KY, Semino-Mora C. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342:770–80.
Goldfarb LG, Park KY, Cervenakova L, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19:402–3.
Bar H, Strelkov SV, Sjoberg G, et al. The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation? J Struct Biol. 2004;148:137–52.
Spaendonck-Zwarts K, van Hessem L, Jongbloed JD, et al. Desmin-related myopathy. Clin Genet. 2011;80:354–66.
Goebel HH. Desmin-related neuromuscular disorders. Muscle Nerve. 1995;18:1306–20.
Bergman JE, Veenstra-Knol HE, van Essen AJ, et al. Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene. Eur J Med Genet. 2007;50:355–66.
Otten E, Asimaki A, Maass A, et al. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm. 2010;7:1058–64.
van Tintelen JP, Van Gelder IC, Asimaki A, et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009;6:1574–83.
van Tintelen JP, Hofstra RM, Katerberg H, et al. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am Heart J. 2007;154:1130–9.
Arbustini E, Pasotti M, Pilotto A, et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail. 2006;8:477–83.
Bar H, Goudeau B, Walde S, et al. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum Mutat. 2007;28:374–86.
Pica EC, Kathirvel P, Pramono ZA, et al. Characterization of a novel S13F desmin mutation associated with desmin myopathy and heart block in a Chinese family. Neuromuscul Disord. 2008;18:178–82.
Jennekens FG, Busch HF, van Hemel NM, et al. Inflammatory myopathy in scapulo-ilio-peroneal atrophy with cardiopathy. A study of two families. Brain. 1975;98:709–22.
Dalakas MC, Dagvadorj A, Goudeau B, et al. Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord. 2003;13:252–8.
Luethje LG, Boennemann C, Goldfarb L, et al. Prophylactic implantable cardioverter defibrillator placement in a sporadic desmin related myopathy and cardiomyopathy. Pacing Clin Electrophysiol. 2004;27:559–60.
Acknowledgements
We thank the patients who participated in this study and Frans G.I. Jennekens (who reported families O and U in 1975) and Alexander F.J.E. Vrancken for referring patients and providing clinical data. We thank Pieter G. Postema for mapping the geographical distribution of carriers of the two mutations, Marian M.J. Kraak for technical assistance in DNA analysis, Eric Hennekam for genealogical investigations, and Jackie Senior for editing the manuscript. Finally, we thank Wim G.M. Spliet and Eleonora Aronica for their histopathological analysis (not included in this manuscript).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Esm 1
(DOCX 16 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Spaendonck-Zwarts, K.Y., van der Kooi, A.J., van den Berg, M.P. et al. Recurrent and founder mutations in the Netherlands: the cardiac phenotype of DES founder mutations p.S13F and p.N342D. Neth Heart J 20, 219–228 (2012). https://doi.org/10.1007/s12471-011-0233-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12471-011-0233-y