
Abstract
The mechanism underlying the progressive deterioration of left ventricular (LV) dysfunction after myocardial infarction (MI) towards overt heart failure remains incompletely understood, but may involve impairments in coronary blood flow regulation within remodelled myocardium leading to intermittent myocardial ischemia. Blood flow to the remodelled myocardium is hampered as the coronary vasculature does not grow commensurate with the increase in LV mass and because extravascular compression of the coronary vasculature is increased. In addition to these factors, an increase in coronary vasomotor tone, secondary to neurohumoral activation and endothelial dysfunction, could also contribute to the impaired myocardial oxygen supply. Consequently, we explored, in a series of studies, the alterations in regulation of coronary resistance vessel tone in remodelled myocardium of swine with a 2 to 3-week-old MI. These studies indicate that myocardial oxygen balance is perturbed in remodelled myocardium, thereby forcing the myocardium to increase its oxygen extraction. These perturbations do not appear to be the result of blunted β-adrenergic or endothelial NO-mediated coronary vasodilator influences, and are opposed by an increased vasodilator influence through opening of KATP channels. Unexpectedly, we observed that despite increased circulating levels of noradrenaline, angiotensin II and endothelin-1, α-adrenergic tone remained negligible, while the coronary vasoconstrictor influences of endogenous endothelin and angiotensin II were virtually abolished. We conclude that, early after MI, perturbations in myocardial oxygen balance are observed in remodelled myocardium. However, adaptive alterations in coronary resistance vessel control, consisting of increased vasodilator influences in conjunction with blunted vasoconstrictor influences, act to minimize the impairments of myocardial oxygen balance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
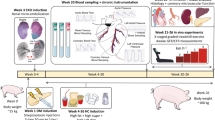
Heart failure constitutes a major cardiovascular disorder of which the incidence and prevalence are increasing, principally due to an increased survival of acute myocardial infarction (MI) in conjunction with an ageing population. The mechanism underlying the progressive deterioration of left ventricular (LV) dysfunction towards overt heart failure remains incompletely understood, but may involve (1) loss of cardiomyocytes through apoptosis [75], (2) a primary reduction in contractile function of the surviving myocardium [97], and/or (3) alterations in extracellular matrix leading to progressive LV dilation [87]. In addition, myocardial blood flow (MBF) abnormalities, resulting in impaired myocardial O2 delivery to the non-infarcted regions (leading to secondary contractile dysfunction and/or enhanced apoptosis), have been suggested to contribute to the progression of LV dysfunction after MI [99]. For example, in vivo studies in rats [54, 55] and swine [108] indicate a reduction in MBF reserve of up to 35% in the surviving remodelled LV myocardium, 3–8 weeks after infarction. Furthermore, in patients with overt heart failure [94], but also in patients with only asymptomatic LV dysfunction [95], flow reserve is reduced in the non-stenotic myocardial regions. In line with these clinical observations, we observed in a porcine model of post-infarct remodelling that during increased O2-demand induced by exercise, the increase in coronary blood flow (CBF) is impaired resulting in perturbations in oxygen delivery [43, 70]. The reduction in flow reserve and the perturbed oxygen delivery during exercise are caused, at least in part, by insufficient growth of the coronary vasculature to maintain flow capacity commensurate with myocardial hypertrophy, in conjunction with a decrease in diastolic pressure time index resulting from the elevated heart rate and particularly elevated LV diastolic pressures [43]. In addition, coronary vasomotor tone may also be increased secondary to neurohumoral activation and endothelial dysfunction, further adding to the perturbations in CBF. However, little is known about the alterations in vasomotor control in coronary resistance vessels within remodelled myocardium (Fig. 1). For this reason we undertook a series of studies to determine whether neurohumoral (autonomic nervous system and renin-angiotensin system), local metabolic, and endothelial control mechanisms of coronary resistance vessel tone are altered in swine with remodelled myocardium produced by a recent MI.

Alterations in determinants of oxygen supply and demand in remodelled myocardium in swine with a 3-week-old myocardial infarction (MI). The net effect of these alterations is a decrease in oxygen supply/demand ratio
2 Characteristics of LV remodelling and dysfunction after MI in swine
Left ventricular remodelling was produced by permanent ligation of the left circumflex coronary artery. This ligation results in a circumscribed transmural infarction of the lateral LV wall, comprising 20–25% of the total LV [82, 96]. LV dysfunction in awake resting MI swine is characterised by 20–30% decreases in cardiac output, stroke volume and LVdP/dt max and a tripling of LV filling pressure. This difference between LVdP/dt max, cardiac output and stroke volume in normal and MI swine remained constant between ∼10 and ∼32 days, indicating that the degree of LV dysfunction and the circulatory adaptations in MI were stable during this observation period [43]. Similarly, we observed that already during the first week after infarction significant LV remodelling occurs, consisting of LV dilation and hypertrophy that remain fairly stable between 1 and 6 weeks after infarction [98]. During exercise, cardiac output, LV systolic pressure and LVdP/dt max increased almost in parallel in MI and normal animals up to 3 km/h, after which curves diverged (Fig. 2); 4 km/h was also the maximally attainable exercise level for most MI swine.

Cardiovascular responses to exercise in normal swine and swine with a ∼3-week-old MI. 0L = lying, 0S = standing. Data are mean ± SEM; *P < 0.05 versus 0L, †P < 0.05 MI versus normal a corresponding exercise level. Data are from Haitsma et al. [43]
Left ventricular dysfunction produced by MI results in neurohumoral activation, characterized by a trend towards elevated plasma levels of catecholamines, but normal circulating levels of renin, angiotensin II and aldosterone at rest. The latter may have been due to the increments in atrial natriuretic peptide (ANP) and endothelin, which can suppress renin and aldosterone release [83]. In resting MI swine, ANP doubled within 24 h, recovered to 50% above normal values within 2 weeks and remained stable between 2 and 6 weeks after infarction, while renin and norepinephrine levels remained normal under resting conditions [98]. In contrast to the discrete neurohumoral activation in resting swine with MI, exercise resulted in exaggerated increases in catecholamines and ANP and increases in endothelin, angiotensin II, and aldosterone (Fig. 3). While resting circulating levels of norepinephrine were still normal and the relative sympathetic drive in response to exercise was preserved in MI, the cardiac responsiveness to exercise (both heart rate and LVdP/dt max) was already blunted 3 weeks after infarction [43], likely due to β-adrenoceptor desensitization and/or downregulation [100].

Neurohumoral responses in exercising swine with a ∼3-week-old MI. In the norepinephrine, epinephrine and dopamine panels, data shown for N represent 0L, 1, 2, 3, 4 and 5 km/h and data for MI represent 0L, 1, 2, 3 and 4 km/h. In all other panels, data shown for N represent 0L, 1, 3 and 5 km/h and data for MI represent 0L, 1, 3 and 4 km/h. Mean data points were fitted with second order curves. VO2 = O2-consumption. Data are mean ± SEM; *P < 0.05 vs 0L, † P < 0.05 MI versus Normal. Data are from Haitsma et al [43]
3 Myocardial O2 balance in remodelled myocardium
Marked decreases in myocardial perfusion occur in pacing-induced severe heart failure in swine [88] and dogs [85, 90], especially in the more vulnerable subendocardial layers. Although one study in dogs indicated that the lower MBF is principally the result of a lower myocardial O2 consumption (MVO2) [90], studies in swine suggest that the impaired perfusion is, at least in part, responsible for the deterioration of LV function because the interstitial edema and disruption of collagen fibers in the subendocardium resemble the ultrastuctural changes that occur with recurrent ischemia [49, 89]. In animal models of severe pressure-overload induced LV hypertrophy, selective underperfusion of the subendocardium can produce myocardial ischemia during exercise and result in post-exercise myocardial stunning [2, 102]. The contribution of perfusion abnormalities in the remote surviving myocardium to LV dysfunction after a MI remains unclear. Studies in rats demonstrated a 25–40% reduction in coronary flow reserve in the surviving myocardium at four [55] and eight [54] weeks after MI. Similarly, maximum subendocardial blood flow was blunted by 40% in anesthetized swine with heart failure 3 weeks after a MI [108]. We hypothesized that the decreased flow reserve could limit the increase in MBF to the hypertrophied myocardium during exercise when hemodynamic abnormalities and neurohumoral activation are exacerbated, thereby impairing myocardial O2-supply.
Three weeks after infarction, blood flow per gram of myocardium in the (remote) LV anterior wall of resting swine with MI was similar to that in normal animals (Fig. 4), confirming previous studies in rats and swine [54, 55, 108]. Interestingly, we observed a trend towards slightly higher blood flows in the outer two layers (P = 0.09), suggesting that despite hypertrophy of the surviving myocardium, metabolic demand in the outer, but not the inner, layers was still slightly elevated, 3 weeks after infarction [43]. During exercise, MBF increased but was redistributed in favor of the subepicardium in MI compared to normal swine. These perturbations were most likely due to increased extravascular compressive forces, resulting from a reduction in diastolic time fraction (secondary to impaired relaxation and increased heart rate) and elevated LV filling pressures [43, 97], that impede MBF, particularly in the subendocardial layers. The decreased MBF necessitated a small increase in O2 extraction that resulted in a slightly lower coronary venous O2 tension (Fig. 4), which actually may have been underestimated as the lower myocardial capillary density in swine with MI possibly prevented a greater increase in O2 extraction [43, 70]. The observation that O2 extraction was forced to increase indicates that increases in extravascular forces are not fully compensated by a concomitant lowering of coronary vasomotor tone in remodelled myocardium during exercise. These observations prompted us to further investigate the control of coronary vasomotor tone in remodelled myocardium during exercise.

Myocardial blood flow and O2-balance in the left ventricular anterior wall of N and MI ∼3 weeks after myocardial infarction. Epi subepicardial, OM outer mid; IM inner mid; Endo subendocardial. MVO2 = myocardial O2-consumption; MEO2 = myocardial O2 extraction; CVPO2 = coronary venous O2 tension In the top left panel data myocardial blood flow data are shown for resting (Rest, lying) conditions, and during maximum exercise (Ex, 5 km/h in N and 4 km/h in MI). Data are mean ± SEM; *P < 0.05 versus 0L, † P < 0.05 MI versus Normal. Data are from Haitsma et al [43]
4 Vasomotor control of the coronary microcirculation in remodelled myocardium
4.1 Neurohumoral control
Cardiac dysfunction is accompanied by a hemodynamic defense reaction consisting of salt and water retention, peripheral vasoconstriction and cardiac stimulation, which serves to partially restore cardiac output and to increase systemic vascular resistance in order to maintain arterial pressure [56]. An integral part of this defense reaction involves alterations in autonomic balance, consisting of an increase in sympathetic activity and a decrease in parasympathetic activity [56].
4.1.1 Sympathetic control
In patients with advanced heart failure plasma noradrenaline levels are already increased under resting conditions [11, 36]. These increased levels result principally from increased sympathetic nerve activity although impaired reuptake may also contribute [56]. Prolonged exposure to elevated noradrenaline levels results in desensitization and downregulation of the β-adrenergic receptors [8, 32, 101]. During exercise, the increases in catecholamine levels are exaggerated in patients with heart failure as compared to healthy controls [35], which is aimed at maintaining chronotropic and inotropic responses to exercise [34].
Also in swine with LV dysfunction produced by a 2–3-week-old MI, we observed exaggerated increases in arterial and coronary venous catecholamine levels during treadmill exercise [23, 43], at a time when resting catecholamine levels were still in the normal range [43, 98]. The exaggerated exercise-induced increases in catecholamine levels reflect increased sympathetic activity, which acts to maintain the chronotropic and inotropic responses to acute exercise. This concept is supported by the observation that β-adrenoceptor blockade produced slightly greater decreases in the chronotropic response during exercise in MI as compared to normal swine. In contrast, β-adrenoceptor blockade in MI swine resulted in a smaller decrease in global LV contractility compared to normal swine, in particular during exercise. The latter findings are consistent with a reduced left ventricular myocardial β-adrenoceptor responsiveness [100].
In normal swine and dogs, β-adrenergic receptor activation contributes to coronary vasodilatation during exercise [27, 39]. The β-adrenergic coronary vasodilatation results in an increase in myocardial oxygen delivery that is commensurate with the increase in oxygen consumption, so that myocardial oxygen extraction and hence coronary venous O2 tension remain constant (Fig. 5). In MI swine, the net β-adrenergic vasodilator influence on the coronary circulation is maintained. However, in view of the exaggerated increases in catecholamine levels during exercise, these findings suggest a diminished β-adrenergic responsiveness of the coronary resistance vessels after MI.

Effect of saline, α-adrenoceptor blockade (phentolamine, 1 mg/kg iv) and/or β-adrenoceptor blockade (propranolol, 0.5 mg/kg iv) and of muscarinic receptor blockade (atropine, 30 μg/kg/min, iv) and/or β-adrenoceptor blockade (propranolol, 0.5 mg/kg iv) on the response of coronary venous O2 tension (CVPO2) plotted as a function of myocardial O2 consumption (MVO2) in seven normal and 7 MI swine. *P < 0.05 versus corresponding Control; † P < 0.05 Atropine + Propranolol versus corresponding Atropine or Phentolamine + Propranolol versus corresponding Phentolamine; Data are mean ± SEM. Data are from Duncker et al. [23]
In dogs, α-adrenoceptor activation limits the exercise-induced increase in CBF, thereby necessitating an increase in myocardial O2 extraction, which leads to a decrease in coronary venous O2 tension [39, 51]. In contrast, α-adrenoceptors do not contribute to regulation of coronary blood flow in normal swine during exercise [27]. We found that, in accordance with our findings in normal swine, administration of the α-adrenoceptor blocker phentolamine had also no effect on coronary venous O2 tension of MI swine (Fig. 5). These findings indicate that even in the presence of exaggerated increments in catecholamine levels in MI swine during exercise, α-adrenoceptors do not contribute to regulation of tone in porcine coronary resistance vessels [23].
4.1.2 Parasympathetic control
The shift in the sympathovagal balance, with an increased sympathetic activity [12, 33] and a blunted parasympathetic activity is reflected in reduced heart rate variability and reduced baroreceptor reflex sensitivity [6, 12, 30, 76] in patients with advanced heart failure. We observed that a maximal dose of the muscarinic receptor blocker atropine produced a similar increase in resting heart rate in swine with MI as compared to normal swine, suggesting preserved parasympathetic activity under resting conditions [23]. In contrast, the atropine-induced increase in heart rate during exercise (particularly at higher exercise levels) was blunted in MI swine, while the increase in LVdP/dt max was abolished [23]. These results are consistent with the concept that gradual inhibition of parasympathetic influence on the heart during exercise was more pronounced in swine with MI compared to normal swine. Importantly, these findings suggest that after MI, at a time when parasympathetic tone under basal resting conditions is normal, a more pronounced inhibition of parasympathetic tone occurs with increasing exercise intensity. Since parasympathetic activity can presynaptically modulate sympathetic activity [1], it is likely that the greater degree of withdrawal of parasympathetic tone during exercise contributed to the exaggerated increase in sympathetic activity during exercise. This is also supported by the observation that in the presence of propranolol, the effects of atropine were no longer different between MI and normal swine [23].
In resting dogs, parasympathetic activity exerts a direct vasodilator influence on coronary resistance vessels that is mediated via nitric oxide [109]. In contrast, in resting swine parasympathetic activity exerts an indirect vasoconstrictor effect on the coronary resistance vessels (which wanes at increasing exercise intensity) that is mediated via inhibition of β-adrenergic vasodilatation [27]. In contrast to the loss of the inhibitory influence of the parasympathetic system on β-adrenoceptor mediated cardiostimulation, we observed that its effects on the coronary circulation were maintained in MI compared to normal swine (Fig. 5) [23]. These findings indicate that at this stage of LV dysfunction, parasympathetic control of β-adrenoceptor-mediated coronary vasodilatation is unimpaired.
4.1.3 Angiotensin II
The renin-angiotensin system plays an important role in cardiovascular homeostasis by contributing to the regulation of blood volume, blood pressure and vascular tone. Angiotensin II (ANG-II) exerts its effects on vascular tone through binding to the AT1-receptor, resulting in vasoconstriction, as well as binding to the AT2-receptor, evoking vasodilation [14]. Both receptor subtypes have been identified in the coronary microcirculation [4, 107], suggesting that endogenous ANG-II may contribute to the regulation of coronary vascular tone and to the regulation of myocardial perfusion. Under pathological circumstances, i.e. after MI, the renin-angiotensin system is activated, resulting in increased plasma levels of ANG-II, particularly during exercise [34, 43]. Moreover, there is evidence that AT1-receptor density in the viable region of the myocardium is increased early after MI [62, 98], suggesting that its vasoconstrictor influence on the coronary vasculature could be increased, which may limit myocardial perfusion thereby exacerbating LV dysfunction.
Contrary to our hypothesis, we observed a loss of ANG-II induced vasoconstrictor influence, reflected by the lack of increase in coronary venous O2 tension (Fig. 6), despite increased plasma ANG-II levels and maintained AT1 receptor densities in coronary arterioles isolated from remodelled myocardium of MI swine [68]. It is unlikely that a generalized loss of vasodilator capacity in the remote myocardium contributed to the blunted vasodilator response to the AT1 receptor blocker irbesartan, as we have previously shown that vasodilation produced by nitroprusside is unperturbed [44]. Although an increased AT2-receptor expression could have acted to limit ANG-II induced vasoconstriction [84] this is unlikely as AT2-mRNA was not altered in coronary arteries from patients with ischemic heart disease [103]. Moreover, the dramatic increases in ANG-II levels that we observed after irbesartan did not result in enhanced, but rather blunted, coronary vasodilation [68]. Therefore, the observation of a reduced vasoconstrictor influence of endogenous ANG-II is best explained by AT1-receptor-desensitization, which is in accordance with studies in dogs with pacing-induced heart failure [78], in rats with pressure-overload LV hypertrophy [63], and in rats with LV remodeling after MI [84], that demonstrated blunted vasoconstrictor responses to exogenous ANG-II.

Effect of AT1-receptor blockade with irbesartan (1 mg/kg iv) on the relation between myocardial O2 consumption (MVO2) and coronary venous O2 tension (CVPO2) in normal swine and swine with a 2–3-week-old myocardial infarction. Data are means ± SE; *P < 0.05 versus corresponding control relation, † P < 0.05 effect of irbesartan different in MI versus Normal animals. Data are from Merkus et al. [68]
4.2 Local metabolic control
4.2.1 Adenosine
Adenosine has been proposed to be a metabolic messenger that regulates coronary resistance vessel tone in response to changes in metabolic needs of the myocardium [5]. However, adenosine receptor blockade with 8PT and/or augmenting adenosine catabolism with intracoronary adenosine deaminase had either no effect [3] or produced a small decrease [28, 67, 93] in basal coronary venous O2 tension (reflecting vasoconstriction), but did not interfere with the normal exercise-induced increase in CBF and O2 delivery [3, 28, 67, 93], indicating that adenosine is not critical for the exercise-induced coronary vasodilation or that loss of adenosine-mediated vasodilation can be compensated for by increased contribution of other vasodilator pathways to maintain adequate metabolic vasodilation.
In contrast to the lack of evidence for an essential role of adenosine in regulation of CBF under physiological conditions, endogenously released adenosine does contribute to coronary vasodilation when there is insufficient supply of O2 [61]. Similarly, adenosine production could be increased in the remodelled myocardium after MI as a result of the perturbations in the myocardial O2 balance [43, 70, 108]. However, we found no evidence for an increased contribution of adenosine to regulation of coronary resistance vessel tone in remodelled myocardium of swine with a recent MI [71], as adenosine receptor blockade with 8PT caused a similar decrease in coronary venous O2 tension in post-infarct remodelled hearts and normal hearts both at rest and during exercise (Fig. 7). These findings are in agreement with observations in dogs with pressure-overload LV hypertrophy, in which adenosine receptor blockade did not affect CBF either at rest or during exercise [65].

Effect of the adenosine receptor antagonist 8-phenyltheophylline (8PT, 5 mg/kg iv), the KATP channel blocker glibenclamide (Glib, 3 mg/kg iv) or their combination on myocardial O2 balance in the LV anterior free wall of normal swine and swine with a recent MI. MVO2 = myocardial O2 consumption; CVPO2 = coronary venous O2 tension; Data are mean ± SEM; Data are mean ± SEM; *P ≤ 0.05 versus corresponding control; † P ≤ 0.05 effect of Glib was blunted at higher MVO2 levels (Glib × MVO2); ‡ P ≤ 0.05 effect of Glib different after MI (Glib × MVO2 × MI). Data are from Merkus et al. [71]
Several reasons could be forwarded for the failure to observe a larger contribution of adenosine to regulation of coronary resistance vessel tone. First, it is possible that the perturbations in the myocardial O2 balance were too mild to increase adenosine production. Second, the activity of enzymes that regulate tissue adenosine levels may have been altered [16, 17]. For example, the activity of adenosine deaminase, the enzyme responsible for breakdown of adenosine to inosine, was found to be elevated in LV hypertrophy [10, 13, 15]. Furthermore, there is evidence that the activity of cytosolic 5′ nucleotidase, which converts 5′AMP to adenosine, is lower in certain models of pressure-overload [13] and volume-overload [10] induced LV hypertrophy, which could be related to intermittent hypoperfusion of the hypertrophic myocardium [41]. Together these enzymatic alterations, which act to decrease myocardial levels of adenosine may have prevented a significant increase in myocardial adenosine levels in the post MI remodelled hearts.
Finally, it is possible that an increased role of adenosine in hypertrophied myocardium is masked by an increased contribution of other vasodilator systems during adenosine receptor blockade, as the process of metabolic vasodilation is thought to be mediated through multiple parallel or redundant pathways. Thus, KATP channel activity may have increased in response to adenosine receptor blockade to compensate for the loss of adenosine-mediated vasodilation. Hence, we evaluated the interactions between these vasodilator pathways.
4.2.2 KATP channels
In addition to the role of KATP channels in the regulation of CBF under physiological conditions [24, 25, 31], there is evidence for an increased KATP channel activity in the coronary circulation of remodelled hearts. For example, in anesthetized dogs with pacing-induced severe heart failure [106], the KATP channel blocker glibenclamide resulted in an exaggerated vasoconstrictor response as compared to normal dogs. Interestingly, in dogs subjected to only 1 week of pacing (when LV function was still normal), KATP channel activity was not different from normal dogs [106], suggesting that KATP channel activity in the basal resting state was only enhanced in the presence of overt heart failure. Similarly, in awake dogs with compensated pressure-overload induced LV hypertrophy, the reduction in CBF produced by glibenclamide was similar to that in normal dogs [65]. During exercise, however, glibenclamide produced a greater reduction in CBF in hypertrophied hearts, indicating increased KATP-channel contribution to coronary vasodilation when O2 requirements of the hypertrophied heart were augmented [65].
In swine with MI-induced moderate LV remodeling and dysfunction, glibenclamide caused a marked decrease in coronary venous O2 tension in remodelled left ventricle under resting conditions, that was similar to the decrease in coronary venous O2 tension in normal hearts (Fig. 7; [71]). Although the vasoconstriction under resting conditions in response to KATP channel blockade was similar in normal and post-MI remodelled hearts, the responses to exercise were different. Thus in normal swine, the effects of KATP channel blockade waned during exercise, suggesting that other vasodilator systems compensated for the loss of KATP channels during exercise. In contrast, in the post-MI remodelled hearts the effects of KATP channel blockade were maintained during exercise. Our findings, which are consistent with the observations in dogs [65], support the hypothesis that KATP channel opening is of greater importance in resistance vessel dilation during exercise in hypertrophied than in normal hearts. It is likely that with the progression from LV dysfunction to overt heart failure, increased KATP channel activity may also become important under resting conditions [106].
4.2.3 Interaction between KATP channels and adenosine
In contrast to the canine heart in which adenosine can act as a back-up system [29, 81], adenosine and KATP channels appear to exert additive vasodilator influences on coronary vasomotor tone in the normal porcine heart [67]. Thus, the coronary vasoconstriction that occurs in response to combined adenosine receptor blockade and KATP channel blockade equalled the sum of the vasoconstriction induced by blockade of the individual pathways. Adenosine mediates its vasodilator effect on porcine coronary resistance vessels via KATP, KCa and Kv channels [9, 46–48] It is therefore possible that following KATP channel blockade adenosine maintained its vasodilator influence via KCa and/or Kv channels.
In contrast to the normal porcine heart, the magnitude of the constriction induced in remodelled hearts by combined blockade of adenosine receptors and KATP channels was virtually identical to that produced by blockade of KATP channels alone [71]. These findings could be interpreted to suggest that in remodelled myocardium the vasodilator influence of endogenous adenosine was entirely mediated through opening of KATP channels, observations that are corroborated by findings in pressure-overload hypertrophied canine hearts [65]. Taken together these observations in the porcine and canine coronary circulations suggest that, although the magnitude of the vasodilator influence exerted by endogenous adenosine was similar in normal and remodelled hearts, its effector pathway was different.
4.3 Endothelial control
Several studies have indicated that endothelial dysfunction, in particular a decreased production of NO and an increased production of endothelin, could aggravate LV dysfunction due to the peripheral vasoconstriction-induced increase in LV afterload, coronary vasoconstriction, and increased myocardial O2 consumption [26, 59, 80, 105].
4.3.1 Nitric oxide
Clinical studies indicate that chronic heart failure is accompanied by blunted vasodilator responses to endothelium-dependent receptor mediated vasodilators (particularly acetylcholine) in the microcirculation of the LV myocardium [92], leg [45, 57], and forearm [19, 52, 57, 64]. In the canine model of pacing-induced end-stage congestive heart failure, attenuated vasodilator responses of resistance vessels to acetylcholine in vivo have also been observed in the microvasculature of the hindleg circulation [22, 64] and the coronary circulation [105]. In swine with a 2–3-week-old MI, we observed reduced vasodilator responses in the systemic and coronary microvasculature to ATP, in doses which we have previously shown to be completely abolished by pretreatment with the eNOS-inhibitor NLA [26]. The findings of a blunted ATP-induced vasodilation are in agreement with the hypothesis that agonist-induced eNOS-mediated NO production is blunted 2–3 weeks after MI.
A loss of NO-mediated vasodilation could enhance the progression of LV dysfunction to heart failure. This is supported by studies in dogs with pacing-induced dilated cardiomyopathy, in which the loss of basal NO production in the LV myocardium coincides with the progression from LV dysfunction to overt heart failure [80, 105]. However, in swine with a 2–3-week-old MI, we did not find any evidence of a reduced coronary vasodilator influence of endogenous NO as decreases in coronary venous O2 tension produced by NLA in resting and exercising MI swine were similar to those of normal swine (Fig. 8). In heart failure patients, studies on the contribution of NO to basal microvascular tone in the forearm, leg, or total systemic bed have yielded equivocal results with responses varying from blunted [45, 58, 64], to maintained [60], and even enhanced [20, 42] increases in vascular tone following NO synthase inhibition. It is possible that a maintained or increased NO production as observed in some studies, was the result of increased iNOS expression [21, 79], as part of a generalized inflammatory response in end-stage heart failure, that occurred in the presence of either a decreased [21, 86] or increased [37, 50] eNOS expression. Since NLA can block all three isoforms of NOS [7], we performed additional experiments, in which we blocked iNOS with aminoguanidine, to determine whether an upregulation of iNOS-mediated NO production masked a reduction in eNOS activity. iNOS blockade had no effect on coronary vasomotor tone, demonstrating that NO production via iNOS does not contribute significantly to vascular tone in MI swine, and, consequently, that basal and exercise-induced endothelial NO production is maintained early after MI [44].

Effect of inhibition of NO synthase by NLA (20 mg/kg iv) on myocardial O2 extraction and coronary venous PO2 at rest (lying) and during treadmill exercise in MI and N. Data are mean ± SEM. *P < 0.05 NLA versus corresponding Control; there were no significant differences in the responses to NLA between MI and N either at rest or during exercise. Data are from Haitsma et al. [44]
The reason for the maintained basal and exercise-induced NO production in the presence of a blunted vasodilator response to ATP is unclear. However, Traverse et al. [91] have shown that the amount of NO, produced after stimulation with an agonist is larger than the amount of NO produced during moderate exercise (60% increase in heart rate). Hence, the maximal capacity of NO production may be reduced, whereas the capacity of eNOS is sufficient to maintain basal and exercise-induced NO production. This explanation is unlikely since agonist-induced dilation is already affected at the lowest dose of administered ATP (which probably releases less NO than strenuous exercise). Moreover, higher doses of ATP still produce more dilation. Another explanation for the divergent results between ATP and exercise-induced increases in NO-production may be that ATP activates eNOS through a different mechanism than shear stress. ATP-induced activation of eNOS is mediated through a calcium-calmodulin dependent pathway [74] whereas shear stress activates eNOS through Akt-mediated phosphorylation [18], resulting in calcium-independent activation of eNOS. Hence, it is possible that perturbations in the calcium homeostasis of endothelial cells contributed to the selective impairment of ATP-induced vasodilation in swine with MI.
4.3.2 Endothelin
Despite the increased plasma levels of endogenous ET in swine with a 2–3 week old MI, its vasoconstrictor influence on the coronary circulation was reduced (Fig. 9 [69, 70]). To determine whether this was the result of blunted receptor responsiveness or reduced local ET-production, we studied the vasoconstriction induced by exogenous ET. The coronary vasoconstrictor influence to exogenous ET-1 in vivo was reduced after MI, indicating a reduced coronary vascular responsiveness to ET. Paradoxically, a recent study showed that ischemic heart disease results in upregulation of ETA and ETB receptor mRNA in human coronary arteries [104]. This is in accordance with our measurements in isolated coronary arterioles obtained from sham-operated swine and swine with a MI, which showed that the ET responsiveness in vessels from animals with a MI was actually increased [70]. The discrepancy between the in vivo and the in vitro findings suggests that ET receptor sensitivity is modulated in vivo, and that this modulation is apparently lost in vitro. Possible modulators of ET-receptor sensitivity are adenosine and NO, which have been shown to desensitize ET-receptors on the coronary vasculature [72, 73], and which may have been lost in the in vitro set-up due to lack of surrounding myocardium and intravascular blood flow. However, since we observed similar vasoconstrictor responses to blockade of adenosine receptors and NO production in MI and normal swine, these vasodilators would seem unlikely explanations for the observed reduced ET receptor sensitivity in MI swine in vivo.

Effect of ETA receptor blockade with EMD (3 mg/kg iv) on myocardial oxygen balance in normal swine and swine with a recent MI. Data are means ± SE; *P < 0.05 versus corresponding Control relation, † P < 0.05 effect of EMD waned during exercise. Data are from Merkus et al. [70]
In conclusion, our observations suggest that when additional coronary vasodilation is required in the hypertrophied myocardium after MI, withdrawal of the ET-mediated vasoconstrictor influence contributes to a shift in vasomotor tone towards vasodilation. These findings may also explain in part why clinical trials of ET-receptor antagonists in heart failure have failed to show therapeutic value of these compounds [40, 77].
4.4 Conclusions and physiological relevance
Under physiological circumstances, the heart matches its blood supply to the demand of the myocardium by altering the balance of vasodilator and vasoconstrictor influences, i.e. an increase in myocardial O2 demand results in an increased influence of vasodilators (opening of KATP channels, NO, adenosine [53], β-adrenergic vasodilation [27]) and a decreased influence of the potent vasoconstrictor endothelin-1 [66, 69]. Studies from our laboratory performed during the past 10 years have shown that following MI myocardial O2 balance in the remodelled LV is perturbed, necessitating vasodilation and recruitment of coronary flow reserve, as evidenced by increased opening of KATP channels [71]. Importantly, however, not only an increased KATP channel vasodilator influence, but also blunting of the coronary vasoconstrictor influences of endogenous endothelin [70] and angiotensin II [68], aid in preventing a more significant impairment of oxygen supply to the remodelled myocardium (Figs. 10, 11). Our studies suggest that generalized blunting of vasoconstrictor influences is one of the first adaptive mechanisms to reduce coronary resistance vessel tone and increase myocardial blood supply. This adaptation, which occurs in the healthy porcine heart during acute exercise, also appears operative in remodelled myocardium. Blunting of vasoconstrictor influences may be a physiologically favorable strategy, since it is more energy-efficient to blunt vasoconstrictor influences than to synthesize vasodilators.

Myocardial oxygen balance in normal and MI swine. Shown are the actual relations between MVO2 and CVPO2 in 30 normal swine (open circles) and 20 MI swine (open triangles) under control conditions. In addition, we have depicted the computed relations in MI swine if the ET (solid diamonds) and ANG II (solid squares) vasoconstrictor influences (which were both attenuated in MI swine) and the KATP (solid triangles) vasodilator influences (which were enhanced in MI swine) would have been identical to those in normal swine. The graph clearly illustrates that the adaptations in coronary vasomotor control act to blunt perturbations in oxygen balance in remodelled myocardium of swine with a recent MI

Alterations in vasomotor balance in the coronary resistance vessels within remodelled myocardium in swine with a 2–3-week-old myocardial infarction
Although the shift in coronary tone towards vasodilatation acts to blunt the flow perturbations in the remodelled heart, it does not appear to fully restore myocardial oxygen balance. This is also supported by the observation that KATP channel activation is increased, consistent with the presence of metabolic distress. Whether these perturbations in myocardial oxygen delivery blood are sufficiently severe to contribute to the progressive deterioration of contractile function of the remodelled left ventricle remains to be established, but it is of interest to note that we previously found that troponin I proteolysis occurred in remodelled myocardium, which was associated with a loss of myofilament force development [97]. Intermittent myocardial ischemia, as may occur in remodelled hearts during exercise or excitement, has been shown to be able to promote troponin I proteolysis [38] and could thereby mediate the flow perturbation-induced progressive deterioration of LV function after MI. Definitive proof of our hypothesis must await future studies demonstrating that prevention of such flow perturbations will indeed prevent progressive loss of contractile function of the remodelled porcine left ventricle.
References
Azevedo ER, Parker JD (1999) Parasympathetic control of cardiac sympathetic activity: Normal ventricular function versus congestive heart failure. Circulation 100:274–279
Bache RJ (1988) Effects of hypertrophy on the coronary circulation. Prog Cardiovasc Dis 30:403–440
Bache RJ, Dai XZ, Schwartz JS et al (1988) Role of adenosine in coronary vasodilation during exercise. Circ Res 62:846–853
Batenburg WW, Garrelds IM, Bernasconi CC et al (2004) Angiotensin II type 2 receptor-mediated vasodilation in human coronary microarteries. Circulation 109:2296–2301
Berne RM, Rubio R (1974) Regulation of coronary blood flow. Adv Cardiol 12:303–317
Binkley PF, Nunziata E, Haas GJ et al (1991) Parasympathetic withdrawal is an integral component of autonomic imbalance in congestive heart failure: Demonstration in human subjects and verification in a paced canine model of ventricular failure. J Am Coll Cardiol 18:464–472
Boer R, Ulrich WR, Klein T et al (2000) The inhibitory potency and selectivity of arginine substrate site nitric-oxide synthase inhibitors is solely determined by their affinity toward the different isoenzymes. Mol Pharmacol 58:1026–1034
Bristow MR, Ginsburg R, Umans V et al (1986) Beta1- and beta2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: Coupling of both receptor subtypes to muscle contraction and selective beta1-receptor down-regulation in heart failure. Circ Res 59:297–309
Cabell F, Weiss DS, Price JM (1994) Inhibition of adenosine-induced coronary vasodilation by block of large-conductance Ca2+-activated K+ channels. Am J Physiol 267:H1455–1460
Cavallini G, De Tata V, Fierabracci V et al (1992) the enzymatic activity of adenosine metabolism in the rat heart. The effects of cardiac hypertrophy. Cardiologia 37:659–661
Chidsey CA, Harrison DC, Braunwald E (1962) Augmentation of plasma norepinephrine response to exercise in patients with congestive heart failure. New Engl J Med 267:650–654
Colucci WS, Braunwald E (2001) Pathophysiology of heart failure. In: Braunwald E, Zipes DP (eds) Heart disease, 6th edn. W.B. Saunders company, Libby P. Philadelphia, pp 503–533
Czarnowski D, Wojcik B, Langfort J et al (1996) 5′-Nucleotidase and adenosine deaminase activities in hypertrophied rat heart. The effect of tachycardia. Rocz Akad Med Bialymst 41:334–340
de Gasparo M, Catt KJ, Inagami T et al (2000) International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52:415–472
De Tata V, Gini S, Simonetti I et al (1989) The regional distribution of adenosine-regulating enzymes in the left and right ventricle walls of control and hypertrophic heart. Basic Res Cardiol 84:597–605
Deussen A (2000) Metabolic flux rates of adenosine in the heart. Naunyn Schmiedebergs Arch Pharmacol 362:351–363
Deussen A (2000) Quantitative integration of different sites of adenosine metabolism in the heart. Ann Biomed Eng 28:877–883
Dimmeler S, Fleming I, Fisslthaler B et al (1999) Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399:601–605
Drexler H, Hablawetz E, Lu W et al (1992) Effects of inhibition of nitric oxide formation on regional blood flow in experimental myocardial infarction. Circulation 86:255–262
Drexler H, Hayoz D, Munzel T et al (1992) Endothelial function in chronic congestive heart failure. Am J Cardiol 69:1596–1601
Drexler H, Kastner S, Strobel A et al (1998) Expression, activity and functional significance of inducible nitric oxide synthase in the failing human heart. J Am Coll Cardiol 32:955–963
Drexler H, Lu W (1992) Endothelial dysfunction of hindquarter resistance vessels in experimental heart failure. Am J Physiol 262:H1640–H1645
Duncker DJ, Haitsma DB, Liem DA et al (2005) Exercise unmasks autonomic dysfunction in swine with a recent myocardial infarction. Cardiovasc Res 65:889–896
Duncker DJ, Laxson DD, Lindstrom P et al (1993) Endogenous adenosine and coronary vasoconstriction in hypoperfused myocardium during exercise. Cardiovasc Res 27:1592–1597
Duncker DJ, Oei HH, Hu F et al (2001) Role of K +ATP channels in regulation of systemic, pulmonary, and coronary vasomotor tone in exercising swine. Am J Physiol 280:H22–H33
Duncker DJ, Stubenitsky R, Tonino PA et al (2000) Nitric oxide contributes to the regulation of vasomotor tone but does not modulate O2-consumption in exercising swine. Cardiovasc Res 47:738–748
Duncker DJ, Stubenitsky R, Verdouw PD (1998) Autonomic control of vasomotion in the porcine coronary circulation during treadmill exercise: evidence for feed-forward beta-adrenergic control. Circ Res 82:1312–1322
Duncker DJ, Stubenitsky R, Verdouw PD (1998) Role of adenosine in the regulation of coronary blood flow in swine at rest and during treadmill exercise. Am J Physiol 275:H1663–1672
Duncker DJ, van Zon NS, Pavek TJ et al (1995) Endogenous adenosine mediates coronary vasodilation during exercise after K +ATP channel blockade. J Clin Invest 95:285–295
Eckberg DL, Drabinsky M, Braunwald E (1971) Defective cardiac parasympathetic control in patients with heart disease. N Engl J Med 285:877–883
Farouque HM, Worthley SG, Meredith IT et al (2002) Effect of ATP-sensitive potassium channel inhibition on resting coronary vascular responses in humans. Circ Res 90:231–236
Fowler MB, Laser JA, Hopkins GL et al (1986) Assessment of the beta-adrenergic receptor pathway in the intact failing human heart: progressive receptor down-regulation and subsensitivity to agonist response. Circulation 74:1290–1302
Francis GS (1991) Heart failure in 1991. Cardiology 78:81–94
Francis GS (2001) Pathophysiology of chronic heart failure. Am J Med 110(Suppl 7A):37S–46S
Francis GS, Goldsmith SR, Ziesche S et al (1985) Relative attenuation of sympathetic drive during exercise in patients with congestive heart failure. J Am Coll Cardiol 5:832–839
Francis GS, Goldsmith SR, Ziesche SM et al (1982) Response of plasma norepinephrine and epinephrine to dynamic exercise in patients with congestive heart failure. Am J Cardiol 49:1152–1156
Fukuchi M, Hussain SN, Giaid A (1998) Heterogeneous expression and activity of endothelial and inducible nitric oxide synthases in end-stage human heart failure: their relation to lesion site and beta-adrenergic receptor therapy. Circulation 98:132–139
Gao WD, Atar D, Liu Y et al (1997) Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ Res 80:393–399
Gorman MW, Tune JD, Richmond KN et al (2000) Feedforward sympathetic coronary vasodilation in exercising dogs. J Appl Physiol 89:1892–1902
Gottlieb SS (2003) The neurohormonal paradigm: Have we gone too far? J Am College Cardiol 41:1458–1459
Gustafson LA, Kroll K (1998) Downregulation of 5′-nucleotidase in rabbit heart during coronary underperfusion. Am J Physiol 274:H529–538
Habib F, Dutka D, Crossman D et al (1994) Enhanced basal nitric oxide production in heart failure: another failed counter-regulatory vasodilator mechanism? Lancet 344:371–373
Haitsma DB, Bac D, Raja N et al (2001) Minimal impairment of myocardial blood flow responses to exercise in the remodeled left ventricle early after myocardial infarction, despite significant hemodynamic and neurohumoral alterations. Cardiovasc Res 52:471–428
Haitsma DB, Merkus D, Vermeulen J et al (2002) Nitric oxide production is maintained in exercising swine with chronic left ventricular dysfunction. Am J Physiol 282:H2198–H2209
Hambrecht R, Fiehn E, Weigl C et al (1998) Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation 98:2709–2715
Heaps CL, Bowles DK (2002) Gender-specific K+-channel contribution to adenosine-induced relaxation in coronary arterioles. J Appl Physiol 92:550–558
Hein TW, Belardinelli L, Kuo L (1999) Adenosine A2A receptors mediate coronary microvascular dilation to adenosine: role of nitric oxide and ATP-sensitive potassium channels. J Pharmacol Exp Ther 291:655–664
Hein TW, Kuo L (1999) cAMP-independent dilation of coronary arterioles to adenosine: role of nitric oxide, G proteins, and KATP channels. Circ Res 85:634–642
Helmer GA, McKirnan MD, Shabetai R et al (1996) Regional deficits of myocardial blood flow and function in left ventricular pacing-induced heart failure. Circulation 94:2260–2267
Heymes C, Vanderheyden M, Bronzwaer JG et al (1999) Endomyocardial nitric oxide synthase and left ventricular preload reserve in dilated cardiomyopathy. Circulation 99:3009–3016
Heyndrickx GR, Muylaert P, Pannier JL (1982) Alpha-adrenergic control of oxygen delivery to myocardium during exercise in conscious dogs. Am J Physiol 242:H805–H809
Hirooka Y, Imaizumi T, Tagawa T et al (1994) Effects of l-arginine on impaired acetylcholine-induced and ischemic vasodilation of the forearm in patients with heart failure. Circulation 90:658–668
Ishibashi Y, Duncker DJ, Zhang J et al (1998) ATP-sensitive K+ channels, adenosine, and nitric oxide-mediated mechanisms account for coronary vasodilation during exercise. Circ Res 82:346–359
Kalkman EA, Bilgin YM, van Haren P et al (1996) Determinants of coronary reserve in rats subjected to coronary artery ligation or aortic banding. Cardiovasc Res 32:1088–1095
Karam R, Healy BP, Wicker P (1990) Coronary reserve is depressed in postmyocardial infarction reactive cardiac hypertrophy. Circulation 81:238–246
Katz AM (2000) Heart failure, pathophysiology, molecular biology, and clinical management. Lippincott Williams & Wilkins, Philadelphia
Katz SD, Biasucci L, Sabba C et al (1992) Impaired endothelium-mediated vasodilation in the peripheral vasculature of patients with congestive heart failure. J Am Coll Cardiol 19:918–925
Katz SD, Krum H, Khan T et al (1996) Exercise-induced vasodilation in forearm circulation of normal subjects and patients with congestive heart failure: Role of endothelium-derived nitric oxide. J Am Coll Cardiol 28:585–590
Kiowski W, Sutsch G, Schalcher C et al (1998) Endothelial control of vascular tone in chronic heart failure. J Cardiovasc Pharmacol 32:S67–S73
Kubo SH, Rector TS, Bank AJ et al (1994) Lack of contribution of nitric oxide to basal vasomotor tone in heart failure. Am J Cardiol 74:1133–1136
Laxson DD, Homans DC, Bache RJ (1993) Inhibition of adenosine-mediated coronary vasodilation exacerbates myocardial ischemia during exercise. Am J Physiol 265:H1471–1477
Lefroy DC, Wharton J, Crake T et al (1996) Regional changes in angiotensin II receptor density after experimental myocardial infarction. J Mol Cell Cardiol 28:429–440
Lopez JJ, Lorell BH, Ingelfinger JR et al (1994) Distribution and function of cardiac angiotensin AT1- and AT2-receptor subtypes in hypertrophied rat hearts. Am J Physiol 267:H844–852
Maguire SM, Nugent AG, McGurk C et al (1998) Abnormal vascular responses in human chronic cardiac failure are both endothelium dependent and endothelium independent. Heart 80:141–145
Melchert PJ, Duncker DJ, Traverse JH et al (1999) Role of K +ATP channels and adenosine in regulation of coronary blood flow in the hypertrophied left ventricle. Am J Physiol 277:H617–H625
Merkus D, Duncker DJ, Chilian WM (2002) Metabolic regulation of coronary vascular tone: Role of endothelin-1. Am J Physiol 283:H1915–H1921
Merkus D, Haitsma DB, Fung TY et al (2003) Coronary blood flow regulation in exercising swine involves parallel rather than redundant vasodilator pathways. Am J Physiol 285:H424–H433
Merkus D, Haitsma DB, Sorop O et al (2006) Coronary vasoconstrictor influence of angiotensin II is reduced in remodeled myocardium after myocardial infarction. Am J Physiol 291:H2082–2089
Merkus D, Houweling B, Mirza A et al (2003) Contribution of endothelin and its receptors to the regulation of vascular tone during exercise is different in the systemic, coronary and pulmonary circulation. Cardiovasc Res 59:745–754
Merkus D, Houweling B, van den Meiracker AH et al (2005) Contribution of endothelin to coronary vasomotor tone is abolished after myocardial infarction. Am J Physiol 288:H871–H880
Merkus D, Houweling B, van Vliet M et al (2005) Contribution of K +ATP channels to coronary vasomotor tone regulation is enhanced in exercising swine with a recent myocardial infarction. Am J Physiol Heart Circ Physiol 288:H1306–H1313
Merkus D, Sorop O, Houweling B et al (2006) NO and prostanoids blunt endothelin-mediated coronary vasoconstrictor influence in exercising swine. Am J Physiol 291:H2075–H2081
Merkus D, Stepp DW, Jones DW et al (2000) Adenosine preconditions against endothelin-induced constriction of coronary arterioles. Am J Physiol 279:H2593–H2597
Motte S, Communi D, Pirotton S et al (1995) Involvement of multiple receptors in the actions of extracellular ATP: the example of vascular endothelial cells. Int J Biochem Cell Biol 27:1–7
Narula J, Haider N, Arbustini E et al (2006) Mechanisms of disease: apoptosis in heart failure-seeing hope in death. Nat Clin Pract Cardiovasc Med 3:681–688
Nolan J, Flapan AD, Capewell S et al (1992) Decreased cardiac parasympathetic activity in chronic heart failure and its relation to left ventricular function. Br Heart J 67:482–485
O’Connor CM, Gattis WA, Adams KF Jr et al (2003) Tezosentan in patients with acute heart failure and acute coronary syndromes: results of the randomized intravenous tezosentan study (RITZ-4). J Am Coll Cardiol 41:1452–1457
Oikawa Y, Maehara K, Saito T et al (2001) Attenuation of angiotensin II-mediated coronary vasoconstriction and vasodilatory action of angiotensin-converting enzyme inhibitor in pacing-induced heart failure in dogs. J Am Coll Cardiol 38:1188–1194
Orus J, Heras M, Morales-Ruiz M et al (2000) Nitric oxide synthase II mRNA expression in cardiac tissue of patients with heart failure undergoing cardiac transplantation. J Heart Lung Transplant 19:139–144
Recchia FA, McConnell PI, Bernstein RD et al (1998) Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure in the conscious dog. Circ Res 83:969–979
Samaha FF, Heineman FW, Ince C et al (1992) ATP-sensitive potassium channel is essential to maintain basal coronary vascular tone in vivo. Am J Physiol 262:C1220–C1227
Schaper W, Remijsen P, Xhonneux R (1969) The size of myocardial infarction after experimental coronary artery ligation. Z Kreislaufforsch 58:904–909
Schrier RW, Abraham WT (1999) Hormones and hemodynamics in heart failure. N Engl J Med 341:577–585
Schuijt MP, Basdew M, van Veghel R et al (2001) AT2 receptor-mediated vasodilation in the heart: effect of myocardial infarction. Am J Physiol Heart Circ Physiol 281:H2590–2596
Shannon RP, Komamura K, Shen YT et al (1993) Impaired regional subendocardial coronary flow reserve in conscious dogs with pacing-induced heart failure. Am J Physiol 265:H801–H809
Smith CJ, Sun D, Hoegler C et al (1996) Reduced gene expression of vascular endothelial no synthase and cyclooxygenase-1 in heart failure. Circ Res 78:58–64
Spinale FG (2007) Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 87:1285–1342
Spinale FG, Grine RC, Tempel GE et al (1992) Alterations in the myocardial capillary vasculature accompany tachycardia-induced cardiomyopathy. Basic Res Cardiol 87:65–79
Spinale FG, Hendrick DA, Crawford FA et al (1990) Chronic supraventricular tachycardia causes ventricular dysfunction and subendocardial injury in swine. Am J Physiol 259:H218–H229
Traverse JH, Melchert P, Pierpont GL et al (1999) Regulation of myocardial blood flow by oxygen consumption is maintained in the failing heart during exercise. Circ Res 84:401–408
Traverse JH, Wang YL, Du R et al (2000) Coronary nitric oxide production in response to exercise and endothelium-dependent agonists. Circulation 101:2526–2531
Treasure CB, Vita JA, Cox DA et al (1990) Endothelium-dependent dilation of the coronary microvasculature is impaired in dilated cardiomyopathy. Circulation 81:772–779
Tune JD, Richmond KN, Gorman MW et al (2000) Adenosine is not responsible for local metabolic control of coronary blood flow in dogs during exercise. Am J Physiol 278:H74–H84
van den Heuvel AF, Bax JJ, Blanksma PK et al (2002) Abnormalities in myocardial contractility, metabolism and perfusion reserve in non-stenotic coronary segments in heart failure patients. Cardiovasc Res 55:97–103
van den Heuvel AF, Blanksma PK, Siebelink HM et al (2001) Impairment of myocardial blood flow reserve in patients with asymptomatic left ventricular dysfunction: effects of ACE-inhibition with perindopril. Int J Cardiovasc Imaging 17:353–359
Van der Giessen WJ, Van Woerkens LJ, Duncker DJ et al (1989) Acute hemodynamic effects of nisoldipine and pimobendan in conscious pigs with chronic heart failure. J Cardiovasc Pharmacol 14:653–658
van der Velden J, Merkus D, Klarenbeek BR et al (2004) Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ Res 95:e85–e95
Van Kats JP, Duncker DJ, Haitsma DB et al (2000) Angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade prevent cardiac remodeling in pigs after myocardial infarction: role of tissue angiotensin II. Circulation 102:1556–1563
van Veldhuisen DJ, van den Heuvel AF, Blanksma PK et al (1998) Ischemia and left ventricular dysfunction: a reciprocal relation? J Cardiovasc Pharmacol 32(Suppl 1):S46–51
van Woerkens LJ, van der Giessen WJ, Verdouw PD (1993) Cardiovascular effects of dopamine and dobutamine in conscious pigs with chronic heart failure. Crit Care Med 21:420–424
Vatner DE, Asai K, Iwase M et al (1999) Beta-adrenergic receptor-G protein-adenylyl cyclase signal transduction in the failing heart. Am J Cardiol 83:80H–85H
Vatner SF, Hittinger L (1993) Myocardial perfusion dependent and independent mechanisms of regional myocardial dysfunction in hypertrophy. Basic Res Cardiol 88:81–95
Wackenfors A, Emilson M, Ingemansson R et al (2004) Ischemic heart disease down-regulates angiotensin type 1 receptor mRNA in human coronary arteries. Eur J Pharmacol 503:147–153
Wackenfors A, Emilson M, Ingemansson R et al (2004) Ischemic heart disease induce upregulation of endothelin receptor mrna in human coronary arteries. Eur J Pharmacol 484:103–109
Wang J, Seyedi N, Xu XB et al (1994) Defective endothelium-mediated control of coronary circulation in conscious dogs after heart failure. Am J Physiol 266:H670–680
Yamamoto M, Egashira K, Arimura K et al (2000) Coronary vascular K+ ATP channels contribute to the maintenance of myocardial perfusion in dogs with pacing-induced heart failure. Jpn Circ J 64:701–707
Zhang C, Hein TW, Wang W et al (2003) Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circ Res 92:322–329
Zhang J, Wilke N, Wang Y et al (1996) Functional and bioenergetic consequences of postinfarction left ventricular remodeling in a new porcine model. MRI and 31P-MRS study. Circulation 94:1089–1100
Zhao G, Shen W, Xu X et al (1995) Selective impairment of vagally mediated, nitric oxide-dependent coronary vasodilation in conscious dogs after pacing-induced heart failure. Circulation 91:2655–2663
Acknowledgments
Dr Merkus is supported by a post-doc stipend from the Netherlands Heart Foundation (2000T042).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Duncker, D.J., de Beer, V.J. & Merkus, D. Alterations in vasomotor control of coronary resistance vessels in remodelled myocardium of swine with a recent myocardial infarction. Med Biol Eng Comput 46, 485–497 (2008). https://doi.org/10.1007/s11517-008-0315-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11517-008-0315-1