
Abstract
Antiretroviral therapy has greatly extended the lifespan of people living with human immunodeficiency virus (PLHIV). As a result, the long-term effects of HIV infection, in particular those originating in the central nervous system (CNS), such as HIV associated depression, have gained importance. Animal models for HIV infection have proved very useful for understanding the disease and developing treatment strategies. However, HIV associated depression remains poorly understood and so far there is neither a fully satisfactory animal model, nor a pathophysiologically guided treatment for this condition. Here we review the neuroimmunological, neuroendocrine, neurotoxic and neurodegenerative basis for HIV depression and discuss strategies for employing HIV animal models, in particular humanized mice which are susceptible to HIV infection, for the study of HIV depression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the combined antiretroviral therapy (CART) era, the human immunodeficiency virus (HIV) epidemic has been stabilized and the life expectancy of people living with HIV (PLHIV) has been prolonged (Kaul 2009). However, depression, as well as other central nervous system (CNS) comorbidities, including HIV-associated neurocognitive disorders (HAND) remain a significant concern (Heaton et al. 2011; Woods et al. 2009), and at least mild neurologic disease is found in approximately 30 % of such individuals (Boisse et al. 2008; Heaton et al. 1995). Approximately one half of PLHIV will develop a neuropsychiatric disorder (Kopnisky et al. 2007; Kraft-Terry et al. 2010) and about two thirds of these patients will develop a depressive disorder (Fulk et al. 2004; Pieper and Treisman 2005). Depression in turn has a large negative impact on quality of life and on treatment because it impairs adherence to CART (Schuster et al. 2012), being associated with increased viral load (Sumari-de Boer et al. 2012). Moreover, there is evidence that depressive symptoms adversely affect the clinical response to CART, even in adherent patients (Gibbie et al. 2006; Vlassova et al. 2009).
The treatment for depression is still frustrating, with patients often only achieving partial remission of the symptoms (Khin et al. 2011; Kirsch et al. 2008). In the case of depression in PLHIV, one recent study based on retrospective chart review of patients in an urban psychiatric clinic reported rates of remission and response at 36.4 % and 50.7 %, respectively (Primeau et al. 2013). More studies are needed to evaluate treatment effectiveness better (Hill and Lee 2013). There are strong reasons to believe that in PLHIV, depression is at least partially related to the virus itself. Animal models for depression associated with HIV are therefore crucial to understanding these mechanisms and for developing new pharmacological therapies.
This paper discusses the requirements of animal models for HIV associated depression, based on their capacity to answer questions related to its current proposed physiopathology. First, we briefly discuss possible mechanisms associated with HIV depression. Second we present the current available animal models for depression in general. Next, we present the already developed models for reproducing HIV infection in rodents, and discuss the adaptation of these models for the investigation of depression associated with HIV, the limitations and the possibilities for improvement for this application. We conclude with suggestions for validation of the models and for future HIV depression rodent model research.
Mechanisms of HIV associated depression
HIV enters the CNS early in the course of infection, and initially resides primarily in microglia and macrophages (Gras and Kaul 2010; Kramer-Hammerle et al. 2005), which express the necessary chemokine receptors (Cartier et al. 2005), for HIV cellular entry. The virus crosses the blood–brain barrier (BBB) and enters the CNS within monocyte-derived macrophages via a “Trojan Horse” mechanism. Once within the CNS, the virus, although not infecting neurons, can produce synaptodendritic neuron injury and neuron death, leading to damage of a variety of neural systems (Hult et al. 2008). As the BBB impairs CART CNS penetration, the chronic neuroinflammatory state persists in HIV infected individuals, even after CART-induced serum viral suppression (Gartner 2000; Kaul et al. 2007; Lindl et al. 2010), and for this reason, the CNS is regarded as a virus reservoir. For instance, the Sydney Blood Bank Cohort consists of individuals infected with a common nef-defective strain of HIV after being transfused with blood products from a common donor (Churchill et al. 2006). Infection with this attenuated strain resulted in either low or absent viral replication in vivo for up to 29 years (Rhodes et al. 2000; Zaunders et al. 2011). Nonetheless, one individual of this cohort developed HIV-associated dementia and showed genetically different viral sequences in the CSF and in the peripheral blood (Churchill et al. 2004). These observations demonstrate that even nef-defective strains of HIV-1 viruses with reduced pathogenic potential can penetrate the BBB and undergo compartmentalized evolution in the CNS, generating new clones with enhanced transcriptional activity, leading to neurologic disease.
PLHIV exhibit a 2-fold increase in prevalence of depression compared with HIV-uninfected individuals (Ciesla and Roberts 2001). The contributions of immuno-inflammatory, monoaminergic, neurodegenerative and neurotrophic pathways to HIV-associated depression have recently been reviewed (Del Guerra et al. 2013).
The cytokinergic hypothesis of depression posits that the depression is caused by the actions of cytokines. This hypothesis arose from three key observations: first, depressed patients showed elevated inflammatory markers, including cytokines, chemokines, and acute phase reactant in serum and cerebral spinal fluid (CSF) (Dowlati et al. 2010; Raison and Miller 2011). Studies of human disease show a close link between elevated CNS tumor necrosis factor (TNF)-α and depressive symptoms (Liu et al. 2012). A recent meta-analysis of immune abnormalities in patients with major depression by Zorrilla et al. showed the existence of an overall leukocytosis, increased prostaglandin E2, (PGE2) and interleukin (IL)-6 concentrations and reduced lymphocyte proliferative responses to mitogens (Zorrilla et al. 2001). Second, people with inflammatory illness are more susceptible to development of depressive symptoms (Steptoe 2007). Third, acute inflammation in healthy mammals is related to behavioral changes, called “sickness behavior”, which show many similarities to depression. Sickness behavior was described by Hart et al. (Hart 1988) and involves decreases in a number of activities, most notably feeding, exploration and sexual activity (interpreted as reduced ability to experience pleasure from natural rewards, or anhedonia), as well as increased time spent sleeping, hyperalgesia and lethargy (hypomotility) (Dantzer 2001).
Although both depression and sickness behavior share elevated inflammatory markers, they are not identical phenomena and might reflect distinct neuroimmunological mechanisms. For instance, depressive behavior induced by IL-1 and lipopolysaccharide (LPS) in rodents may develop after sickness behavior has resolved, since hypomotility in the tail-suspension test and in the forced-swim test, which are used to estimate despair under inescapable stress conditions, persisted 24 h after LPS treatment, a time point, when the motor activity had already returned to normal. Furthermore decrease in preference for a sweet solution (measure of anhedonia), was still apparent after food intake and drinking had already normalized (Dantzer 2001; Dantzer et al. 2008; Frenois et al. 2007). Moreover, patients on interferon (IFN)-α treatment seem to develop early manifestations of psychomotor slowing, and fatigue (neurovegetative syndrome, analogous to sickness behavior), non-responsiveness to antidepressants and in a later stage depressed mood, anxiety and cognitive dysfunction (mood/cognitive syndrome), which is responsive to antidepressants (Capuron and Miller 2004). Despite contradictory results, with some rodent studies based on LPS injection reporting depressive-like behavior at earlier time points (Yirmiya 1996; Zhu et al. 2010), and others showing persistence of sickness-behavior at 24 h (Berg et al. 2004; Godbout et al. 2008), taken together these results indicate that depressive-like behavior could be separated over time from sickness, as depression seems to emerge in a later time point and to have a different neurochemical basis and pharmacological response. This led Biesmans et al. to suggest that, in order to evaluate depressive-like behaviors in animal models without the confounding effect of sickness, a situation of chronic, persistent inflammation should be promoted (Biesmans et al. 2013). As chronic neuroinflammation is a major feature of CNS HIV infection, it seems plausible to assume that cytokinergic mechanisms may play an important role in depression amongst PLHIV.
Next, we briefly consider cytokinergic-inflammation mediated neurobiological changes thought to predispose to depression. These include the effects of cytokines on the hypothalamus-pituitary-adrenal (HPA) axis, reduced production of trophic factors, especially brain derived neurotrophic factor (BDNF) with impaired hippocampal neurogenesis, glutamate-induced excitotoxicity, activation of the kynurenine pathway and changes in neurotransmission, especially of monoamines (Raison and Miller 2011; Sahay and Hen 2007; Schmidt and Duman 2007).
Dysfunction of the HPA axis with consequent enhancement of cortisol plasma levels is a common aspect of depression (Krishnan and Nestler 2011) and is attributed to reduced expression or decreased functionality of the glucocorticoid receptor, a situation called “glucocorticoid resistance” (Pariante 2003). Indeed, proinflammatory cytokines, including IL-1β and IL-6, are thought to activate the HPA axis and to be related to hypercortisolemia following stress or during depression, which is attributed to glucocorticoid receptor downregulation (Capuron et al. 2003; Maes 1993). Based on this, rodent models were developed that disrupt glucocorticoid homeostasis in the HPA axis. In some models, animals are treated chronically with glucocorticoids (Gourley et al. 2008). In others, mutant mice express abnormal levels of brain glucocorticoid receptors to disrupt the normal HPA axis feedback inhibition. These models display anhedonia that is reversible with antidepressants (Muller and Holsboer 2006).
BDNF has long been implicated in stress and depression-induced behavioral and synaptic plasticity in several brain regions (Krishnan et al. 2007). Berton and colleagues, using a social defeat model and a mesolimbic dopamine pathway-specific knockdown of BDNF, showed that ventral tegmental area-derived BDNF is required in the nucleus accumbens to produce experience-dependent social aversion, an index of depression-like behavior in the Chronic Social Defeat (CSD) Stress model (Berton et al. 2006). Supporting a brain region-specific effect of BDNF levels on depressive behaviors, knockdown of BDNF in dorsal dentate gyrus leads to the emergence of such behaviors (Taliaz et al. 2010). Other studies of human depressed patient samples show increased BDNF protein levels in the nucleus accumbens, and decreased levels in hippocampus, also supporting the neurotrophic model (Berton et al. 2006; Christoffel et al. 2011; Krishnan et al. 2007).
Another pathway, which arises from proinflammatory cytokine action, acts through glutamate-induced excitotoxicity, which has been extensively implicated in psychiatric disorders, including depression (Duman 2009). Inflammatory mediators cause an increase in glutamate neurotransmission, upregulating NMDA receptor function, increasing the release and inhibiting the reuptake of glutamate. Exposure of hippocampal neurons to IL-1β and TNF-α intensifies the excitotoxic neuronal damage induced through NMDA and AMPA receptors. IL-1β has also been found to inhibit the reuptake of glutamate by glial cells (Bernardino et al. 2005).
Cytokines in brain can also lead to depression via the action of indoleamine 2,3 dioxygenase (IDO), an enzyme that degrades tryptophan, an essential amino acid that is the limiting factor for the synthesis of serotonin. Two main pathways metabolize tryptophan: the kynurenine (KYN) pathway, which is initiated by the enzyme IDO (Hirata et al. 1974), and the serotonin (5-HT) pathway, initiated by the enzyme tryptophan 5-monooxygenase (Ichiyama et al. 1970). Release of proinflammatory cytokines such as IL-1, IFN-γ and TNF-α induce glial IDO activity, leading to metabolism of tryptophan to kynurenine acid (KYNA), 3 hydroxy kynurenine (3HK) and quinolinic acid (QUIN), rather than serotonin (5HT) (Christmas et al. 2011). After crossing the BBB, QUIN is neurotoxic through its action as an agonist of the glutamatergic NMDA receptor, and 3HK can induce oxidative damage. Though KYNA is an antagonist of glutamate receptors with antioxidant properties, in inflammatory states increases in 3-HK and QUIN levels in the brain outweigh the elevation in KYNA (Schwarcz et al. 2012). Thus this pathway may not only reduce serotonin, leading to a reduction in the sense of well-being, but also induce neurodegeneration.
Activation of IDO, associated with increased cytokine levels and depressive behavior, has been demonstrated in mice inoculated intraperitoneally with a bacillus (Moreau et al. 2005) and after intracerebroventricular administration of LPS (Fu et al. 2010).
Studies in humans have also revealed a role for the kynurenine pathway in depression. Capuron and colleagues reported that depression elicited by IFN-α therapy was associated with activation of the KYN pathway (Capuron et al. 2002). Winchers and colleagues showed that patients receiving IFN-α therapy have an increase in the serum KYN/KYNA ratio that was significantly associated with the severity of depression (Wichers et al. 2005). In the studies of Myint et al., patients with major depression have decreased plasma KYNA and decreased KYNA/KYN ratio (Myint et al. 2007). Furthermore, the proinflammatory cytokine, IFN-γ, induces IDO expression in hypothalamic and pituitary cells and consequent synthesis of tryptophan metabolites, which may result in neurodegenerative changes in the HPA axis with inhibition of the negative feedback activity of glucocorticoid receptors and hippocampal atrophy, as glucocorticoids are toxic to the hippocampus (Tu et al. 2005).
Acute or chronic activation of the immune system, including immunotherapy, atherosclerosis and even HIV infection, enhance the enzymatic activity of IDO (Werner et al. 1988; Wirleitner et al. 2003). Several soluble factors, including IL-6, IL-1β, and TNFα and inducible nitric oxide synthase mRNAs, are significantly elevated in HIV-demented patients compared with the non-demented ones (Griffin 1997). In the CSF of macaques with simian acquired immune deficiency syndrome (SAIDS) an increase in the QUIN/KYNA ratio was observed (Heyes et al. 1990). Thus, analysis of the levels of IDO and its metabolites may potentially be used in conjunction with behavioral tests as an assay for HIV depression. IDO analysis may also contribute to development of therapeutics (Potula et al. 2005).
During the course of HIV infection, specific HIV proteins also play an important role in the cytokinergic mechanisms of HIV depression. The envelope glycoprotein gp120 is one of the viral antigens that induce an neuroinflammatory response during HIV CNS infection (Louboutin et al. 2010). In this response, TNF-α, IL-1β, IL- 6 and IFN-γ are expressed and may work in synergy with gp120-associated neurotoxic mechanisms (described below) (Conti et al. 2004; Gorse et al. 2006; Lee et al. 2005).
Transgenic mouse studies demonstrate that expression of gp120 on its own is sufficient to elevate TNF-α, although other viral antigens may contribute as well (Toggas et al. 1994). Modification of the effects of exogenous gp120 by growth factors has also been reported, for instance, microinjection of gp120 into striatum induced neuron toxicity, which could be blocked by expression of BDNF from a viral vector (Mocchetti et al. 2007). The neuroprotective activity of BDNF may result from its ability to down regulate the chemokine receptor CXCR4R, activated by gp120 (Nosheny et al. 2007).
In addition to mechanisms of depression dependent upon inflammatory cytokines, HIV proteins may also contribute to depression through direct induction of neuron death. Many studies have demonstrated the toxicity of gp120 for neurons (Mocchetti et al. 2012; Regulier et al. 2004). Working through multiple mechanisms, including elevation of reactive oxygen species, disruption of Ca2+ homeostasis and direct interaction with the CXCR4R, gp120 kills neurons (Mattson et al. 2005; Mocchetti et al. 2012). Gp120 may also overstimulate NMDA receptors by acting on astrocytes to produce nitric oxide synthase (iNOS), inhibiting Na+-dependent glutamate influx in astrocytes, releasing arachidonic acid from astrocytes, which then inhibits the reuptake of glutamate by neurons and astrocytes (Lipton 1994a; Patton et al. 2000). Indeed, administration of gp120 for 1-2 h to acute hippocampal slices in culture caused a decrease in long-term potentiation (LTP) dependent upon gp120 interaction with the CXCR4R (Dong and Xiong 2006). Significantly, a gp120 transgenic HIV mouse model demonstrated that expression of the viral peptide produces neurodegenerative changes compatible with abnormalities in brains of HIV infected patients (Toggas et al. 1994).
The gp120 protein preferentially attacks neurons of the dopaminergic system, which regulate fronto-striatal circuits (Agrawal et al. 2010; Bachis et al. 2010; Hu et al. 2009). As the fronto-striatal loops, involving the caudate, putamen (CP) and nucleus accumbens (NA), are direct regulators of motion, cognitive associations, motivated behavior and reward, disruption of their functions results in motor and cognitive impairments, lethargy, anhedonia and depressive mood, all of which are frequently experienced by HIV/AIDS patients (Ciesla and Roberts 2001; Gibbie et al. 2006). Autopsy and functional imaging studies show that HIV attacks basal ganglia, causing decreases in volume and function that result from neuron death (Aylward et al. 1993; Berger and Arendt 2000; Theodore et al. 2007). Furthermore, gp120 may induce the death of striatal dopamine neurons by downregulating neurotrophic factors (Nosheny et al. 2007).
The HIV-1 regulatory transactivator of transcription protein (Tat) is crucial for viral replication and like gp120 is also released by infected microglia and is neurotoxic as well (Li et al. 2009). Tat may enhance the neurotoxicity of the NMDA receptor (Haughey et al. 2001), induce Ca2+ dependent neuronal apoptosis (Bonavia et al. 2001; Kruman et al. 1998; Perez et al. 2001) and may disrupt Ca2+ homeostasis (Cheng et al. 1998) by altering L-type Ca2+ channel expression (Wayman et al. 2012). When coinjected, Tat and gp120 may synergize in the death of striatal neurons (Bansal et al. 2000).
Of special relevance is the finding that long-term CART itself might be neurotoxic and for this reason indirectly contribute to depression. Neurotoxicity of CART components varies considerably and depends on the substance involved and the levels of CNS penetration (Husstedt et al. 2009). Schweinsburg demonstrated that stavudine and didanosine are toxic to brain mitochondria (Schweinsburg et al. 2005). Zidovudine has been associated with hallucinations, psychosis, mania, and depression, which were thought to be side effects of monotherapy at the drug high doses administered in the past. Depression, hallucinations, and suicidal ideation have been reported in patients on abacavir therapy (Cespedes and Aberg 2006). Tovar-Y-Romo provide evidence that the main metabolite of efavirenz (EFV), 8-OH-EFV, is a neurotoxin that deregulates neuronal calcium homeostasis, and damages dendritic spines (Tovar-y-Romo et al. 2012). CART may also increase β-amyloid protein (Aβ) generation, by preventing microglial phagocytosis (the normal mechanism of Aβ clearance), and enhancing Aβ aggregation and deposition, which itself is neurotoxic (Giunta et al. 2011).
Nevertheless, drugs with good CNS penetration were found to be neuroprotective, as they reduced the risk of developing neurocognitive impairments (Ellis et al. 2007; McCutchan et al. 2007; Robertson et al. 2004). Indeed, CART may stabilize, or even improve cognitive function by reversing infection related dysfunctional brain changes, which have not yet induced structural damage. Decreased viral load induced by CART may reverse the IDO overactivation, decreasing kynurenine levels and the kyn/trp ratio (Zangerle et al. 2002).
Psychological, social and biographical factors interact with inflammatory and neurotoxic, virally induced changes to play a crucial role in depression amongst PLHIV. The relationships between psychosocial stressors and neuroimmunological and endocrine processes are complex and reciprocal (Lutgendorf et al. 1999; McDade et al. 2006). Their contribution to neuropsychiatric disorders is neither additive, nor mutually exclusive, but dynamically interacting and potentiating each other in as yet unclear ways (Anisman 2009; Gibb et al. 2008; Glaser et al. 2003). For example, exposure to stress is associated with an increase in IL-6 serum levels, which is more pronounced in patients who are already depressed (Kiecolt-Glaser et al. 2005; Pace et al. 2006).
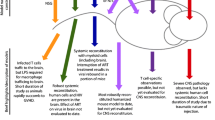
In animal models, external stress-induced, depression-like behaviors are associated with increased IL-1β, TNF-α, IL-6, nuclear factor κB, cyclooxygenase-2, expression of Toll-like receptors and lipid peroxidation (Kubera et al. 2011). These and other observations lead to the proposal that the brain translates psychosocial stress into immunological activation, but, on the other hand, by means of the reverse pathway, also translates immunological activation into perceived stress (Anisman 2009). HIV-related stigma, homophobia, racism, non-disclosure, social isolation, fear of premature death, uncertainty about the future, concerns about the appearance (the disease could become visible) have all been shown to be associated with higher depression rates (Simoni et al. 2011). Moreover, socially disadvantaged and marginalized people are over-represented amongst PLHIV (for example, the economically disadvantaged, ethnic minorities, sexual minorities, drug users, sex professionals) and they are at greater risk for depression even before contracting the disease (Gonzalez et al. 2009). For these reasons, depression among PLHIV should be understood as multifactorial, implying complex interactions between constitutional factors (for example, genetic polymorphisms), early-life experiences (which are related to epigenetic and neurodevelopmental changes) (Provencal et al. 2012), psychosocial circumstances and neurobiological, virally induced changes. Some of these elements, such as genetic polymorphisms, may not be changeable, but the management of the others could have a strong effect on the entire system, reducing vulnerability for development of the disorder. Despite several limitations for reproducing the complexity of the interplay of these factors, translational research and especially animal models may provide a great resource that deserves further exploration. Possible interactions between psychosocial stressors and HIV infection mediating depression are shown in Fig. 1. The evidence for contributions to HIV depression by the immuno-inflammatory, monoaminergic, neurotoxic and neurotrophic processes listed here has recently been reviewed (Del Guerra et al. 2013).

Hypothetical interactions of genetic, epigenetic, and psychosocial factors with HIV infection resulting in depression. HIV associated depression may result from complex interactions between genetic polymorphisms (not shown in the figure), early life events that lead to neurodevelopmental and epigenetic changes, and exposure to psychosocial stressors. The latter can either be related to the burden of the infection (e. g. having a chronic disease, disclosure, treatment side effects, etc.), or be unrelated to it, in as much as marginalized groups are overrepresented among PLHIV and they are more exposed to stress and more susceptible to depression than the general population (Gonzalez et al. 2009). The virus induces immuno-inflammatory, monoaminergic, neurotoxic and neurotrophic changes, which contribute to depressive symptoms and neurodegeneration (Del Guerra et al. 2013). We hypothesize a synergistic interaction between HIV related neurobiological mechanisms and genetic (not shown in the figure), biographical and psychosocial factors, leading to depression, which could be investigated with animal models
Current animal models for depression
Animal models have contributed to the general study of depression, although reproducing a disease as complex as depression is a challenge (Krishnan and Nestler 2011). For a neuropsychiatric disease animal model to be valid, the model should demonstrate three characteristics: 1) reproducing the features of the disease (face validity); 2) reproducing pathophysiological aspects of the disease (etiological, or construct validity); and also, 3) having its features reversed by the disease therapeutic agents (pharmacological, or predictive validity) (Willner 1986; Willner and Mitchell 2002). However, certain deficiencies are associated with each criterion (Nestler and Hyman 2010). To achieve significant face validity, an animal model of depression must present depressive symptoms equivalent to those found in humans. While symptoms such as guilt, suicidal ideation and sadness are likely to be purely human features, other aspects of the depressive syndrome have been replicated in laboratory rodent models, such as, anhedonia, irritability, cognitive impairments, and neurovegetative symptoms, such as abnormalities in appetite and sleep (Belzung and Lemoine 2011).
Construct validity requires that the model delineate pathophysiological mechanisms of the human disease. Considering that the etiology and pathophysiology of depression have not yet been elucidated, it is difficult for models of depression to fulfill this criterion. However, incorporation of genetic and environmental factors into animal models will increase model construct validity (Schmidt et al. 2011b; Willner 1986). In contrast to construct and face validity, predictive validity is often excellent. Indeed, many models respond well to drugs employed for the treatment of depression, thereby assisting disease evaluation (Berton and Nestler 2006; Willner 1986).
A potential emerging fourth criterion, pathological validity, requires that animal models recapitulate known postmortem pathological or serological changes found in human patients (Krishnan and Nestler 2011).
Mouse models to assess depression through acute stress
Standardized tests may be employed to assess symptoms of depression, and to analyze therapeutic performance of antidepressant treatment. Among these are:
-
1)
Sucrose preference test: mice consume water or a solution of sucrose from two test bottles. Preference is calculated as the percentage of sucrose consumed compared to total fluid intake. The consumption of sucrose decreases in depressed mice, due to anhedonia (Monleon et al. 1995; Nielsen et al. 2000; Willner et al. 1987).
-
2)
Tail suspension test: the mouse is suspended by the tail from a lever and the movements of the animal are recorded. The total duration of the test is divided into periods of agitation and immobility. Immobility is scored as a measure of one aspect of depression (despair associated with inescapable stress). Antidepressant drugs decrease the duration of immobility (Krishnan and Nestler 2011; Cryan et al. 2005).
-
3)
Forced swim test: this test is used to evaluate antidepressant action and also to infer depression-like behavior. Mice are placed in a cylindrical plastic container that is partially filled with water. Following an initial period of struggling, swimming and climbing, the animal eventually displays a floating or immobile posture indicating depression (Krishnan and Nestler 2011; Petit-Demouliere et al. 2005).
-
4)
The learned helplessness model: After exposure to inescapable electric shocks, the animals develop a state of helplessness. When re-exposed to the same shocks, the animal does not try to escape even when provided with an easy escape route, indicating depression (Chourbaji et al. 2005; Krishnan and Nestler 2011)
Generating animal models of depression through chronic and early life stress
In the absence of known genetic factors, many models make use of stress and emotional losses as a means to develop depressive symptoms (Nestler and Hyman 2010).
Several chronic stress procedures have been employed that attempt to achieve a measure of construct validity (Nestler and Hyman 2010). In the case of chronic mild or chronic unpredictable stress (CMS), normal rats or mice are exposed, over a period of weeks, to a series of repeated physical stressors, for example, restraint, foot shock or cold temperature. The resulting depressive symptoms are tracked by the sucrose preference test. The model shows good predictive validity in that behavioral changes are reversed by treatment with a wide variety of antidepressants. It also provides face validity in that almost all demonstrable symptoms of depression are observed. For instance, CMS causes a generalized decrease in responsiveness to rewards, comparable to anhedonia. Drawbacks include the difficulties of reproducibility and the long time required to administer the procedure (Willner 1997).
The CSD model also uses stress to reproduce depressive symptoms. In the male tree shrew test, two adult males are housed together in one cage, leading them to fight and establish a social hierarchy with a dominant and a subordinate. With CSD, a mouse is forced to intrude into the territorial space of a larger mouse of a more aggressive genetic strain. This leads to antagonistic encounters that ultimately subordinate the intruder. With CSD, the rodents show a range of depression-like symptoms, including anhedonia and social withdrawal as observed in depressed human patients (Fuchs and Flugge 2002; Krishnan et al. 2007). Besides use for the study of depression, this model can be employed to study resilience, since some animals, even when subjected to high levels of stress, do not develop the characteristic clinical depression symptoms.
Also included among the stress-induced rodent models is early life stress, such as maternal separation, which induces long-life behavioral and neuroendocrine abnormalities, some of which can be reversed by antidepressant medications (Schmidt et al. 2011b).
Social stress effects on animal’s brain neurotransmitter systems have been extensively investigated, and are most often associated with changes in serotonergic and noradrenergic systems. Morphological changes and alterations of neurogenesis and of cell survival, particularly ones involving the hippocampus and dentate gyrus, have been reported with severe social stress, as have longer-term changes in functioning of the HPA axis (Blanchard et al. 2001).
Animal models for HIV
Animal models for HIV infection have provided a controlled setting for many HIV studies (McCune 1997). One logical beginning for such a model would be chimpanzees, given their genetic similarity to humans. Nonetheless, financial considerations, differences in immune responses as well as societal concerns and other practical factors hinder their use (Van Duyne et al. 2009). HIV-1 rodent models in contrast offer lower cost, ease of handling and housing, short lifetime, and a genome that may be manipulated (Gorantla et al. 2012). Rodent models may be created by several means, including direct injection of HIV proteins either into the CNS or blood via intravenous (IV) route in rats or mice, expression of viral transgenes in rodents and transplantation of HIV-1 infected cells into immunodeficient rodent tissues to overcome HIV-1’s failure to infect rodent cells. However, the use of humanized mice, which are genetically modified to accept human immune system grafts and support HIV infection, presents the best opportunities for a model (Van Duyne et al. 2009). In light of the relevance of HIV associated CNS disorders, animal models for CNS HIV infection may have special importance.
Viral protein and transgenic models
In the early efforts to reproduce the effects of viral infection, purified viral proteins were applied to neuron cultures or injected into the relevant regions of rodent brains. Such studies have demonstrated the toxicity of Tat and other HIV proteins (Agrawal et al. 2012; Chen et al. 1997; Conant et al. 1998; New et al. 1998; Shi et al. 1998; Yao and Buch 2012). In a second approach, transgenic models, HIV genes or genome fragments are inserted directly into the genome of a mouse, leading to an animal expressing one or more viral proteins. Like protein injection models, transgenic models offer the possibility of deducing the effect of individual viral products (Nath and Geiger 1998) (Agrawal et al. 2012; Carey et al. 2012; Toneatto et al. 1999) (Krucker et al. 1998; Lipton 1994b; Toggas et al. 1994) (Ballester et al. 2012). However no virus is produced and viral transgene expression will vary for each mouse line produced. The basic transgenic models may also expose the nervous system to transgenic viral proteins throughout the animal’s lifespan. They may modify development and fail to recapitulate the actual events in HIV-1 induced neurological disorders (Jaeger and Nath 2012). The use of doxycycline-dependent gene promoters can provide regulated, brain-targeted expression of HIV-1 Tat and other viral proteins (Kim et al. 2003; Zucchini et al. 2013). Nonetheless, protein injection and transgenic mouse models are severely limited in their ability to mimic the pathological bases of the human disease. Some immunodeficient animal models employing viral infection or transplantation of infected cells can mimic the early events in viral replication (Van Duyne et al. 2009), but they neither sustain production of virus nor reproduce the natural onset and progression of HIV neuropathogenesis nor peripheral viral replication nor the host immune response. (Gorantla et al. 2012).
Humanized mice
An optimal rodent model would provide relevant host cells for viral infection, including CD4+ T lymphocytes, dendritic cells, monocytes and macrophages, all of which must possess the receptors and co-receptors and cellular machinery required to complete the viral life cycle. The model must have the capacity to create reservoirs of virus, such as seen in the human disease, and the model must modify the permeability of the BBB to enable entry of infected cells into the CNS. The model must also simulate the chronicity of the disease and exhibit an immune response similar to that observed in humans (Gorantla et al. 2012). For these purposes, humanized mice have proved, so far, the best alternative to transgenic models. Briefly, humanized mice are genetically modified, immunodeficient mice that accept human cell grafts, in this case, cells of the immune system (Jaeger and Nath 2012)..
Recently, a mouse model defective in the common γ chain (γc) of the receptors for IL-2, IL-7, IL-15 and other cytokines, was constructed from the recombinase activating gene (Rag) knockout mouse (Ishikawa et al. 2005; Shultz et al. 2005; Traggiai et al. 2004) and the non-obese diabetic (NOD) and “severe combined immunodeficiency” (SCID) mutation in CB-17 scid/scid mice (Bosma et al. 1983). These Rag-/-γc -/- and NOD-SCID γc null (NOG) mice have no functional T, B, or NK cell activity, thus being superior to previous models (Ito et al. 2002; Van Duyne et al. 2009). Due to the presence of an intact human immune system and the ability to support multilineage hematopoiesis, the humanized Rag2-/-γc -/- scaffold provides an excellent system to study HIV pathogenesis and can produce sustained HIV-1 infection (Berges et al. 2006) Infection of Rag2-/-γc -/- animals, 10–28 weeks of age, with CCR5-tropic YU-2 or CXCR4-tropic NL4-3 HIV-1 viral strains, produced a chronic infection lasting up to 190 days, as well as an initial, acute burst phase of viral replication, as detected by assay of plasma viral RNA. At 18 weeks post infection, spleen and lymph nodes of both NL4-3- and YU-2-infected animals showed biomarkers of infection (Baenziger et al. 2006). Using humanized mice, Zou and colleagues concluded that Nef elevates viral replication and leads to CD4+ T cell killing. Furthermore, CD4+CD8+ thymocyte killing was dependent on Nef. This depletion of thymic T cell precursors can be a relevant factor in the elevated pathogenicity of CXCR4 trophic HIV-1 (Zou et al. 2012).
Validation and application of the models
Because virus-specific mechanisms may modulate neuroinflammatory and neurodegenerative processes that lead to depression, in order to establish an optimal rodent model, one must strive to reproduce the complex interaction between the virus, the immune system and the brain faithfully as the immune response to the infection progresses, and cytokines and viral antigens alter brain and immune system function. In this respect, humanized mouse models are most appealing for the study of HIV depression because, of the available strategies, they most faithfully recapitulate the interaction of the virus with the immune system (Gorantla et al. 2012). However, in addition to asking if a model reproduces virus-immune system interaction adequately, any such model should also be evaluated from a behavioral perspective so that its utility for modeling comorbidities of great clinical interest, such as depression, could be assessed. One limitation is that rodent models cannot reproduce emotional and consciousness-based components of depression that are found in humans, such as low self-esteem (LeDoux 2012). Nonetheless, rodent models may be applied to studies of the behavioral manifestations of depression, such as anhedonia, and to the underlying inflammatory and neurodegenerative mechanisms.
Attempts in this direction have been limited, but a number of strategies are possible. First, the assumption that HIV associated depression is, at least partially, induced by the virally related neuroinflammatory and neurotoxic mechanisms could be tested by evaluating depression among HIV-1-infected humanized mice using standardized behavioral measurements. However, to evaluate the quality and adequacy of animal models for HIV depression, we may not just ask if the animals actually get depressed. We may also ask about the influence of psychosocial factors on the course of HIV mediated brain pathology. Humanized mice infected with HIV could be exposed to various depression-inducing influences, such as social conflict using the male tree shrew model adapted for mice, or to learned helplessness protocols or to other forms of stress such as early life stress (maternal separation), to see if they develop depression more rapidly, or if the depression-like symptoms are more pronounced or longer lasting than in control animals.
A further possibility is to assess the correlation of depressive behavioral measurements with endophenotypes, such as deregulation of the HPA axis, production of inflammatory cytokines and of toxic metabolites, including kynurenic and quinolinic acids and peroxynitrites. The latter, together with viral proteins, gp120 and Tat, may be toxic to dopaminergic neurons, leading to depression, or toxic to cortical neurons, leading to memory loss and cognitive decline. Mouse models may be assayed for degeneration of basal ganglia, resulting in loss of hedonic function, and of prefrontal cortex, diminishing executive function, and of cerebral cortex and hippocampus, impairing memory formation and retrieval. Decreases in dopamine and serotonin release may be measured. Changes in the levels of inflammatory cytokines in serum and in CSF could also be correlated with behavioral measurements, monoaminergic dysfunction and with expression or levels of neurotrophic factors in different brain areas.
Models for HIV depression may be employed to address clinically relevant questions. mRNA biomarkers of gene expression could be identified by micro array comparison of rodent model mRNA. Models could be used to screen for protein biomarkers for depression in serum and cerebrospinal fluid. Caution should be taken since not all such biomarkers may be found in humans or have the same significance. These biomarkers could be evaluated for diagnostic validity with the human disease by assaying the protein biomarkers in HIV patient CSF and serum (e.g. cytokines, BDNF), and by assaying the mRNA biomarkers in patient lymphocytes or, if a brain bank and information about the psychiatric status of the patient are available, in autopsy brain tissue. Furthermore, one could assay whether biomarkers identified in humans with major depressive disorder (Schmidt et al. 2011a) are also elevated in the mouse models for HIV depression, and determine if their levels change with CART or antidepressants. These could be clues to mechanisms of depression. Biomarkers identified in MDD patients could also be related to AIDS patient depressive state as assessed by psychiatric clinic evaluation and analyzed mechanistically in rodents.
Rodent models for HIV depression may be applied to test antidepressant and antiretroviral therapies. The efficacy of antidepressants could be evaluated through behavioral tests for despair (Cryan et al. 2005; Petit-Demouliere et al. 2005), anhedonia and hypophagia (Nielsen et al. 2000) and anxiety (Dulawa and Hen 2005; Holmes 2001). The animal models can also be used to determine if an HIV animal model of depression shows a similar response to treatment as other depression animal models. To test these ideas, one could see if the HIV rodent responses to conventional antidepressants, such as tricyclic antidepressants or selective serotonin reuptake inhibitors, are poorer than the responses of controls, and to determine if treatment with potential HIV depression-specific agents, such as antibodies anti-TNF-α, or anti-gp120, improves depression preferentially in the infected animals versus controls. New mood stabilizers or combinations of existing drugs for mood elevation may be evaluated. The models could also be used to determine whether cocaine, amphetamines and antidepressants that elevate dopamine accelerate the infection, as has been suggested (Del Guerra et al. 2013).
The influence of psychosocial factors on the levels of cytokines and other protein biomarkers in the cerebral spinal fluid, or mRNA biomarkers could be monitored. Similar measurements could be made of the effects of psychosocial influences on viral load and CD4 levels in the infected humanized mice. Effects on hippocampal function and learning behavior and molecular markers for neuroplasticity could also be evaluated. For instance, it is already known that BDNF is decreased in HIV infection (Nosheny et al. 2007), and, as described above, is also altered during depression. BDNF enhances the neuron life cycle, is neuroprotective and stimulates neurogenesis in the hippocampus. The association between memory impairment, which is related to neurogenesis, and BDNF levels could be evaluated. By these means, the relationship between depressive behavior and cognitive deficits could be investigated, as it could be assumed that the chronic neuroinflammatory and neurodegenerative virally induced changes lead to both disorders, probably in different stages. Furthermore depression might be itself neurotoxic and further studies could evaluate if psychosocial factors could synergize with viral factors in elevating the extent of cognitive deficit in HIV depression and the effects of HIV on the capacity for neuroplasticity.
Finally, but perhaps most significant, mouse models offer the very powerful opportunity to take advantage of mouse genetics to determine the contributions of specific genes and pathways to the origin of HIV depression. Mouse genetic models have been established to test the roles of monoamines, CRF and BDNF in depression mechanisms (Barkus 2013) and potentially could be applied to the study of HIV depression. Specialized humanized mouse lines with genetic modifications for testing genes, pathways etc. for a role in HIV depression could be constructed. For example, it should be possible to construct model mice with doxycycline inducible CRE recombinase in T cell lineages. By administering doxycycline at different stages of infection, it would be possible to knock out floxed viral and cellular genes. One could determine if replication and new infection are halted if a critical viral gene is excised and if so, whether depression is impeded. In addition, using CRE excision, one could construct a mouse model in which a chemokine receptor gene can be selectively knocked out in a specific T cell lineage. Does this protect that lineage from HIV toxicity and does it protect the mouse from cognitive losses and depression? The models could be used to determine how changes in mouse genetic background affect the treatment of HIV depression. Specific genetic backgrounds in humans have been linked to adverse effects of antidepressants (Lin and Chen 2008). Using the models it would be possible to determine if they have the same effects in mice, and if they do, the system might also be use to understand the basis.
Conclusions and future perspectives
Despite the many advances in the treatment of HIV infection, chronic neuropsychiatric HIV associated disorders, such as depression and HAND, are still a challenge. The underlying pathophysiological mechanisms are not well understood, and there are no effective pathophysiology based treatments. Thus, there is an unmet need for animal models that enable the study of these particularly important comorbities and the testing of new therapeutic approaches to combat them.
The many animal models generated so far have provided numerous findings about HIV disease mechanisms and have contributed significantly to understanding the action of HIV-1 in the CNS. However, little of the information gained so far has been directed to the study of depression. To search for appropriate therapies for treating patients with depressive comorbidities, the construction of an animal model suitable for such translational research becomes important.
Because HIV is a chronic neuroinflammatory disease, HIV associated depression fits very well with the inflammatory-neurodegenerative theory of depression. Furthermore, it would be very important to understand the neuroimmunological process related to HIV infection, but most models so far have studied the systemic immunological process, rather than processes that transpire within the nervous system.
An appropriate model should enable investigation of the persistence of HIV replication in brain microglia, evolution of highly neurovirulent CNS HIV strains, and even long-term CNS toxicity of CART. Such a model could be used to investigate inflammatory biomarkers in CSF related to depression and cognitive decline. Indeed, cerebrospinal fluid (CSF) markers of immune activation and inflammation are commonly detected in individuals with HAND (Gannon et al. 2011) and depression (Zorrilla et al. 2001). By reviewing CSF factors that are related with HIV-1 infection and depression such as neopterin, β-2-microglobulin, PGE2, (QUIN), 1 (MCP-1), IFNs, ILs and other chemokines, it will be possible to distinguish biomarkers that could be useful in clinical practice for diagnosis and treatment follow up. Behavioral evaluations carried out by means of these models may provide access to the origins of depressive symptoms and the steps necessary for the construction of an ideal model. Modulation of these biomarkers by the model could be one test of the model’s validity.
References
Agrawal L, Louboutin JP, Marusich E, Reyes BA, Van Bockstaele EJ, Strayer DS (2010) Dopaminergic neurotoxicity of HIV-1 gp120: reactive oxygen species as signaling intermediates. Brain Res 1306:116–130
Agrawal L, Louboutin JP, Reyes BA, Van Bockstaele EJ, Strayer DS (2012) HIV-1 Tat neurotoxicity: a model of acute and chronic exposure, and neuroprotection by gene delivery of antioxidant enzymes. Neurobiol Dis 45:657–670
Anisman H (2009) Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci: JPN 34:4–20
Aylward EH, Henderer JD, McArthur JC, Brettschneider PD, Harris GJ, Barta PE, Pearlson GD (1993) Reduced basal ganglia volume in HIV-1-associated dementia: results from quantitative neuroimaging. Neurology 43:2099–2104
Bachis A, Cruz MI, Mocchetti I (2010) M-tropic HIV envelope protein gp120 exhibits a different neuropathological profile than T-tropic gp120 in rat striatum. Eur J Neurosci 32:570–578
Baenziger S, Tussiwand R, Schlaepfer E, Mazzucchelli L, Heikenwalder M, Kurrer MO, Behnke S, Frey J, Oxenius A, Joller H, Aguzzi A, Manz MG, Speck RF (2006) Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2-/-gamma c-/- mice. Proc Natl Acad Sci 103(43):15951–15956
Ballester LY, Capo-Velez CM, Garcia-Beltran WF, Ramos FM, Vazquez-Rosa E, Rios R, Mercado JR, Melendez RI, Lasalde-Dominicci JA (2012) Up-regulation of the neuronal nicotinic receptor alpha7 by HIV glycoprotein 120: potential implications for HIV-associated neurocognitive disorder. J Biol Chem 287:3079–3086
Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM (2000) Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res 879:42–49
Barkus C (2013) Genetic mouse models of depression. Curr Top Behav Neurosci 14:55–78
Belzung C, Lemoine M (2011) Criteria of validity for animal models of psychiatric disorders: focus on anxiety disorders and depression. Biol Mood Anxiety Disord 1:9
Berg BM, Godbout JP, Kelley KW, Johnson RW (2004) Alpha-tocopherol attenuates lipopolysaccharide-induced sickness behavior in mice. Brain Behav Immun 18:149–157
Berger JR, Arendt G (2000) HIV dementia: the role of the basal ganglia and dopaminergic systems. J Psychopharmacol 14:214–221
Berges BK, Wheat WH, Palmer BE, Connick E, Akkina R (2006) HIV-1 infection and CD4 T cell depletion in the humanized Rag2-/-gamma c-/- (RAG-hu) mouse model. Retrovirology 3:76
Bernardino L, Xapelli S, Silva AP, Jakobsen B, Poulsen FR, Oliveira CR, Vezzani A, Malva JO, Zimmer J (2005) Modulator effects of interleukin-1beta and tumor necrosis factor-alpha on AMPA-induced excitotoxicity in mouse organotypic hippocampal slice cultures. J Neurosci : Off J Soc Neurosci 25:6734–6744
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M et al (2006) Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311:864–868
Berton O, Nestler EJ (2006) New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci 7:137–151
Biesmans S, Meert TF, Bouwknecht JA, Acton PD, Davoodi N, De Haes P, Kuijlaars J, Langlois X, Matthews LJ, Ver Donck L et al (2013) Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediat Inflamm 2013:271359
Blanchard RJ, McKittrick CR, Blanchard DC (2001) Animal models of social stress: effects on behavior and brain neurochemical systems. Physiol Behav 73:261–271
Boisse L, Gill MJ, Power C (2008) HIV infection of the central nervous system: clinical features and neuropathogenesis. Neurol Clin 26:799–819
Bonavia R, Bajetto A, Barbero S, Albini A, Noonan DM, Schettini G (2001) HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Biophys Res Commun 288:301–308
Bosma GC, Custer RP, Bosma MJ (1983) A severe combined immunodeficiency mutation in the mouse. Nature 301:527–530
Capuron L, Miller AH (2004) Cytokines and psychopathology: lessons from interferon-alpha. Biol Psychiatry 56:819–824
Capuron L, Raison CL, Musselman DL, Lawson DH, Nemeroff CB, Miller AH (2003) Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry 160:1342–1345
Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R (2002) Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry 7:468–473
Carey AN, Sypek EI, Singh HD, Kaufman MJ, McLaughlin JP (2012) Expression of HIV-Tat protein is associated with learning and memory deficits in the mouse. Behav Brain Res 229:48–56
Cartier L, Hartley O, Dubois-Dauphin M, Krause KH (2005) Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Rev 48:16–42
Cespedes MS, Aberg JA (2006) Neuropsychiatric complications of antiretroviral therapy. Drug Saf : An Int J Med Toxic Drug Experience 29:865–874
Chen P, Mayne M, Power C, Nath A (1997) The Tat protein of HIV-1 induces tumor necrosis factor-alpha production. Implications for HIV-1-associated neurological diseases. J Biol Chem 272:22385–22388
Cheng J, Nath A, Knudsen B, Hochman S, Geiger JD, Ma M, Magnuson DS (1998) Neuronal excitatory properties of human immunodeficiency virus type 1 Tat protein. Neuroscience 82:97–106
Chourbaji S, Zacher C, Sanchis-Segura C, Dormann C, Vollmayr B, Gass P (2005) Learned helplessness: validity and reliability of depressive-like states in mice. Brain Res Protoc 16:70–78
Christmas DM, Potokar J, Davies SJ (2011) A biological pathway linking inflammation and depression: activation of indoleamine 2,3-dioxygenase. Neuropsychiatr Dis Treat 7:431–439
Christoffel DJ, Golden SA, Russo SJ (2011) Structural and synaptic plasticity in stress-related disorders. Rev Neurosci 22:535–549
Churchill M, Sterjovski J, Gray L, Cowley D, Chatfield C, Learmont J, Sullivan JS, Crowe SM, Mills J, Brew BJ et al (2004) Longitudinal analysis of nef/long terminal repeat-deleted HIV-1 in blood and cerebrospinal fluid of a long-term survivor who developed HIV-associated dementia. J Infect Dis 190:2181–2186
Churchill MJ, Rhodes DI, Learmont JC, Sullivan JS, Wesselingh SL, Cooke IR, Deacon NJ, Gorry PR (2006) Longitudinal analysis of human immunodeficiency virus type 1 nef/long terminal repeat sequences in a cohort of long-term survivors infected from a single source. J Virol 80:1047–1052
Ciesla JA, Roberts JE (2001) Meta-analysis of the relationship between HIV infection and risk for depressive disorders. Am J Psychiatry 158:725–730
Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO (1998) Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci U S A 95:3117–3121
Conti L, Fantuzzi L, Del Corno M, Belardelli F, Gessani S (2004) Immunomodulatory effects of the HIV-1 gp120 protein on antigen presenting cells: implications for AIDS pathogenesis. Immunobiology 209:99–115
Cryan JF, Mombereau C, Vassout A (2005) The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev 29:571–625
Dantzer R (2001) Cytokine-induced sickness behavior: mechanisms and implications. Ann N Y Acad Sci 933:222–234
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56
Del Guerra FB, Fonseca JLI, Figueiredo VM, Ziff EB, Castelon Konkiewitz E (2013) Human immunodeficiency virus associated depression: contributions of immuno-inflammatory Monoaminergic, Neurodegenerative and Neurotrophic pathways. J Neurovirol
Dong J, Xiong H (2006) Human immunodeficiency virus type 1 gp120 inhibits long-term potentiation via chemokine receptor CXCR4 in rat hippocampal slices. J Neurosci Res 83:489–496
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctot KL (2010) A meta-analysis of cytokines in major depression. Biol Psychiatry 67:446–457
Dulawa SC, Hen R (2005) Recent advances in animal models of chronic antidepressant effects: the novelty-induced hypophagia test. Neurosci Biobehav Rev 29:771–783
Duman RS (2009) Neuronal damage and protection in the pathophysiology and treatment of psychiatric illness: stress and depression. Dialogues Clin Neurosci 11:239–255
Ellis R, Langford D, Masliah E (2007) HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci 8:33–44
Frenois F, Moreau M, O’Connor J, Lawson M, Micon C, Lestage J, Kelley KW, Dantzer R, Castanon N (2007) Lipopolysaccharide induces delayed FosB/DeltaFosB immunostaining within the mouse extended amygdala, hippocampus and hypothalamus, that parallel the expression of depressive-like behavior. Psychoneuroendocrinology 32:516–531
Fu X, Zunich SM, O’Connor JC, Kavelaars A, Dantzer R, Kelley KW (2010) Central administration of lipopolysaccharide induces depressive-like behavior in vivo and activates brain indoleamine 2,3 dioxygenase in murine organotypic hippocampal slice cultures. J Neuroinflammation 7:43
Fuchs E, Flugge G (2002) Social stress in tree shrews: effects on physiology, brain function, and behavior of subordinate individuals. Pharmacol, Biochem Behav 73:247–258
Fulk LJ, Kane BE, Phillips KD, Bopp CM, Hand GA (2004) Depression in HIV-infected patients: allopathic, complementary, and alternative treatments. J Psychosom Res 57:339–351
Gannon P, Khan MZ, Kolson DL (2011) Current understanding of HIV-associated neurocognitive disorders pathogenesis. Curr Opin Neurol 24:275–283
Gartner S (2000) HIV infection and dementia. Science 287:602–604
Gibb J, Hayley S, Gandhi R, Poulter MO, Anisman H (2008) Synergistic and additive actions of a psychosocial stressor and endotoxin challenge: Circulating and brain cytokines, plasma corticosterone and behavioral changes in mice. Brain, Behavior, and Immunity 22:573–589
Gibbie T, Mijch A, Ellen S, Hoy J, Hutchison C, Wright E, Chua P, Judd F (2006) Depression and neurocognitive performance in individuals with HIV/AIDS: 2-year follow-up. HIV Med 7:112–121
Giunta B, Ehrhart J, Obregon DF, Lam L, Le L, Jin J, Fernandez F, Tan J, Shytle RD (2011) Antiretroviral medications disrupt microglial phagocytosis of beta-amyloid and increase its production by neurons: implications for HIV-associated neurocognitive disorders. Mol Brain 4:23
Glaser R, Robles TF, Sheridan J, Malarkey WB, Kiecolt-Glaser JK (2003) Mild depressive symptoms are associated with amplified and prolonged inflammatory responses after influenza virus vaccination in older adults. Arch Gen Psychiatry 60:1009–1014
Godbout JP, Moreau M, Lestage J, Chen J, Sparkman NL, O’Connor J, Castanon N, Kelley KW, Dantzer R, Johnson RW (2008) Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharm : Off Publ Am Coll Neuropsychopharm 33:2341–2351
Gonzalez JS, Hendriksen ES, Collins EM, Duran RE, Safren SA (2009) Latinos and HIV/AIDS: examining factors related to disparity and identifying opportunities for psychosocial intervention research. AIDS Behav 13:582–602
Gorantla S, Poluektova L, Gendelman HE (2012) Rodent models for HIV-associated neurocognitive disorders. Trends Neurosci 35:197–208
Gorse GJ, Simionescu RE, Patel GB (2006) Cellular immune responses in asymptomatic human immunodeficiency virus type 1 (HIV-1) infection and effects of vaccination with recombinant envelope glycoprotein of HIV-1. Clin Vaccine Immunol : CVI 13:26–32
Gourley SL, Kiraly DD, Howell JL, Olausson P, Taylor JR (2008) Acute hippocampal brain-derived neurotrophic factor restores motivational and forced swim performance after corticosterone. Biol Psychiatry 64:884–890
Gras G, Kaul M (2010) Molecular mechanisms of neuroinvasion by monocytes-macrophages in HIV-1 infection. Retrovirology 7:30
Griffin DE (1997) Cytokines in the brain during viral infection: clues to HIV-associated dementia. J Clin Investig 100:2948–2951
Hart BL (1988) Biological basis of the behavior of sick animals. Neurosci Biobehav Rev 12:123–137
Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD (2001) HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem 78:457–467
Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP et al (2011) HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 17:3–16
Heaton RK, Grant I, Butters N, White DA, Kirson D, Atkinson JH, McCutchan JA, Taylor MJ, Kelly MD, Ellis RJ et al (1995) The HNRC 500–neuropsychology of HIV infection at different disease stages. HIV Neurobehavioral research center. J Int Neuropsychol Soc : JINS 1:231–251
Heyes MP, Mefford IN, Quearry BJ, Dedhia M, Lackner A (1990) Increased ratio of quinolinic acid to kynurenic acid in cerebrospinal fluid of D retrovirus-infected rhesus macaques: relationship to clinical and viral status. Ann Neurol 27:666–675
Hill L, Lee KC (2013) Pharmacotherapy considerations in patients with HIV and psychiatric disorders: focus on antidepressants and antipsychotics. Ann Pharmacotherapy 47:75–89
Hirata F, Hayaishi O, Tokuyama T, Seno S (1974) In vitro and in vivo formation of two new metabolites of melatonin. J Biol Chem 249:1311–1313
Holmes A (2001) Targeted gene mutation approaches to the study of anxiety-like behavior in mice. Neurosci Biobehav Rev 25:261–273
Hu S, Sheng WS, Lokensgard JR, Peterson PK, Rock RB (2009) Preferential sensitivity of human dopaminergic neurons to gp120-induced oxidative damage. J Neurovirol 15:401–410
Hult B, Chana G, Masliah E, Everall I (2008) Neurobiology of HIV. Int Rev Psychiatry 20:3–13
Husstedt IW, Reichelt D, Neuen-Jakob E, Hahn K, Kastner F, von Einsiedel R, Vielhaber B, Arendt G, EversS (2009) Highly active antiretroviral therapy of neuro-AIDS. Side effects on the nervous system and interactions. Der Nervenarzt 80:1133-1134, 1136-1138, 1140-1132
Ichiyama A, Nakamura S, Nishizuka Y, Hayaishi O (1970) Enzymic studies on the biosynthesis of serotonin in mammalian brain. J Biol Chem 245:1699–1709
Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, Watanabe T, Akashi K, Shultz LD, Harada M (2005) Development of functional human blood and immune systems in NOD/SCID/IL2 receptor gamma chain(null) mice. Blood 106:1565–1573
Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K et al (2002) NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 100:3175–3182
Jaeger LB, Nath A (2012) Modeling HIV-associated neurocognitive disorders in mice: new approaches in the changing face of HIV neuropathogenesis. Dis Mod Mech 5:313–322
Kaul M (2009) HIV-1 associated dementia: update on pathological mechanisms and therapeutic approaches. Curr Opin Neurol 22:315–320
Kaul M, Ma Q, Medders KE, Desai MK, Lipton SA (2007) HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ 14:296–305
Khin NA, Chen YF, Yang Y, Yang P, Laughren TP (2011) Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of new drug applications. J Clin Psychiatry 72:464–472
Kiecolt-Glaser JK, Loving TJ, Stowell JR, Malarkey WB, Lemeshow S, Dickinson SL, Glaser R (2005) Hostile marital interactions, proinflammatory cytokine production, and wound healing. Arch Gen Psychiatry 62:1377–1384
Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ (2003) Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol 162:1693–1707
Kirsch I, Deacon BJ, Huedo-Medina TB, Scoboria A, Moore TJ, Johnson BT (2008) Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med 5:e45
Kopnisky KL, Bao J, Lin YW (2007) Neurobiology of HIV, psychiatric and substance abuse comorbidity research: workshop report. Brain, Behavior, and Immunity 21:428–441
Kraft-Terry SD, Stothert AR, Buch S, Gendelman HE (2010) HIV-1 neuroimmunity in the era of antiretroviral therapy. Neurobiol Dis 37:542–548
Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R (2005) Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res 111:194–213
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC et al (2007) Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131:391–404
Krishnan V, Nestler EJ (2011) Animal models of depression: molecular perspectives. Curr Top Behav Neurosci 7:121–147
Krucker T, Toggas SM, Mucke L, Siggins GR (1998) Transgenic mice with cerebral expression of human immunodeficiency virus type-1 coat protein gp120 show divergent changes in short- and long-term potentiation in CA1 hippocampus. Neuroscience 83:691–700
Kruman II, Nath A, Mattson MP (1998) HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol 154:276–288
Kubera M, Obuchowicz E, Goehler L, Brzeszcz J, Maes M (2011) In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Prog Neuro-Psychopharmacol Biol Psychiatry 35:744–759
LeDoux J (2012) Rethinking the emotional brain. Neuron 73:653–676
Lee C, Tomkowicz B, Freedman BD, Collman RG (2005) HIV-1 gp120-induced TNF-{alpha} production by primary human macrophages is mediated by phosphatidylinositol-3 (PI-3) kinase and mitogen-activated protein (MAP) kinase pathways. J Leukocyte Biol 78:1016–1023
Li W, Li G, Steiner J, Nath A (2009) Role of Tat protein in HIV neuropathogenesis. Neurotoxicity Res 16:205–220
Lin E, Chen PS (2008) Pharmacogenomics with antidepressants in the STAR*D study. Pharmacogenomics 9:935–946
Lindl KA, Marks DR, Kolson DL, Jordan-Sciutto KL (2010) HIV-associated neurocognitive disorder: pathogenesis and therapeutic opportunities. J Neuroimmune Pharm : Off J Soc NeuroImmune Pharm 5:294–309
Lipton SA (1994a) AIDS-related dementia and calcium homeostasis. Ann N Y Acad Sci 747:205–224
Lipton SA (1994b) HIV-related neuronal injury. Potential therapeutic intervention with calcium channel antagonists and NMDA antagonists. Mol Neurobiol 8:181–196
Liu Y, Ho RC, Mak A (2012) Interleukin (IL)-6, tumour necrosis factor alpha (TNF-alpha) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: a meta-analysis and meta-regression. J Affect Disord 139:230–239
Louboutin JP, Reyes BA, Agrawal L, Van Bockstaele EJ, Strayer DS (2010) HIV-1 gp120-induced neuroinflammation: relationship to neuron loss and protection by rSV40-delivered antioxidant enzymes. Exp Neurol 221:231–245
Lutgendorf SK, Garand L, Buckwalter KC, Reimer TT, Hong SY, Lubaroff DM (1999) Life stress, mood disturbance, and elevated interleukin-6 in healthy older women. J Gerontol Series A, Biol Sci Med Sci 54:M434–M439
Maes M (1993) A review on the acute phase response in major depression. Rev Neurosci 4:407–416
Mattson MP, Haughey NJ, Nath A (2005) Cell death in HIV dementia. Cell Death Differ 12(Suppl 1):893–904
McCune JM (1997) Animal models of HIV-1 disease. Science 278:2141–2142
McCutchan JA, Wu JW, Robertson K, Koletar SL, Ellis RJ, Cohn S, Taylor M, Woods S, Heaton R, Currier J et al (2007) HIV suppression by HAART preserves cognitive function in advanced, immune-reconstituted AIDS patients. Aids 21:1109–1117
McDade TW, Hawkley LC, Cacioppo JT (2006) Psychosocial and behavioral predictors of inflammation in middle-aged and older adults: the Chicago health, aging, and social relations study. Psychosom Med 68:376–381
Mocchetti I, Bachis A, Avdoshina V (2012) Neurotoxicity of human immunodeficiency virus-1: viral proteins and axonal transport. Neurotoxicity Res 21:79–89
Mocchetti I, Nosheny RL, Tanda G, Ren K, Meyer EM (2007) Brain-derived neurotrophic factor prevents human immunodeficiency virus type 1 protein gp120 neurotoxicity in the rat nigrostriatal system. Ann N Y Acad Sci 1122:144–154
Monleon S, D’Aquila P, Parra A, Simon VM, Brain PF, Willner P (1995) Attenuation of sucrose consumption in mice by chronic mild stress and its restoration by imipramine. Psychopharmacology 117:453–457
Moreau M, Lestage J, Verrier D, Mormede C, Kelley KW, Dantzer R, Castanon N (2005) Bacille Calmette-Guerin inoculation induces chronic activation of peripheral and brain indoleamine 2,3-dioxygenase in mice. J Infect Dis 192:537–544
Muller MB, Holsboer F (2006) Mice with mutations in the HPA-system as models for symptoms of depression. Biol Psychiatry 59:1104–1115
Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B (2007) Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord 98:143–151
Nath A, Geiger J (1998) Neurobiological aspects of human immunodeficiency virus infection: neurotoxic mechanisms. Prog Neurobiol 54:19–33
Nestler EJ, Hyman SE (2010) Animal models of neuropsychiatric disorders. Nat Neurosci 13:1161–1169
New DR, Maggirwar SB, Epstein LG, Dewhurst S, Gelbard HA (1998) HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non-N-methyl-D-aspartate receptors by a NFkappaB-independent mechanism. J Biol Chem 273:17852–17858
Nielsen CK, Arnt J, Sanchez C (2000) Intracranial self-stimulation and sucrose intake differ as hedonic measures following chronic mild stress: interstrain and interindividual differences. Behav Brain Res 107:21–33
Nosheny RL, Ahmed F, Yakovlev A, Meyer EM, Ren K, Tessarollo L, Mocchetti I (2007) Brain-derived neurotrophic factor prevents the nigrostriatal degeneration induced by human immunodeficiency virus-1 glycoprotein 120 in vivo. Eur J Neurosci 25:2275–2284
Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, Heim CM (2006) Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry 163:1630–1633
Pariante CM (2003) Depression, stress and the adrenal axis. J Neuroendocrinol 15:811–812
Patton HK, Zhou ZH, Bubien JK, Benveniste EN, Benos DJ (2000) gp120-induced alterations of human astrocyte function: Na(+)/H(+) exchange, K(+) conductance, and glutamate flux. Am J Physiol Cell Physiol 279:C700–C708
Perez A, Probert AW, Wang KK, Sharmeen L (2001) Evaluation of HIV-1 Tat induced neurotoxicity in rat cortical cell culture. J Neurovirol 7:1–10
Petit-Demouliere B, Chenu F, Bourin M (2005) Forced swimming test in mice: a review of antidepressant activity. Psychopharmacology 177:245–255
Pieper AA, Treisman GJ (2005) Drug treatment of depression in HIV-positive patients : safety considerations. Drug Saf : An Int J Med Toxic Drug Experience 28:753–762
Potula R, Poluektova L, Knipe B, Chrastil J, Heilman D, Dou H, Takikawa O, Munn DH, Gendelman HE, Persidsky Y (2005) Inhibition of indoleamine 2,3-dioxygenase (IDO) enhances elimination of virus-infected macrophages in an animal model of HIV-1 encephalitis. Blood 106:2382–2390
Primeau MM, Avellaneda V, Musselman D, St Jean G, Illa L (2013) Treatment of depression in individuals living with HIV/AIDS. Psychosomatics 54:336–344
Provencal N, Suderman MJ, Guillemin C, Massart R, Ruggiero A, Wang D, Bennett AJ, Pierre PJ, Friedman DP, Cote SM et al (2012) The signature of maternal rearing in the methylome in rhesus macaque prefrontal cortex and T cells. J Neurosci : Off J Soc Neurosci 32:15626–15642
Raison CL, Miller AH (2011) Is depression an inflammatory disorder? Curr Psychiatry Rep 13:467–475
Regulier EG, Reiss K, Khalili K, Amini S, Zagury JF, Katsikis PD, Rappaport J (2004) T-cell and neuronal apoptosis in HIV infection: implications for therapeutic intervention. Int Rev Immunol 23:25–59
Rhodes DI, Ashton L, Solomon A, Carr A, Cooper D, Kaldor J, Deacon N (2000) Characterization of three nef-defective human immunodeficiency virus type 1 strains associated with long-term nonprogression. Australian long-term nonprogressor study group. J Virol 74:10581–10588
Robertson KR, Robertson WT, Ford S, Watson D, Fiscus S, Harp AG, Hall CD (2004) Highly active antiretroviral therapy improves neurocognitive functioning. J Acquir Immune Defic Syndr 36:562–566
Sahay A, Hen R (2007) Adult hippocampal neurogenesis in depression. Nat Neurosci 10:1110–1115
Schmidt HD, Duman RS (2007) The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav Pharmacol 18:391–418
Schmidt HD, Shelton RC, Duman RS (2011a) Functional biomarkers of depression: diagnosis, treatment, and pathophysiology. Neuropsychopharm : Off Publ Am Coll Neuropsychopharm 36:2375–2394
Schmidt MV, Wang XD, Meijer OC (2011b) Early life stress paradigms in rodents: potential animal models of depression? Psychopharmacology 214:131–140
Schuster R, Bornovalova M, Hunt E (2012) The influence of depression on the progression of HIV: direct and indirect effects. Behav Modif 36:123–145
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012) Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13:465–477
Schweinsburg BC, Taylor MJ, Alhassoon OM, Gonzalez R, Brown GG, Ellis RJ, Letendre S, Videen JS, McCutchan JA, Patterson TL et al (2005) Brain mitochondrial injury in human immunodeficiency virus-seropositive (HIV+) individuals taking nucleoside reverse transcriptase inhibitors. J Neurovirol 11:356–364
Shi B, Raina J, Lorenzo A, Busciglio J, Gabuzda D (1998) Neuronal apoptosis induced by HIV-1 Tat protein and TNF-alpha: potentiation of neurotoxicity mediated by oxidative stress and implications for HIV-1 dementia. J Neurovirol 4:281–290
Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J et al (2005) Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174:6477–6489
Simoni JM, Safren SA, Manhart LE, Lyda K, Grossman CI, Rao D, Mimiaga MJ, Wong FY, Catz SL, Blank MB et al (2011) Challenges in addressing depression in HIV research: assessment, cultural context, and methods. AIDS Behav 15:376–388
Steptoe A (2007) Depression and physical illness. Cambridge University Press, Cambridge
Sumari-de Boer IM, Sprangers MA, Prins JM, Nieuwkerk PT (2012) HIV stigma and depressive symptoms are related to adherence and virological response to antiretroviral treatment among immigrant and indigenous HIV infected patients. AIDS Behav 16:1681–1689
Taliaz D, Stall N, Dar DE, Zangen A (2010) Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry 15:80–92
Theodore S, Cass WA, Nath A, Maragos WF (2007) Progress in understanding basal ganglia dysfunction as a common target for methamphetamine abuse and HIV-1 neurodegeneration. Curr HIV Res 5:301–313
Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L (1994) Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature 367:188–193
Toneatto S, Finco O, van der Putten H, Abrignani S, Annunziata P (1999) Evidence of blood–brain barrier alteration and activation in HIV-1 gp120 transgenic mice. Aids 13:2343–2348
Tovar-y-Romo LB, Bumpus NN, Pomerantz D, Avery LB, Sacktor N, McArthur JC, Haughey NJ (2012) Dendritic spine injury induced by the 8-hydroxy metabolite of efavirenz. J Pharmacol Exp Ther 343:696–703
Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, Manz MG (2004) Development of a human adaptive immune system in cord blood cell-transplanted mice. Science 304:104–107
Tu H, Rady PL, Juelich T, Smith EM, Tyring SK, Hughes TK (2005) Cytokine regulation of tryptophan metabolism in the hypothalamic-pituitary-adrenal (HPA) axis: implications for protective and toxic consequences in neuroendocrine regulation. Cell Mol Neurobiol 25:673–680
Van Duyne R, Pedati C, Guendel I, Carpio L, Kehn-Hall K, Saifuddin M, Kashanchi F (2009) The utilization of humanized mouse models for the study of human retroviral infections. Retrovirology 6:76
Vlassova N, Angelino AF, Treisman GJ (2009) Update on mental health issues in patients with HIV infection. Curr Infect Dis Reports 11:163–169
Wayman WN, Dodiya HB, Persons AL, Kashanchi F, Kordower JH, Hu XT, Napier TC (2012) Enduring cortical alterations after a single in-vivo treatment of HIV-1 Tat. Neuroreport 23:825–829
Werner ER, Fuchs D, Hausen A, Jaeger H, Reibnegger G, Werner-Felmayer G, Dierich MP, Wachter H (1988) Tryptophan degradation in patients infected by human immunodeficiency virus. Biol Chem Hoppe-Seyler 369:337–340
Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpe S, Maes M (2005) IDO and interferon-alpha-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Mol Psychiatry 10:538–544
Willner P (1986) Validation criteria for animal models of human mental disorders: learned helplessness as a paradigm case. Prog Neuro-Psychopharmacol Biol Psychiatry 10:677–690
Willner P (1997) Validity, reliability and utility of the chronic mild stress model of depression: a 10-year review and evaluation. Psychopharmacology 134:319–329
Willner P, Mitchell PJ (2002) The validity of animal models of predisposition to depression. Behav Pharmacol 13:169–188
Willner P, Towell A, Sampson D, Sophokleous S, Muscat R (1987) Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology 93:358–364
Wirleitner B, Neurauter G, Schrocksnadel K, Frick B, Fuchs D (2003) Interferon-gamma-induced conversion of tryptophan: immunologic and neuropsychiatric aspects. Curr Med Chem 10:1581–1591
Woods SP, Moore DJ, Weber E, Grant I (2009) Cognitive neuropsychology of HIV-associated neurocognitive disorders. Neuropsychol Rev 19:152–168
Yao H, Buch S (2012) Rodent models of HAND and drug abuse: exogenous administration of viral protein(s) and cocaine. J Neuroimmune Pharm : Off J Soc NeuroImmune Pharm 7:341–351
Yirmiya R (1996) Endotoxin produces a depressive-like episode in rats. Brain Res 711:163–174
Zangerle R, Widner B, Quirchmair G, Neurauter G, Sarcletti M, Fuchs D (2002) Effective antiretroviral therapy reduces degradation of tryptophan in patients with HIV-1 infection. Clin Immunol 104:242–247
Zaunders J, Dyer WB, Churchill M (2011) The Sydney Blood Bank Cohort: implications for viral fitness as a cause of elite control. Curr Opin HIV and AIDS 6:151–156
Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA (2010) Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharm : Off Publ Am Coll Neuropsychopharm 35:2510–2520
Zorrilla EP, Luborsky L, McKay JR, Rosenthal R, Houldin A, Tax A, McCorkle R, Seligman DA, Schmidt K (2001) The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain, Behavior, and Immunity 15:199–226
Zou W, Denton PW, Watkins RL, Krisko JF, Nochi T, Foster JL, Garcia JV (2012) Nef functions in BLT mice to enhance HIV-1 replication and deplete CD4+CD8+ thymocytes. Retrovirology 9:44
Zucchini S, Pittaluga A, Brocca-Cofano E, Summa M, Fabris M, De Michele R, Bonaccorsi A, Busatto G, Barbanti-Brodano G, Altavilla G et al (2013) Increased excitability in tat-transgenic mice: role of tat in HIV-related neurological disorders. Neurobiol Dis 55:110–119
Acknowledgments
ECK and EBZ thank the Ciencias Sem Fronteiras (Science Without Borders) program of the Brazilian governmental agency, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), for its support.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barreto, I.C.G., Viegas, P., Ziff, E.B. et al. Animal Models for Depression Associated with HIV-1 Infection. J Neuroimmune Pharmacol 9, 195–208 (2014). https://doi.org/10.1007/s11481-013-9518-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-013-9518-9