
Abstract
Evaluating mitogenic signaling specifically through the human insulin receptor (IR) is relevant for the preclinical safety assessment of developmental insulin analogs. It is known that overexpression of IR sensitizes cells to the mitogenic effects of insulin, but it is essentially unknown how mitogenic responses can be optimized to allow practical use of such recombinant cell lines for preclinical safety testing. We constitutively overexpressed the short isoform of the human insulin receptor (hIR-A, exon 11-negative) in L6 rat skeletal myoblasts. Because the mitogenic effect of growth factors such as insulin is expected to act in G0/G1, promoting S-phase entry, we developed a combined topoinhibition + serum deprivation strategy to explore the effect of G0/G1 synchronization as an independent parameter in the context of serum deprivation, the latter being routinely used to reduce background in mitogenicity assays. G0/G1 synchronization significantly improved the mitogenic responses of L6-hIR cells to insulin, measured by 3H-thymidine incorporation. Comparison with the parental L6 cells using phospho-mitogen-activated protein kinase, phospho-AKT, as well as 3H-thymidine incorporation end points supported that the majority of the mitogenic effect of insulin in L6-hIR cells was mediated by the overexpressed hIR-A. Using the optimized L6-hIR assay, we found that the X-10 insulin analog was more mitogenic than native human insulin, supporting that X-10 exhibits increased mitogenic signaling through the hIR-A. In summary, this study provides the first demonstration that serum deprivation may not be sufficient, and G0/G1 synchronization may be required to obtain optimal responsiveness of hIR-overexpressing cell lines for preclinical safety testing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the major advances in diabetes therapy over the last decade has been the development of short-acting (“prandial”) and long-acting (“basal”) insulin analogs. Such insulin analogs provide the clinical ability to better reproduce the pattern of fasting and mealtime insulin secretion, leading to improved glycemic control (Eckel 2005; Garg 2005; Kurtzhals 2004; Vajo et al. 2001; Zib and Raskin 2006).
Human insulin has, in addition to the classical actions on glucose, fat, and protein metabolism, also less well-explored mitogenic effects. Insulin binds to and activates the cognate insulin receptor (IR) as well as, with a 100–1,000-fold lower affinity, the closely related insulin-like growth factor 1 receptor (IGF-1R). Furthermore, insulin can activate hybrid receptors formed by IR and IGF-1R chains. Traditionally, the IR was thought to mediate metabolic responses, while the IGF-1R was thought to mediate mitogenic and antiapoptotic responses (Dupont and LeRoith 2001; Lammers et al. 1989; Nakae et al. 2001). However, it is becoming increasingly clear that activation of the IR by insulin can also cause mitogenic effects (Alexander-Bridges et al. 1992; Berhanu et al. 1997; Chou et al. 1987; Giorgino et al. 1991; Hofmann et al. 1989; Kaburagi et al. 2004; Lammers et al. 1989; Mur et al. 2008; Randazzo et al. 1990; Shymko et al. 1997, 1999; Urso et al. 2003) and that such mitogenic control by the IR has physiological relevance (Okada et al. 2007). In humans, where two IR isoforms exist (hIR-A and hIR-B), the short isoform (hIR-A) has been described to be more developed towards mitogenic signaling than the hIR-B isoform (Sciacca et al. 2003).
In order to develop insulin analogs with improved pharmacokinetic properties, structural changes are introduced in native human insulin, which could potentially change the mitogenic properties of the molecule. For example, X-10 human insulin is a quick-acting insulin analog which differs from human insulin by a single-amino-acid substitution (Schwartz et al. 1987; Vincent et al. 1995). During preclinical development, suprapharmacological doses of X-10 caused mammary adenocarcinomas in female Sprague-Dawley rats, and further development was therefore discontinued (The European Agency for the Evaluation of Medicinal Products 2001).
Tumors caused by growth factors such as insulin are expected to arise by receptor-mediated (nongenotoxic) mechanisms. Generally, receptor-mediated carcinogenesis is assumed to involve binding of growth factors to cognate receptors on preneoplastic cells, followed by for example mitogenic, antiapoptotic, vasogenic, or migratory effects, all promoting cancer progression. These steps in receptor-mediated carcinogenesis are all very well suited for modeling in mammalian cell cultures in vitro, with mitogenesis appearing to have a predictive value for cancer development (Christov et al. 2007). In fact, in all mammalian cell culture systems where this has been examined, X-10 insulin was between threefold and tenfold more mitogenic than native human insulin (Berti et al. 1998; Bornfeldt et al. 1991; Hamel et al. 1999; Milazzo et al. 1992; Slieker et al. 1997; Vajo et al. 2001; Zib and Raskin 2006). Therefore, determining in vitro mitogenic potency compared to native human insulin constitutes an important part of the preclinical safety assessment of developmental insulin analogs, supplementary to carcinogenicity evaluation in animals (The European Agency for the Evaluation of Medicinal Products 2001).
As mentioned above, insulin and insulin analogs can interact with several receptors (hIR-A, hIR-B, hIGF-1R, and hybrids), all of which can potentially mediate mitogenic signaling. Further complicating matters, the effects of IR and IGF-1R are expected to depend on crosstalk with other membrane receptors, particularly the EGFR (Ahmad et al. 2004; Kreuzer et al. 2004; Ornskov et al. 2006). In the context of preclinical in vitro mitogenicity safety evaluation, this can be addressed by using a panel of different cell lines, allowing evaluation of the mitogenic effects of the insulin analog on different receptors and in different cellular backgrounds (Le 2007). Because the increased mitogenic potency of X-10 insulin has been ascribed to increased occupancy at the IR (De Meyts et al. 1993, 1994; Hansen et al. 1996; Shymko et al. 1997; Shymko et al. 1999), as well as increased affinity for the IGF-1R (Kellerer and Haring 2001; Kurtzhals et al. 2000), cell lines where the mitogenic effect of insulin is mediated mainly by the hIR are very relevant for preclinical safety assessment of new insulin analogs. Yet, while still not completely resolved, there is convincing evidence to suggest that IGF-1R has a stronger mitogenic signaling capability than the IR (Lammers et al. 1989; Mastick et al. 1994; Nakae et al. 2001; Urso et al. 1999). This could explain why despite the low affinity of insulin and its analogs for the IGF-1R, in very few if any nonmodified mammalian cell types naturally coexpressing IR and IGF-1R, are mitogenic responses to insulin mediated primarily by the IR (Eckardt et al. 2007; Milazzo et al. 1997; Ohsugi et al. 2005), and the IGF-1R appears to contribute significantly to the mitogenic effect of even low nanomolar insulin levels in cell lines coexpressing IR and IGF-1R (Milazzo et al. 1997). Therefore, recombinant overexpression of hIR is used to generate cell lines for preclinical safety assessment of developmental insulin analogs (Berti et al. 1998). For preclinical safety assessment, the mitogenic potency of developmental insulin analogs is compared to the mitogenic potency of native human insulin, based on EC50 values determined from full dose–response curves spanning picomolar to micromolar insulin concentrations. However, it is currently unknown how mitogenic responses to insulin might be improved in recombinant hIR-overexpressing cell lines. Therefore, we overexpressed hIR-A in L6 rat skeletal muscle cells and explored the parameters influencing mitogenic responses to human insulin in the recombinant cell line.
Materials and methods
Materials
Materials included the following: native human insulin (S100 reference solution, Novo Nordisk A/S), Dulbecco’s modified Eagle’s medium (DMEM) with 5 mM glucose, fetal calf serum (FCS), penicillin–streptomycin–glutamine mix (10,000 U/ml penicillin, 10,000 μg/ml streptomycin, and 29.2 mg/ml l-glutamine), trypsin–EDTA, phosphate-buffered saline (PBS), and geneticin (50 mg/ml; Gibco). Tissue culture flasks and 96-well microtiter plates (Nunclon Surface, Nunc) were used, as well as 6-3H-thymidine (Amersham Biosciences), BSA, propidium iodide, and RNaseI (Sigma).
Cell culture
L6 rat skeletal muscle cells constitutively overexpressing the human IR under control of the CMV promoter were produced by transfection with a plasmid cDNA clone of the human IR (short isoform, lacking exon 11), followed by selection in geneticin and cloning. The resulting cell line, L6-hIR cells, express approximately 50 × 103 hIR-A per cell. The hIR isotype in the recombinant cell line was confirmed by DNA sequencing.
L6-hIR cells were maintained at 37°C in a 5% CO2-humidified atmosphere in growth medium: DMEM, 5 mM glucose, 10 vol.% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 292 μg/ml glutamine, and 1 mg/ml geneticin. To keep cultures in a proliferative (asynchronous) state, subculturing at a 1:3 split ratio was done every 2–3 days.
Insulin signaling analysis
Exponentially growing L6-hIR and parental L6 rat skeletal muscle cells were washed with PBS, trypsinized, centrifuged (100×g, 5 min), and resuspended in growth medium with 10% FCS at 2.5 × 105 cells per milliliter. The cells were seeded in 12-well plates at 1-ml cell suspension per well, incubated for 48 h, and treated with human insulin or IGF-1 diluted in growth medium without FCS but containing 0.5% BSA as carrier. The concentrations of human insulin and IGF-1 covered the range 0.001–1,000 nM. The cells were incubated for 10 min at 37°C in a 5% CO2-humidified atmosphere, washed with ice-cold PBS, snap-frozen in liquid nitrogen, and scraped off the plate in 100-μl lysis buffer (Cell Extraction Buffer, Bio Source, Invitrogen). The cell lysates were transferred to cold Eppendorf tubes, incubated at room temperature for 20 min, and centrifuged (20,000×g, 15 min, 4°C), and the resulting supernatants were transferred to new cold Eppendorf tubes. The protein content of the supernatants was determined with the BCA method (Thermo Scientific). Western blotting with phosphospecific antibodies was done as described below. Western blots were quantitated using the Science Lab 2001 ImageGauge version 4 software (Fujifilm, Sweden).
Synchronization of L6-hIR cells by combined topoinhibition and serum deprivation
Exponentially growing L6-hIR cells were seeded in growth medium with 10% FCS in six 175-cm2 culture flasks, at six different starting densities: flasks 1, 2, 3, 4, 5, and 6 were seeded with 1 × 105, 5 × 105, 1 × 106, 2.3 × 106, 1.2 × 107, and 2.3 × 107 cells in total, respectively.
After 48-h incubation, monolayers were washed with PBS and refed with starvation medium with 0.1% FCS: DMEM, 5 mM glucose, 0.1 vol.% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 292 μg/ml glutamine, and 1 mg/ml geneticin. After 24 h, the cells were seeded in 96-well plates for insulin mitogenicity assay as described below.
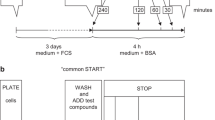
Insulin mitogenicity assay
L6-hIR cells at varying degrees of synchronization in T175 flasks numbers 1 through 6, prepared as described above, were washed with PBS and trypsinized. Cells were centrifuged (100×g, 5 min) and resuspended in starvation medium with 0.1% FCS at 4 × 105 cells per milliliter.
Cells were seeded in 96-well plates at 100-μl suspension per well. Following 1-h incubation to allow cells time to settle, 100 μl per well of recombinantly produced native human insulin (Novo Nordisk A/S reference solution) serially diluted in starvation medium with 0.1% FCS was added. After 18–20-h stimulation with human insulin, 20 μl of 6.25 μCi/ml 3H-thymidine (Amersham Biosciences) was added, and the plates were incubated for 2 h. Scintillation counting of 3H-thymidine incorporated into cellular DNA was done using a TopCount NXT scintillation counter (Packard), as recommended by the manufacturer.
Bivariate flow cytometric analysis of hIR expression and cell cycle distribution
L6-hIR cells were washed with PBS and trypsinized briefly to minimize perturbation of surface receptors. Cells were centrifuged (100×g, 5 min, 4°C) and resuspended at 2 × 106 cells per milliliter in ice-cold growth medium with 10% FCS, supplemented with 0.1% sodium azide.
Cells were incubated with 5 µg/ml murine monoclonal antibody 83-7 against hIR for 1 h on ice (Soos et al. 1986), followed by washing and incubation for 1 h dark on ice with fluorescein isothiocyanate (FITC)-conjugated goat antimouse F(ab′)2 fragment (DAKO F0479). Negative controls included samples without any antibody added, samples added 5 μg/ml isotype control IgG1 murine monoclonal antibody and FITC-conjugated secondary antibody, and samples added only FITC-conjugated secondary antibody.
Following incubation with antibodies, cells were washed, resuspended gently but completely in 200 µL PBS, 1% FCS, and 0.1% sodium azide, and fixed by adding 2 ml 70% ethanol, followed by immediate vortexing. The fixed cells were washed with PBS, 1% FCS, and 0.1% sodium azide and resuspended in 50 μg/ml propidium iodide, 2 mg/ml RNAseI, 1.12% citrate, pH 7.4. Double-stained suspensions were analyzed using a FACSCalibur flow cytometer (Becton Dickinson). Width versus area propidium iodide fluorescence pulse plots (F3W/A plots) were used to exclude cell doublets and aggregates. DNA content and cell cycle position was determined in the F3 channel, and FITC fluorescence (hIR expression) was determined in the F1 channel. Analysis was done with CellQuest Pro software
Immunofluorescent analysis of hIR expression
For immunofluorescent analysis (IFA), L6-hIR cells were seeded on glass chamber slides and allowed to reach 60–80% confluence. To prevent receptor internalization, all staining steps were done at 4°C, using ice-cold growth medium supplemented with 0.1% sodium azide for washes and antibody dilutions.
Monolayers were incubated with 1 µg/ml of murine monoclonal antibody 83-7 (IgG1; Soos et al. 1986), diluted in growth medium supplemented with 0.1% sodium azide, for 1 h at 4°C. For negative control, a murine antiserotonin monoclonal antibody was used (clone 5HT-H209, IgG1 kappa, DAKO, M0758).
Following washing, the monolayers were incubated with FITC-conjugated, affinity-isolated goat antimouse F(ab′)2 fragment (DAKO F0479) for 1 h at 4°C. Following postfixation in 1% paraformaldehyde, counterstain of nuclei with 1 µg/ml 4′,6-diamidino-2-phenylindole (DAPI), and mounting in antifade medium, monolayers were examined by fluorescence microscopy.
Fully denaturing and reducing Western blotting
Unless otherwise stated, subconfluent cell monolayers were directly lysed in sample buffer (LDS buffer, Invitrogen). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrotransfer to polyvinylidene fluoride membranes were done using the NuPage utensil and reagent suite (Invitrogen).
Filters were probed with primary and secondary antibodies and visualized with chemiluminescent substrate (ECLAdvance, GE Healthcare) using a CCD camera (LAS3000, FujiFilm) as recommended by the manufacturers.
The following primary antibodies were used: antihuman/rat/mouse phospho-retinoblastoma protein rabbit immunoglobulin (Ser807/811, Cell Signaling, #9308), antihuman/rat/mouse caspase 3 rabbit immunoglobulin (Cell Signaling #9662), phospho-Akt (Ser473) antibody (Cell Signaling #9271), and anti-ACTIVE® mitogen-activated protein kinase (MAPK) pAb (Promega, V8031). To verify equal protein loading, we used a cocktail of murine monoclonal antibodies to alpha-tubulin (T6199), GAPDH (ab8245), and beta-actin (ab6276) from Sigma and Abcam.
Secondary antibodies (horse-radish-peroxidase-conjugated rabbit antimouse immunoglobulin and horse antirabbit immunoglobulin) were from Cell Signaling.
Results
Characterization of IR expression and insulin signaling in L6-hIR and parental L6 rat skeletal muscle cells
Expression of the human IR on the surface of stably transfected rat L6 cells was confirmed by immunofluorescence, using monoclonal antibody 83-7, which recognizes human but not rat IR (Soos et al. 1986; Fig. 1a). The expression of high levels of IR was confirmed by Western blotting, comparing the IR expression in L6-hIR cells with the parental L6 cells (data not shown). Western blotting also demonstrated that both the L6-hIR and L6 cells expressed very low comparable levels of the rat IGF-1R (data not shown).

Characterization of IR expression and insulin-mediated signaling in L6-hIR cells. a Confirmation of hIR expression on the surface of L6-hIR cells: live, exponentially growing L6-hIR monolayers on glass slides were stained with mAb 83-7, which recognizes a conformational epitope of human but not rat IR (Soos et al. 1986; left double panel) or an antiserotonin mAb as negative isotype control (right double panel). Following postfixation with paraformaldehyde, nuclei were counterstained with DAPI. b, c Stimulation of phosphorylation of MAPK (b) and PKB (c) by human insulin and IGF-1 in L6-hIR and L6 cells. Concentrations of human insulin, X-10, and IGF-1 are shown in nanomolar on a logarithmic scale, i.e., 0 indicates 1 nM, 1 indicates 10 nM, −1 indicates 0.1 nM, etc. Data points represent means of triplicate Western blots; error bars show ±1 standard deviation
Both MAPK and protein kinase B Akt (PKB) have been described to be involved in mitogenic responses to insulin and IGF-1. Based on phosphorylation of MAPK, L6-hIR cells were 355-fold more sensitive than the parental L6 cells to the effect of insulin (Fig. 1b). Based on phosphorylation of PKB, L6-hIR cells were 275-fold more sensitive than the parental L6 cells to the effect of insulin (Fig. 1c), i.e., both MAPK and PKB end points gave essentially the same estimate of the degree to which L6-hIR cells were sensitized to insulin. By contrast, the EC50 values of IGF-1 were comparable between the two cell systems (Fig. 1b, c). This strongly suggested that the increased sensitivity of L6-hIR cells to the effect of insulin was due to the overexpression of the human IR-A.
Graded G0/G1 synchronization of L6-hIR cells independent of serum deprivation
Serum deprivation is routinely used for in vitro mitogenicity assays to reduce background proliferation levels and thus increase sensitivity. To explore the specific effect of G0/G1 synchronization in the context of serum deprivation, we exploited the fact that L6-hIR cells readily arrested in G0/G1 in response to topoinhibition, i.e., under conditions of high confluence. Thus, to coordinately generate L6-hIR cultures at different levels of synchronization, six parallel flasks seeded at different densities were incubated for 2 days in growth medium (with 10% FCS), allowing topoinhibition to occur in some flasks. Then, all flasks were incubated for 1 day in medium with 0.1% serum. This strategy generated six parallel serum-starved monolayers, termed “flask 1” through “flask 6,” spanning cell densities from subconfluent in flasks 1 and 2 confluent in flasks 3 and 4 to highly confluent with many detached cells in flasks 5 and 6.
In flasks 1 through 3, increasing levels of G0/G1 cells and decreasing levels of S-phase and G2/M-phase cells were present (Fig. 2a). The proliferating fraction, defined as cells in S and G2/M, was strongly and significantly reduced from flask 1 to flask 3 and remained at low and comparable levels in flasks 3 through 6. Levels of pRb, a marker for S-phase entry, decreased from flask 3 to 4 (Fig. 2b). Thus, the combined flow cytometric cell cycle and Western blot data showed that, by using topoinhibition, G0/G1 synchronization could be induced in the context of serum deprivation (Fig. 2a, b).

Topoinhibition allows graded G0/G1 synchronization of L6-hIR cells. a G0/G1 synchronization of L6-hIR cells: different levels of topoinhibition across six tissue culture flasks with L6-hIR cells were accomplished as described in “Materials and methods.” “Flask number 1” corresponded to low-confluence cells and “flask 6” to overconfluent cells with beginning degeneration of the monolayer. From flasks 1 through 6, L6-hIR monolayers were trypsinized; cell suspensions were fixed with ethanol and stained with propidium iodide and analyzed by flow cytometry. Cell cycle distributions were determined by mathematical modeling of DNA histograms with ModFit software (Verity Software House). Values are means from five independent experiments; error bars represent one standard deviation. Apoptotic (sub-G0/G1) fractions were observed in increasing amounts through flasks 1–6 and were excluded from analysis, i.e., in all cases, percentages of G0/G1, S, and G2/M cells add up to 100%. Proliferative fractions were determined based on the data in the bar diagram, as (S + G2M)/(G0G1), mean of five independent experiments, ±1 SD. Asterisks, the reduced proliferative fraction in flask 3, compared to flask 1, was highly statistically significant (P = 0.00146, two-tailed t test). b, c Detection of phospho-retinoblastoma protein and activated caspase-3: monolayers of L6-hIR cells in flasks 1 through 6 were lysed in situ. The Western blots were probed with antibodies against phospho-retinoblastoma protein (b) or caspase 3 (c). The 17- and 19-kDa bands represent cleaved (activated) caspase 3. To verify equal protein loading, blots were probed with a cocktail of three murine monoclonal antibodies against alpha-tubulin, GAPDH, and beta-actin. Lanes 1–6 correspond to flasks 1–6, respectively (low- through high-confluency L6-hIR monolayers)
In flasks 4 through 6, the G0/G1 percentages were comparable, but S-phase percentages appeared to increase (Fig. 2a). The S-phase data for flasks 4 through 6 were variable, likely reflecting a beginning degeneration of the monolayers. Perhaps for this reason, levels of pRb were not increased in flasks 4 through 6 (Fig. 2b).
A gradual increase in apoptosis was observed in flasks 3 through 6, by Western blotting for caspase 3 (Fig. 2c). Thus, it is possible that the slightly increased S-phase levels in flasks 5 and 6 (Fig. 2c) represented reduced topoinhibition effect due to loss of adherent cells by apoptosis.
Correlation between G0/G1 synchronization and mitogenic response to human insulin in L6-hIR cells
L6-hIR cells at six different degrees of synchronization were used for six parallel insulin mitogenicity assays, as described in “Materials and methods.” A mitogenic response to human insulin was observed in L6-hIR cells regardless of level of synchronization (Fig. 3a). However, cells at a low degree of synchronization exhibited flat dose–response curves, with maximal fold responses around fourfold (Fig. 3a, “flask 1”) whereas cells at a higher degree of synchronization responded with a maximum response above eightfold. Furthermore, G0/G1 synchronization increased the steepness of the dose–response curves (Fig. 3a, compare for example “flask 1” and “flask 3”) as well as the maximal fold response to 100 nM insulin (Fig. 3b). Maximal fold responses were increased from flasks 1 to 3 and were reduced from flasks 3 to 6 (P = 0.0187, one-way analysis of variance (ANOVA)). Thus, a clear correlation was observed between the degree of topoinhibition of cells used for the mitogenicity assay and maximal fold responses in the mitogenicity assay (Fig. 3a, b).

The mitogenic response of L6-hIR cells to insulin is improved by G0/G1 synchronization and mediated mainly by the IR-A: a Mitogenic responses (3H-thymidine incorporation) of L6-hIR cultures at different levels of G0/G1 synchronization. Concentrations of human insulin, X-10, and IGF-1 are shown in nanomolar on a logarithmic scale, i.e., 0 indicates 1 nM, 1 indicates 10 nM, −1 indicates 0.1 nM, etc. The study was repeated on four independent occasions, and results from a typical experiment are shown. b Correlation between mitogenic responses and G0/G1 synchronization in L6-hIR cells. Four independent experiments were carried out. Each experiment comprised graded G0/G1 synchronization of L6-hIR cells in six flasks, which were used for six parallel mitogenicity experiments using native human insulin as mitogen. Levels of G0/G1 cells, from Fig. 2a are shown on the x-axis. Normalized mitogenic response values, expressed as maximal fold mitogenic response compared to flask 1 (serum-deprived but essentially asynchronous conditions), are shown on the y-axis. Data points represent mean and SD from four independent experiments. c, d Mitogenic response to human insulin in L6-hIR cells is mediated mainly by the human insulin receptor. L6-hIR cells (c) and parental L6 cells (d) were synchronized in G0/G1 under optimal conditions (flask 3 conditions) and seeded in starvation medium in 96-wells plates for mitogenicity assays, as described in “Materials and methods.” Cells were exposed to human insulin, human IGF-1, and X-10 human insulin for 18 h, and mitogenic effects were measured by a 2-h pulse label with 3H-thymidine. Data points represent means of triplicate wells. Concentrations of human insulin, X-10, and IGF-1 are shown in nanomolar on a logarithmic scale, i.e., 0 indicates 1 nM, 1 indicates 10 nM, −1 indicates 0.1 nM, etc. Error bars, one SD. In the parental L6 cells, the response curves for insulin and IGF-1 overlap
The mitogenic response to human insulin in L6-hIR cells is mainly mediated through the hIR, activated at subnanomolar insulin concentrations, and associated with high fold responses
L6-hIR and the parental L6 cells were synchronized in G0/G1 under optimal conditions as described in “Materials and methods” for flask 3 and exposed to tenfold dilution series of human insulin, IGF-1, and X-10 (Fig. 3c, d).
For human insulin, potency was increased 48-fold in L6-hIR cells (Fig. 3c, d, compare EC50 values for human insulin in c and d). In contrast, the potency of IGF-1 was decreased by sixfold in L6-hIR cells (Fig. 3c, d, compare EC50 values for IGF-1 in L6-hIR and parental L6 cells). These findings paralleled the high (human) IR expression in L6-hIR cells (Fig. 1a), the comparable (rat) IGF-1R expression between L6-hIR cells and the parental L6 cell line (data not shown), and the increased sensitivity to human insulin-mediated signaling (Fig. 1b, c). The slight decrease in IGF-1 potency in L6-hIR cells could be due to titration of (rat) IGF-1R into rat/human IR/IGF-1R hybrids or titration of intracellular signaling molecules by the overexpressed hIR-A. In any case, these data indicated that the increased mitogenic response in L6-hIR cells was specific for the cognate receptor ligand (insulin) and also that the great majority (approximately 1 − 1/48 = 98%) of the mitogenic response to insulin in L6-hIR cells was mediated by the hIR-A. Interestingly, X-10 insulin was more mitogenic than native human insulin in L6-hIR cells (Fig. 3c), in agreement with IR binding characteristics being relevant for the well-known increased mitogenicity of this analog (De Meyts et al. 1993; Hansen et al. 1996; Shymko et al. 1997, 1999).
Reduced hIR-A expression levels correlate with a reduced mitogenic response to human insulin in strongly topoinhibited L6-hIR cells
In flasks 3 through 6, apoptosis was observed at the culture level (Fig. 2c). To examine whether nonapoptotic cells were also affected by exaggerated topoinhibition, we double-stained serum-starved and topoinhibited L6-hIR cells for hIR-A surface expression and nuclear DNA content, followed by flow cytometric analysis (Fig. 4a). Apoptotic cells, defined as cells with sub-G0/G1 DNA content, were excluded from the analysis. Interestingly, surface hIR expression in nonapoptotic L6-hIR cells was reduced in flasks 4 through 6. Thus, cellular stress affecting surface expression of hIR likely explained the strongly reduced mitogenic responses in flasks 4 through 6 (Fig. 4a).

Surface expression of hIR-A positively correlates with proliferation under growth-limiting as well as exponential proliferation conditions in L6-hIR cells: L6-hIR cultures differing inG0/G1 synchronization levels were produced by topoinhibition, as in Fig. 3c. Flask 1 (low-confluency cells) did not provide sufficient cells for flow cytometry. Serum-starved and topoinhibited L6-hIR cells from flasks 2–6, as well as an asynchronous populations of L6-hIR and parental L6 cells (exponentially proliferating, in medium with 10% FCS), were stained with the anti-IR antibody 83-7 (left panel) or mouse IgG1 isotype control (right panel) and FITC-conjugated secondary antibody. Following immunolabeling for surface hIR, cells were ethanol-fixed and stained with propidium iodide, allowing bivariate flow analysis of surface hIR expression and cell cycle position at the single-cell level. Apoptotic cells (defined as cells with <G0/G1 DNA content) were excluded from analysis. a hIR expression is reduced in strongly confluent L6-hIR monolayers. Percentages of cells positive for surface hIR expression are given in each plot. The histogram marker was set identically in all plots. b hIR-A expression correlates with a higher proliferative fraction in L6-hIR cells. Cells exhibiting surface expression of the human IR-A (hIRPos cells) were defined by the histogram marker shown in a. Cells without surface expression of the human IR-A (hIRNeg) cells were defined as NOT hIRPos cells. Cell cycle distributions are shown for gated hIRPos and hIRNeg cells in the same flask (left and right panels, respectively). The histogram marker indicates the percentage of S and G2/M-phase cells, i.e., the proliferative fraction. The proliferative S + G2/M fraction was on average 2.02-fold higher in hIRPos than in hIRNeg cells, across flasks 2–6 (n = 5, SD = 0.08, P < 0.0001, one sample t test). Flask number as well as hIR expression significantly affected cell cycle distributions (two-way ANOVA, P = 0.0360 for flasks, P = 0.0006 for hIR expression)
hIR-A expression positively correlates with proliferation in the absence of exogenously added insulin
In the flow experiment described above to examine surface hIR expression in flasks 2 through 6 (Fig. 4a), following immunolabeling for surface hIR, cells were ethanol-fixed and stained with propidium iodide, as described in “Materials and methods.” This allowed bivariate flow analysis of surface hIR expression and cell cycle position at the single-cell level (Fig. 4b). Paralleling the previously observed synchronization in G0/G1 (Fig. 2a), proliferative fractions were reduced from flasks 2 through 4 for hIRPos as well as hIRNeg cells. For example, for hIRPos cells, proliferative fractions were reduced from 32% to 20% from flask 2 to 6 (Fig. 4b, left panels), and for hIRNeg cells, proliferative fractions were reduced from 16% to 9% from flasks 2 to 6 (Fig. 4b, right panels). However, intriguingly, we found that irrespective of topoinhibition levels, cells positive for surface hIR expression exhibited twofold higher proliferating fractions than did cells negative for surface hIR expression (Fig. 4b, P < 0.0001). The overrepresentation of proliferating cells in the hIRPos fraction was also observed, albeit less pronounced, in asynchronous L6-hIR cells under exponential growth in medium with 10% FCS (Fig. 4b, bottom panels).
Discussion
Insulin is thought to exert mitogenic effect by promoting cell growth in the G0/G1 phase of the cell cycle, thus facilitating S-phase entry (Derenzini et al. 2005; Goberdhan and Wilson 2003; Kimura et al. 2001; Reid and Reid 1987; Roche et al. 1998; Straus 1984). Thus, cell cultures synchronized in G0/G1 would be expected to be most sensitive to the mitogenic effect of insulin, but this has never been directly explored. To specifically examine the effect of G0/G1 arrest, we generated L6-hIR monolayers that were all serum-starved to the same extent but nevertheless contained different levels of G0/G1 cells. We found that L6-hIR cells that were serum-deprived but not topoinhibited exhibited cell cycle distributions essentially identical to proliferating cultures in medium with 10% FCS (Fig. 2a, flask 1). Such serum-deprived but not G0/G1-synchronized L6-hIR cultures exhibited relatively weak mitogenic responses to human insulin, with EC50 values in the 1–10-nM range (Fig. 3a). With increasing levels of G0/G1 synchronization, the maximal mitogenic responses increased significantly (Fig. 3a, compare flasks 1 through 3), with a significant dose–response relationship between G0/G1 synchronization levels and maximal mitogenic responses (Fig. 3a, b, flasks 1 through 3). Excepting “oversynchronized” cultures where degeneration of the monolayers occurred, synchronization was not associated with changes in hIR-A expression (Fig. 4a, compare flasks 2 and 3 and asynchronous L6-hIR cells). Thus, our data unequivocally showed that cells synchronized in G0/G1 became more sensitive to the mitogenic effects of human insulin. This finding might explain the modest insulin mitogenicity response seen in some systems (Bahr et al. 1997; Berti et al. 1998; Liefvendahl et al. 2008; Staiger et al. 2007)
The mitogenic response of L6-hIR cells to insulin was mediated mainly by the hIR-A. This was demonstrated first by comparing the parental L6 cells with the L6-hIR cells, with the latter being 48-fold more sensitive to the mitogenic effects of insulin (Fig. 3a, compare insulin EC50 values in L6-hIR cells with parental L6 cells). This finding paralleled the increased sensitivity of the L6-hIR cells to human insulin-mediated signaling (Fig. 1b, c). Also, our data correspond well with the literature, where 10–100-fold increases in the mitogenic potency of insulin were observed in NIH3T3 and Chinese hamster ovary (CHO) cells following overexpression of hIR-A or hIR-B (Chou et al. 1987; Hofmann et al. 1989). Second, the drop in mitogenic response that was observed with too severe topoinhibition of L6-hIR cells (Fig. 3a, flask 3–6) correlated with a reduced hIR-A expression on the surface of nonapoptotic cells in the cultures (Fig. 4a, compare percentage of hIR-A-expressing cells in flasks 3 through 6. Apoptotic cells, defined by sub-G0/G1 DNA content, were excluded from the analysis). Third, in L6-hIR cells, human insulin exhibited a 275-fold stronger mitogenic effect than did IGF-1 (Fig. 3c). This ranking of mitogenic potencies of insulin and IGF-1 is characteristic of recombinant cells overexpressing hIR (Hofmann et al. 1989), and the opposite of what is seen in most mammalian cells, where IGF-1 is at least as strong or stronger mitogen than insulin.
In contrast to the 48-fold increased mitogenic potency of insulin in the L6-hIR cells compared to the parental L6 cells, the mitogenic potency of IGF-1 was reduced by fivefold in L6-hIR cells compared to parental L6 cells (Fig. 3c, d, compare IGF-1 EC50 values in L6-hIR cells with parental L6 cells). This suggested that hIR-A overexpression inhibited signaling by the rat IGF-1R. The reduced potency of IGF-1 in L6-hIR cells could be due to recruitment of endogenous (rat) IGF-1R in hybrid receptors or titration of intracellular signaling partners by the overexpressed hIR-A.
Interestingly, in L6-hIR cells, the mitogenic potency of X-10 insulin was still higher than the mitogenic potency of native human insulin (Fig. 3c). Because the mitogenic response in L6-hIR cells was mainly hIR-driven, this is in accordance with the enhanced IR binding of X-10 being relevant for the mitogenic effect of this analog (De Meyts et al. 1993; Hansen et al. 1996; Kellerer and Haring 2001; Kurtzhals et al. 2000; Shymko et al. 1997, 1999). Importantly, this does not contradict that the increased mitogenic effect of X-10 is mediated by IGF-1R in some systems (De Meyts et al. 1993; Hansen et al. 1996; Kellerer and Haring 2001; Kurtzhals et al. 2000; Shymko et al. 1997, 1999). Rather, the combined data suggest that the enhanced IR as well as IGF-1R binding effects of X-10 may both be relevant to the mitogenic effect of the analog, depending on cellular context in which in vitro mitogenicity assays are performed (De Meyts et al. 1993; Hansen et al. 1996; Kellerer and Haring 2001; Kurtzhals et al. 2000; Shymko et al. 1997, 1999).
Surprisingly, in L6-hIR cells that had been serum-starved for 24 h and without exogenously added insulin, cells with surface hIR-A expression were twofold more likely to be cycling (in S or G2/M) than cells without hIR-A expression (Fig. 4b). This positive correlation between hIR-A surface expression and cycling fraction was maintained in cultures with increasing levels of confluency (Fig. 4b, flasks 2 through 6). Thus, there was a positive correlation between hIR-A surface expression and cycling in L6-hIR cultures under serum-deprived and confluent conditions, in the absence of exogenously added insulin (Fig. 4b). At least two explanations could account for this: first, it is possible that L6-hIR cells secreted paracrine factors able to activate hIR-A, such as IGF-II. Second, overexpression of hIR-A might allow for some ligand-independent activity. Here, it should be mentioned that, at 50,000 per cell, hIR-A overexpression in the L6-hIR cells is moderate, fivefold to 15-fold lower than what was reported from NIH3T3 cells (Lammers et al. 1989; Riedel et al. 1989), and fourfold higher than what was reported from CHO cells (Chou et al. 1987). Importantly, with increased stringency of topoinhibition, apoptosis was induced in the L6-hIR cells (Fig. 2c). However, apoptotic cells (defined as cells with below G0/G1 DNA content) were excluded from the analysis in Fig. 4a, b. Also, the correlation between hIR-A expression and cycling was also observed in flask 2, where apoptosis was not detected (Fig. 4b), and the correlation between hIR-A expression and cycling was constant in flasks 2 through 6, despite increasing levels of apoptosis. The correlation between hIR-A expression and cycling was therefore independent of apoptosis levels in the L6-hIR cultures (Fig. 4b).
As the G0/G1 synchronization can be accomplished by topoinhibition, it might be considered whether in vitro mitogenicity assays should be performed under serum-free conditions or with serum present. In fact, it has in some cases been argued that “serum starvation does not represent a condition that occurs physiologically to cancer cells in vivo” (Erbel et al. 2008). However, while serum is used in cell culture media at typically 5–10% for its high content of growth factors, cells in vivo are normally exposed to plasma where clotting-associated growth factors are not present in high levels, i.e., it could be argued that having serum in cell culture media represents an artifactual situation compared to the in vivo situation. In our experience, serum starvation is generally likely to reduce background proliferation levels in in vitro mitogenicity assays and is likely to improve the sensitivity of in vitro mitogenicity assays. Also, by performing mitogenicity assays under serum-free conditions, interactions between the compound under examination and serum growth factors are minimized. We therefore believe that in vitro mitogenicity assays should generally be performed under completely or essentially serum-free conditions.
In summary, we found that synchronization in G0/G1 significantly improved the sensitivity of L6 cells overexpressing the A isoform of the human insulin receptor to the mitogenic effect of insulin. Our data strongly support that the mitogenic effect of insulin in the L6-hIR cell line is mediated mainly by the human insulin receptor. Under optimal synchronization conditions, the combination of sensitivity and fold responses of the L6-hIR mitogenicity assay compared very favorably to the most sensitive insulin in vitro mitogenicity cell systems described, natural (Allegra and Lippman 1978; Koontz and Iwahashi 1981; Liefvendahl et al. 2008; Osborne et al. 1976) as well as recombinant (Chou et al. 1987; Lammers et al. 1989; Riedel et al. 1989). Because it is known from a number of cell systems and for many growth factors that mitogenic effects are exerted in G0/G1, we believe that the findings in our study may be generally applicable to developing optimized in vitro mitogenicity assays for growth factor biopharmaceuticals.
Abbreviations
- BSA:
-
Bovine serum albumin
- EGF:
-
Epidermal growth factor
- EGFR:
-
EGF receptor
- hIR:
-
Human insulin receptor
- hIR-A, IR-A:
-
Human insulin receptor, A (exon 11Minus) isoform
- hIR-B, IR-B:
-
Human insulin receptor, B (exon 11Plus) isoform
- hIGF-1R:
-
Human insulin-like growth factor receptor
- MAPK:
-
Mitogen-activated protein kinase
- PKB:
-
Protein kinase B Akt
- pRb:
-
Retinoblastoma protein
- AspB10 human insulin:
-
X-10
References
Ahmad T, Farnie G, Bundred NJ, Anderson NG. The mitogenic action of insulin-like growth factor I in normal human mammary epithelial cells requires the epidermal growth factor receptor tyrosine kinase. J Biol Chem. 2004;279:1713–9.
Alexander-Bridges M, Buggs C, Giere L, Denaro M, Kahn B, White M, et al. Models of insulin action on metabolic and growth response genes. Mol Cell Biochem. 1992;109:99–105.
Allegra JC, Lippman ME. Growth of a human breast cancer cell line in serum-free hormone-supplemented medium. Cancer Res. 1978;38:3823–9.
Bahr M, Kolter T, Seipke G, Eckel J. Growth promoting and metabolic activity of the human insulin analogue [GlyA21, ArgB31, ArgB32]insulin (HOE 901) in muscle cells. Eur J Pharmacol. 1997;320:259–65.
Berhanu P, Anderson C, Hickman M, Ciaraldi TP. Insulin signal transduction by a mutant human insulin receptor lacking the NPEY sequence. Evidence for an alternate mitogenic signaling pathway that is independent of Shc phosphorylation. J Biol Chem. 1997;272:22884–90.
Berti L, Kellerer M, Bossenmaier B, Seffer E, Seipke G, Haring HU. The long acting human insulin analog HOE 901: characteristics of insulin signalling in comparison to Asp(B10) and regular insulin. Horm Metab Res. 1998;30:123–9.
Bornfeldt KE, Gidlof RA, Wasteson A, Lake M, Skottner A, Arnqvist HJ. Binding and biological effects of insulin, insulin analogues and insulin-like growth factors in rat aortic smooth muscle cells. Comparison of maximal growth promoting activities. Diabetologia. 1991;34:307–13.
Chou CK, Dull TJ, Russell DS, Gherzi R, Lebwohl D, Ullrich A, et al. Human insulin receptors mutated at the ATP-binding site lack protein tyrosine kinase activity and fail to mediate postreceptor effects of insulin. J Biol Chem. 1987;262:1842–7.
Christov K, Grubbs CJ, Shilkaitis A, Juliana MM, Lubet RA. Short-term modulation of cell proliferation and apoptosis and preventive/therapeutic efficacy of various agents in a mammary cancer model. Clin Cancer Res. 2007;13:5488–96.
De Meyts P. The structural basis of insulin and insulin-like growth factor-I receptor binding and negative cooperativity, and its relevance to mitogenic versus metabolic signalling. Diabetologia. 1994;37(Suppl 2):S135–48.
De Meyts P, Christoffersen CT, Urso B, Ish-Shalom D, Sacerdoti-Sierra N, Drejer K, et al. Insulin’s potency as a mitogen is determined by the half-life of the insulin-receptor complex. Exp Clin Endocrinol. 1993;101(Suppl. 2):22–3.
Derenzini M, Montanaro L, Chilla A, Tosti E, Vici M, Barbieri S, et al. Key role of the achievement of an appropriate ribosomal RNA complement for G1-S phase transition in H4-II-E-C3 rat hepatoma cells. J Cell Physiol. 2005;202:483–91.
Dupont J, LeRoith D. Insulin and insulin-like growth factor I receptors: similarities and differences in signal transduction. Horm Res. 2001;55(Suppl 2):22–6.
Eckardt K, May C, Koenen M, Eckel J. IGF-1 receptor signalling determines the mitogenic potency of insulin analogues in human smooth muscle cells and fibroblasts. Diabetologia. 2007;50:2534–43.
Eckel J. Insulin analogs revisited. Curr Med Chem Immunol Endocr Metab Agents. 2005;5:475–84.
Erbel S, Reers C, Eckstein VW, Kleeff J, Buchler MW, Nawroth PP, et al. Proliferation of Colo-357 pancreatic carcinoma cells and survival of patients with pancreatic carcinoma is not altered by insulin glargine. Diabetes Care. 2008;31:1105–11.
Garg SK. New insulin analogues. Diabetes Technol Ther. 2005;7:813–7.
Giorgino F, Belfiore A, Milazzo G, Costantino A, Maddux B, Whittaker J, et al. Overexpression of insulin receptors in fibroblast and ovary cells induces a ligand-mediated transformed phenotype. Mol Endocrinol. 1991;5:452–9.
Goberdhan DC, Wilson C. The functions of insulin signaling: size isn’t everything, even in Drosophila. Differentiation. 2003;71:375–97.
Hamel FG, Siford GL, Fawcett J, Chance RE, Frank BH, Duckworth WC. Differences in the cellular processing of AspB10 human insulin compared with human insulin and LysB28ProB29 human insulin. Metabolism. 1999;48:611–7.
Hansen BF, Danielsen GM, Drejer K, Sorensen AR, Wiberg FC, Klein HH, et al. Sustained signalling from the insulin receptor after stimulation with insulin analogues exhibiting increased mitogenic potency. Biochem J. 1996;315(Pt 1):271–9.
Hofmann C, Goldfine ID, Whittaker J. The metabolic and mitogenic effects of both insulin and insulin-like growth factor are enhanced by transfection of insulin receptors into NIH3T3 fibroblasts. J Biol Chem. 1989;264:8606–11.
Kaburagi Y, Yamashita R, Ito Y, Okochi H, Yamamoto-Honda R, Yasuda K, et al. Insulin-induced cell cycle progression is impaired in Chinese hamster ovary cells overexpressing insulin receptor substrate-3. Endocrinology. 2004;145:5862–74.
Kellerer M, Haring HU. Insulin analogues: impact of cell model characteristics on results and conclusions regarding mitogenic properties. Exp Clin Endocrinol Diabetes. 2001;109:63–4.
Kimura T, Van KA, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocr Rev. 2001;22:631–56.
Koontz JW, Iwahashi M. Insulin as a potent, specific growth factor in a rat hepatoma cell line. Science. 1981;211:947–9.
Kreuzer J, Nurnberg B, Krieger-Brauer HI. Ligand-dependent autophosphorylation of the insulin receptor is positively regulated by Gi-proteins. Biochem J. 2004;380:831–6.
Kurtzhals P. Engineering predictability and protraction in a basal insulin analogue: the pharmacology of insulin detemir. Int J Obes Relat Metab Disord. 2004;28(Suppl 2):S23–8.
Kurtzhals P, Schaffer L, Sorensen A, Kristensen C, Jonassen I, Schmid C, et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes. 2000;49:999–1005.
Lammers R, Gray A, Schlessinger J, Ullrich A. Differential signalling potential of insulin- and IGF-1-receptor cytoplasmic domains. EMBO J. 1989;8:1369–75.
Le RD. Insulin glargine and receptor-mediated signalling: clinical implications in treating type 2 diabetes. Diabetes Metab Res Rev. 2007;23:593–9.
Liefvendahl E, Arnqvist HJ. Mitogenic effect of the insulin analogue glargine in malignant cells in comparison with insulin and IGF-I. Horm Metab Res. 2008;40:369–74.
Mastick CC, Kato H, Roberts CT Jr, LeRoith D, Saltiel AR. Insulin and insulin-like growth factor-I receptors similarly stimulate deoxyribonucleic acid synthesis despite differences in cellular protein tyrosine phosphorylation. Endocrinology. 1994;135:214–22.
Milazzo G, Giorgino F, Damante G, Sung C, Stampfer MR, Vigneri R, et al. Insulin receptor expression and function in human breast cancer cell lines. Cancer Res. 1992;52:3924–30.
Milazzo G, Sciacca L, Papa V, Goldfine ID, Vigneri R. ASPB10 insulin induction of increased mitogenic responses and phenotypic changes in human breast epithelial cells: evidence for enhanced interactions with the insulin-like growth factor-I receptor. Mol Carcinog. 1997;18:19–25.
Mur C, Valverde AM, Kahn CR, Benito M. Increased insulin sensitivity in IGF-1 receptor-deficient brown adipocytes. Diabetes. 2008;51(3):743–54.
Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001;22:818–35.
Ohsugi M, Cras-Meneur C, Zhou Y, Bernal-Mizrachi E, Johnson JD, Luciani DS, et al. Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. J Biol Chem. 2005;280:4992–5003.
Okada T, Liew CW, Hu J, Hinault C, Michael MD, Krtzfeldt J, et al. Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci U S A. 2007;104:8977–82.
Ornskov D, Nexo E, Sorensen BS. Insulin-induced proliferation of bladder cancer cells is mediated through activation of the epidermal growth factor system. FEBS J. 2006;273:5479–89.
Osborne CK, Bolan G, Monaco ME, Lippman ME. Hormone responsive human breast cancer in long-term tissue culture: effect of insulin. Proc Natl Acad Sci U S A. 1976;73:4536–40.
Randazzo PA, Morey VA, Polishook AK, Jarett L. Characterization of the growth of murine fibroblasts that express human insulin receptors. I. The effect of insulin in the absence of other growth factors. Exp Cell Res. 1990;190:25–30.
Reid TW, Reid WA. The labile nature of the insulin signal(s) for the stimulation of DNA synthesis in mouse lens epithelial and 3T3 cells. J Biol Chem. 1987;262:229–33.
Riedel H, Dull TJ, Honegger AM, Schlessinger J, Ullrich A. Cytoplasmic domains determine signal specificity, cellular routing characteristics and influence ligand binding of epidermal growth factor and insulin receptors. EMBO J. 1989;8:2943–54.
Roche S, Downward J, Raynal P, Courtneidge SA. A function for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in fibroblasts during mitogenesis: requirement for insulin- and lysophosphatidic acid-mediated signal transduction. Mol Cell Biol. 1998;18:7119–29.
Schwartz GP, Burke GT, Katsoyannis PG. A superactive insulin: [B10-aspartic acid]insulin(human). Proc Natl Acad Sci U S A. 1987;84:6408–11.
Sciacca L, Prisco M, Wu A, Belfiore A, Vigneri R, Baserga R. Signaling differences from the A and B isoforms of the insulin receptor (IR) in 32D cells in the presence or absence of IR substrate-1. Endocrinology. 2003;144:2650–8.
Shymko RM, De MP, Thomas R. Logical analysis of timing-dependent receptor signalling specificity: application to the insulin receptor metabolic and mitogenic signalling pathways. Biochem J. 1997;326(Pt 2):463–9.
Shymko RM, Dumont E, De MP, Dumont JE. Timing-dependence of insulin-receptor mitogenic versus metabolic signalling: a plausible model based on coincidence of hormone and effector binding. Biochem J. 1999;339(Pt 3):675–83.
Slieker LJ, Brooke GS, DiMarchi RD, Flora DB, Green LK, Hoffmann JA, et al. Modifications in the B10 and B26–30 regions of the B chain of human insulin alter affinity for the human IGF-I receptor more than for the insulin receptor. Diabetologia. 1997;40(Suppl 2):S54–61.
Soos MA, Siddle K, Baron MD, Heward JM, Luzio JP, Bellatin J, et al. Monoclonal antibodies reacting with multiple epitopes on the human insulin receptor. Biochem J. 1986;235:199–208.
Staiger K, Hennige AM, Staiger H, Haring HU, Kellerer M. Comparison of the mitogenic potency of regular human insulin and its analogue glargine in normal and transformed human breast epithelial cells. Horm Metab Res. 2007;39:65–7.
Straus DS. Growth-stimulatory actions of insulin in vitro and in vivo. Endocr Rev. 1984;5:356–69.
The European Agency for the Evaluation of Medicinal Products. Points to consider document on the non-clinical assessment of the carcinogenic potential of insulin analogues. (CPMP/SWP/372/01). 2001.
Urso B, Cope DL, Kalloo-Hosein HE, Hayward AC, Whitehead JP, O’Rahilly S, et al. Differences in signaling properties of the cytoplasmic domains of the insulin receptor and insulin-like growth factor receptor in 3T3-L1 adipocytes. J Biol Chem. 1999;274:30864–73.
Urso B, Ilondo MM, Holst PA, Christoffersen CT, Ouwens M, Giorgetti S, et al. IRS-4 mediated mitogenic signalling by insulin and growth hormone in LB cells, a murine T-cell lymphoma devoid of IGF-I receptors. Cell Signal. 2003;15:385–94.
Vajo Z, Fawcett J, Duckworth WC. Recombinant DNA technology in the treatment of diabetes: insulin analogs. Endocr Rev. 2001;22:706–17.
Vincent MT, Carroll RJ, Hammer RE, Chan SJ, Guz Y, Steiner DF, et al. A transgene coding for a human insulin analog has a mitogenic effect on murine embryonic beta cells. Proc Natl Acad Sci U S A. 1995;92:6239–43.
Zib I, Raskin P. Novel insulin analogues and its mitogenic potential. Diabetes Obes Metab. 2006;8:611–20.
Acknowledgments
Anders Sørensen is thanked for stimulating discussions and expert advice on dose–response curve modeling.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bonnesen, C., Nelander, GM., Hansen, B.F. et al. Synchronization in G0/G1 enhances the mitogenic response of cells overexpressing the human insulin receptor A isoform to insulin. Cell Biol Toxicol 26, 293–307 (2010). https://doi.org/10.1007/s10565-009-9142-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-009-9142-x