
Abstract
Purpose
To investigate the effects of coadministration of paroxetine or fluvoxamine on the pharmacokinetics of aripiprazole in healthy adult Japanese with different CYP2D6 genotypes.
Methods
Fourteen CYP2D6 extensive metabolizer (EM) and 14 CYP2D6 intermediate metabolizer (IM) subjects were coadministered a single oral dose of aripiprazole 3 mg after steady-state plasma concentrations of the SSRIs paroxetine (20 mg/day) or fluvoxamine (100 mg/day) were reached by repeated oral doses for 6–7 days. The pharmacokinetics of aripiprazole with and without coadministration of SSRIs were compared according to CYP2D6 genotypes.
Results
Coadministration of paroxetine, a potent CYP2D6 inhibitor, decreased systemic clearance (CL/F) of aripiprazole by 58 and 23% in CYP2D6 EMs and IMs, respectively, demonstrating that the percentage inhibition of CYP2D6 activity by coadministration of paroxetine was apparently greater in CYP2D6 EMs than in IMs. Coadministration of fluvoxamine, a less potent CYP3A4 inhibitor, decreased the CL/F of aripiprazole by 39% in CYP2D6 EMs and 40% in IMs, indicating the same inhibitory effect on CYP enzymes, regardless of the CYP2D6 genotype. Percent contribution of CYP2D6 to total CL/F (CYP2D6 plus CYP3A4) of aripiprazole estimated as a reduced percentage of CL/F by CYP enzyme inhibition was 62% for CYP2D6 EMs and 24% for IMs in paroxetine coadministration, and 40% for CYP2D6 EMs and 18% for IMs in fluvoxamine coadministration.
Conclusions
There were marked differences in the degree of influence of paroxetine coadministration on the pharmacokinetics of aripiprazole between CYP2D6 EMs and IMs, but no apparent differences were found between two CYP2D6 genotypes in fluvoxamine coadministration. Aripiprazole can be used safely in combination with SSRIs that have a CYP enzyme-inhibitory action.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aripiprazole (APZ) is a novel atypical antipsychotic that acts as a partial agonist at dopamine D2 receptors and the serotonin 5-HT1A receptor, and as an antagonist at the serotonin 5-HT2A receptor [1]. APZ is now widely used for the treatment of schizophrenia in many countries worldwide including the European Union, Japan, and the United States. It is currently approved as an adjunctive therapy for major depressive disorder (MDD) in the United States.
Results of an in vitro metabolism study demonstrated that APZ was metabolized only by human cytochrome P450 isozymes CYP3A4 and CYP2D6 to dehydroaripiprazole (DAPZ; main metabolite in blood) and several other metabolites, and DAPZ was also metabolized only by CYP3A4 and CYP2D6 into several components, and those metabolites are excreted in urine or feces. There was almost no APZ or DAPZ excreted in urine and feces [2]. Because of this metabolic fate of APZ, co-administration of APZ with CYP3A4 or CYP2D6 inhibitors may potentially affect the pharmacokinetics of APZ.
For the pharmacotherapy of schizophrenia, coadministration with antidepressants, mainly selective serotonin reuptake inhibitors (SSRIs), has been widely used as adjunctive therapy depending on the patient’s symptoms. Additionally, coadministration of APZ with SSRIs is used as an adjunctive therapy in patients with MDD. However, since many SSRIs have inhibitory effects on the activities of hepatic CYP enzymes, possible drug interactions caused by the combination of SSRIs and other drugs have been of concern [3, 4].
Among the SSRIs, paroxetine (PRX) is a potent inhibitor of CYP2D6 but has no inhibitory effect on the activities of other CYP enzymes [5]. It has been reported that 93% of CYP2D6 enzymes were inactivated at the steady-state blood concentrations (35–43 ng/mL) of clinical doses of PRX (20–30 mg q.d.) [6]. In contrast, fluvoxamine (FLV) has been reported to be a less potent inhibitor but inhibits both CYP3A4 and CYP2D6 [5, 7]. Moreover, FLV is considered to potently inhibit CYP1A2 and CYP2C19 and to mildly inhibit CYP3A4, CYP2C9, and CYP2D6. However, the drug-drug interaction studies of the coadministration of FLV (100 mg q.d.) showed that FLV increased the blood concentration of CYP3A4 substrates several-fold [8–10], without significantly affecting the PK of desipramine and metoprolol, both of which are CYP2D6 substrates [11, 12], suggesting that FLV has an extremely weak or no inhibitory effect on CYP2D6.
To date, two studies have been conducted to assess the drug interaction of APZ with quinidine (CYP2D6 inhibitor), and with ketoconazole and itraconazole (CYP3A4 inhibitors) in healthy subjects [2, 13].
Based on these findings, we investigated the effects of coadministration of APZ and PRX or FLV on the pharmacokinetics of APZ in healthy subjects. In addition, since DAPZ has similar pharmacological activities as APZ, and both CYP3A4 and CYP2D6 are involved in the generation and elimination of DAPZ, we also investigated the pharmacokinetics of DAPZ [14]. CYP2D6 reportedly has genetic polymorphisms that affect metabolic activity [15, 16]. Our previous pharmacokinetic study of a single dose of 6 mg APZ in healthy subjects demonstrated that the pharmacokinetics (PK) of APZ were different between CYP2D6 extensive metabolizer (EM) and intermediate metabolizer (IM) subjects [17]. Similarly, it is considered that CYP2D6 polymorphism may affect the pharmacokinetics of APZ and its main metabolite DAPZ. Thus, in this study, we performed CYP2D6 genotyping and investigated the effects of coadministration of APZ with SSRIs on the pharmacokinetics of APZ and DAPZ according to CYP2D6 genotypes.
Methods
Study design and subjects
This study was designed as an open-label add-on study in reference to the Guidance for Drug Interaction Studies (notification no. 813 issued by the Pharmaceutical Affairs Bureau, Ministry of Health, Labour and Welfare of Japan, on June 4, 2001). The study was conducted in accordance with the ethical principles that have their origins in the Declaration of Helsinki, the Pharmaceutical Affairs Law, and “Good Clinical Practice” (GCP). This study was conducted with the approval of the ethics committee of the clinical study institution.
Healthy Japanese male volunteers aged 20–40 years at the time of informed consent with a body mass index (BMI) ranging from 18.5 to 25.0 and with a CYP2D6 genotype classified as EM or IM were eligible for the study.
Sample size was determined with reference to the results of our previously published study [13]. Assuming that the geometric mean ratios (GMR) for maximum concentration (Cmax) and area under the curve (AUC)336h of APZ with and without coadministration of SSRI were all 1.3, a sample size of seven subjects per group would be enough to provide 80% power to detect a difference between APZ with and without coadministration of SSRI. Based on the results of a screening test, seven eligible CYP2D6 EM subjects and seven CYP2D6 IM subjects were randomly selected from each PRX and FLV coadministration group using a table of random numbers. A total of 28 subjects (EM 14, IM 14) were selected.
The following subjects were excluded from the study: those who were taking other medications; those who had taken St. John’s wort products within 2 weeks prior to the initial dose of the study drug; those who had taken grapefruit and its processed products within 1 week prior to the initial dose of the study drug; or those who had smoked within 1 month prior to the initial dose of the study drug.
Administration
APZ was administered as a single dose under fasting conditions (period I), followed by a 34-day washout period. After the subjects were pretreated with PRX for 7 days or FLV for 6 days, a single dose of APZ was coadministered with PRX or FLV under fasting conditions at the steady-state plasma concentration of these SSRIs, then repeated oral doses of SSRIs were administered for 14 consecutive days (period II) (Fig. 1).

Dosing schedule
One tablet containing 3 mg APZ (Abilify®, Otsuka Pharmaceutical) was used for APZ administration. In the PRX coadministration group, one tablet of PRX hydrochloride hydrate (Paxil Tablets® 20 mg, GlaxoSmithKline) was administered once daily after breakfast. In the FLV coadministration group, one tablet of FLV maleate (Depromel® tablets 25 mg, Meiji Seika Kaisha) was administered for 3 days, and subsequently two tablets were administered for 17 days as repeated oral doses after breakfast and dinner (twice daily), respectively. Subjects were admitted to the study site from the day before APZ administration to day 4 of period I (5 days), and from the day before SSRI administration in SSRI pretreatment period to the day after completion of SSRI administration in period II (22 or 23 days).
CYP2D6 genotyping
With the QIAamp DNA Blood Mini Kit (Qiagen, Tokyo, Japan), DNA was extracted from 2 mL of venous blood that was collected using a blood collection tube containing EDTA-2Na. To detect CYP2D6-mutated alleles, the Invader method (for *2, *3, *4, *10, *14, *18, *21, *41) and the Long-PCR method (for *5 gene-deleted) were performed. Based on the results of CYP2D6 genotyping, the CYP2D6 genotypes of subjects were classified into three categories, namely, PM (poor metabolizer: homozygotes with the non-activity allele), IM (homozygotes with the deceased activity allele), EM (homozygotes with the normal activity allele and heterozygotes including the normal activity allele), according to the Web site of CYP2D6 allele nomenclature (http://www.cypalleles.ki.se/cyp2d6.htm).
The frequency of the PM genotype is generally reported to be below 1% in the Japanese population [18–20]. Therefore, inclusion of an adequate number of PM subjects in this study was considered to be difficult, and the study was conducted with EM and IM subjects.
Drug concentration analysis and pharmacokinetic analysis
Venous blood sampling (5 mL) was performed at the following times both in periods I and II: within 2 h before and 1, 2, 3, 4, 5, 6, 8, 12, 24, 48, 72, 144, 240, and 336 h after APZ administration. Each blood sample was collected using a heparinized plasma separation tube and centrifuged for 10 min (4°C, 1,581× g) to obtain the plasma. Plasma samples were stored frozen at −20°C until assay. Plasma concentrations of APZ and DAPZ were determined by liquid chromatography tandem mass spectrometry (LC-MS/MS) [21]. The lower limit of quantitation for both analytes was 0.1 ng/mL.
For the concentration range of 0.1–100 ng/mL, the intra-assay precision was less than 5.1% CV (coefficient of variation) for APZ and less than 15.9% CV for DAPZ. The accuracy was less than ±12.0% CV for APZ and less than ±14.0% CV for DAPZ.
PK parameters of APZ [Cmax, \( {\text{AU}}{{\text{C}}_{\infty }} \), maximum concentration time point (tmax ), elimination half life (t1/2,z), and systemic clearance (CL/F )] and DAPZ (Cmax, \( {\text{AU}}{{\text{C}}_{\infty }} \), tmax, and t1/2,z) were calculated for each subject by a noncompartmental method using the PK analysis software WinNonlin Professional (version 5.2, Pharsight).
Statistical analysis
Descriptive statistics (mean and standard deviation) of the plasma concentrations of APZ and DAPZ at each blood drawing time point in periods I and II were determined for CYP2D6 genotype of each coadministration group. In addition, descriptive statistics of the PK parameters of APZ and DAPZ in periods I and II were also calculated and summarized for CYP2D6 genotype of each coadministration group. The values of GMR (period II/I) for PK parameters (mean difference was calculated for tmax) were also individually calculated and summarized for CYP2D6 genotype of each coadministration group.
Results
Summary of demographics at baseline
Of the 28 subjects treated in the study, 27 subjects, excluding one subject who discontinued from the study in period II, were included in the analysis of drug interaction. The CYP2D6 genotypes in each coadministration group were as follows: seven EM subjects (six for *1/*1 and one for *1/*10) and seven IM subjects (six for *10/*10 and one for *10/*21) in the PRX coadministration group; six EM subjects (one for *1/*1, one for *1/*5, three for *1/*10, and one for *1/*21) and seven IM subjects (one for *5/*10 and six for *10/*10) in the FLV coadministration group.
The homozygote of CYP2D6 *1 allele was identified in most of the EM subjects (six out of seven subjects) in the PRX coadministration group, but only in one EM subject (of seven) in the FLV coadministration group; thus an incidental bias in the distribution of the CYP2D6 allele was observed. There was no biased distribution of CYP2D6 allele in the IM subjects.
There were no marked differences in age, height, body weight, and BMI between the coadministration groups or between the EM and IM groups (Table 1).
Plasma concentration profiles
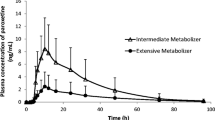
The time courses of the mean plasma concentrations of APZ and DAPZ with and without PRX following a single oral 3 mg dose of APZ in CYP2D6 EM and IM subjects are shown in Fig. 2, and those for FLV are shown in Fig. 3. The major PK parameters of APZ and DAPZ in terms of GMR (period II/I) for these parameters are shown in Table 2 (PRX group) and Table 3 (FLV group).

Time courses of plasma concentrations of aripiprazole (APZ) and dehydroaripiprazole (DAPZ) in CYP2D6 EMs and IMs with and without paroxetine (PRX) following a single oral dose of 3 mg APZ (mean + standard deviation, semilogarithmic scale). Figure at the top right is shows the 0–24 h plasma concentrations of APZ (mean + standard deviation, actual scale). EM Extensive metabolizer, IM intermediate metabolizer

Time courses of the plasma concentrations of aripiprazole (APZ) and dehydroaripiprazole (DAPZ) in CYP2D6 EMs and IMs with and without of fluvoxamine (FLV) following a single oral dose of 3 mg APZ (mean + standard deviation, semilogarithmic scale). Figure at the top right shows the 0–24 h plasma concentrations of APZ (mean + standard deviation, actual scale). EM Extensive metabolizer, IM intermediate metabolizer
PRX coadministration group
The tmax of APZ was approximately 3 h after a single dose of APZ in period I and approximately 2 h after coadministration of APZ with PRX in period II. After that the plasma APZ concentrations gradually decreased. The plasma APZ concentrations of the EM subjects in the terminal phase were consistently higher in period II than in period I, while the plasma concentration-time profiles of APZ in the IM subjects were similar in the two periods.
Among the PK parameters of APZ in the EM subjects, coadministration of PRX resulted in an increase in t1/2,z, a 58% decrease in CL/F, a 39% increase in Cmax, and a 140% increase in \( {\text{AU}}{{\text{C}}_{\infty }} \). In the IM subjects, coadministration of PRX resulted in a 23% decrease in CL/F, a 27% increase in Cmax, and a 30% increase in \( {\text{AU}}{{\text{C}}_{\infty }} \), showing that the effects of coadministration with PRX on the pharmacokinetics of APZ were smaller in the IM subjects than in the EM subjects.
In addition, coadministration of PRX had no effect on the plasma concentration-time profiles of DAPZ in the IM subjects, whereas in the EM subjects, the Cmax of DAPZ was decreased as opposed to the result obtained in APZ, the t1/2,z was increased similarly to the result obtained in APZ. The plasma concentration-time profiles of DAPZ in period II were similar between the EM and IM subjects.
FLV coadministration group
The plasma APZ concentration reached its maximum approximately 2–3 h after a single dose of APZ both in period I and period II and then remained at a higher level in period II than in period I in both the EM and IM subjects.
The values of GMR (period II/I) for PK parameters of APZ were comparable between the IM and EM subjects. Compared with period I (administration of APZ alone), the CL/F of APZ was decreased by approximately 40%, and the \( {\text{AU}}{{\text{C}}_{\infty }} \) was increased by approximately 60% in period II in both the IM and EM subjects.
Coadministration of FLV increased both Cmax and t1/2,z of DAPZ in the EM subjects. The results were similar to those observed in APZ. In contrast, in the IM subjects, no apparent changes were found in the plasma concentration-time profiles of DAPZ because of an inability to detect the elimination phase of DAPZ in period II in these IM subjects.
Calculations of the contribution of CYP enzyme to APZ metabolism
In this study, we investigated the changes in the PK parameters of APZ and its main metabolite DAPZ according to CYP2D6 genotypes after administration of APZ alone and after coadministration with clinical doses of SSRIs.
The changes in CL/F, one of the parameters to indicate the metabolic activities of hepatic CYP enzymes, before and after coadministration of PRX or FLV, are shown in Fig. 4.

Changes in systemic clearance of aripiprazole (APZ) by coadministration of clinical doses of paroxetine (PRX) and of fluvoxamine (FLV) in CYP2D6 EMs and IMs for each subject. Extensive metabolizers (EM) and intermediate metabolizers (IM) are shown, respectively, with solid lines and dashed lines
The extent of change in the CL/F after coadministration of PRX (a potent CYP2D6 inhibitor) was comparable between the EM and the IM subjects. In contrast, coadministration of FLV, a less potent CYP3A4 inhibitor, showed no such marked difference in the extent of decrease in the CL/F between CYP2D6 EM and IM subjects.
Considering that most APZ disappeared from the body by the metabolism via liver CYP enzymes (CYP2D6 and CYP3A4), we calculated the contribution of CYP2D6 and CYP3A4 to the hepatic metabolism of APZ expressed as CL/F, based on the assumptions as follows:
-
1.
Coadministration of APZ and PRX inhibits the metabolic activity of CYP2D6 by 93% (i.e., the amount of reduced CL/F by coadministration of PRX is equivalent to 93% of CL/F corresponding to the CYP2D6 [6]).
-
2.
Coadministration of FLV inhibits the metabolic activity of CYP3A4 by a similar percentage in all subjects regardless of their CYP2D6 genotype.
-
3.
Mean CL/F values corresponding to the CYP3A4 for EM and IM subjects after administration of APZ alone (before coadministration of FLV) in the FLV coadministration group are similar to those in the PRX coadministration group.
As a result, in terms of CYP3A4, the mean CL/F value calculated from the amount of reduced CL/F by coadministration of PRX was 1.98 L/h for CYP2D6 EM subjects and 1.74 L/h for CYP2D6 IM subjects. If the mean CL/F values corresponding to CYP3A4 for the EM and IM subjects in the FLV coadministration group are equal to those in the PRX coadministration group, the percent inhibition of CYP3A4 by coadministration of FLV was calculated to be 64% for CYP2D6 EM subjects and 48% for CYP2D6 IM subjects.
Estimated values of CL/F (mean and standard deviation) related to CYP2D6 and CYP3A4 for each subject, calculated according to the above-mentioned assumptions and procedure, for each SSRI coadministration group are shown in Fig. 5. The percent contribution of CYP2D6 to total metabolic activity of APZ before the inhibition of CYP enzymes by the SSRIs was calculated to be 62 and 24% in the EM and IM subjects of the PRX coadministration group, respectively, and 40 and 18% in the EM and IM subjects of the FLV coadministration group, respectively.

Change in the estimated systemic clearance (CL/F) of aripiprazole (APZ) by CYP enzyme inhibition after coadministration with SSRIs. The error bars represent the standard deviation of CYP2D6 (lower), CYP3A4 (middle) and the total (upper). EM Extensive metabolizer, IM intermediate metabolizer
Discussion
APZ is mainly metabolized into its main metabolite, DAPZ, by CYP3A4 and CYP2D6 [2], both of which are involved in the generation and elimination of DAPZ [14]. CYP2D6 exhibits extensive genetic polymorphism. More than 20 CYP2D6 alleles have been identified in Japanese subjects [22, 23], many of which are reported to show reduced activity or a loss of activity that may affect drug pharmacokinetics. Racial differences in the frequency of each genotype have also been reported.
It has also been reported that there is large individual variability in CYP3A4 metabolic activity due to differences in the amount of enzymatic protein, although no genetic polymorphism of CYP3A4 has been found that affects its metabolic activity [24]. For APZ metabolism, the intensity of metabolic activities of CYP3A4 and CYP2D6 corresponds to the amount of systemic clearance (CL/F).
DAPZ exposure in blood (AUC) following repeated administration at clinical levels was about 25% of that of APZ [25], regardless of CYP2D6 genotype. It was considered that the change in the AUC of DAPZ by SSRI coadministration would have little influence on overall exposure (APZ plus DAPZ) and the efficacy and safety of APZ. Therefore, this study mainly investigated the influence of SSRI coadministration on the CL/F of APZ.
In the present study, the large interindividual variability in CL/F (23% CV) observed in the EM subjects of PRX coadministration group was reduced (14% CV) by coadministration of PRX due to nearly complete inhibition of CYP2D6. However, in the IM subjects, the interindividual variability in CL/F (14% CV) was much smaller compared to the EM subjects and did not differ greatly from that (11% CV) after coadministration of PRX. Both the CL/F values and the interindividual variability after coadministration of PRX were similar in CYP2D6 EM and IM subjects. In contrast, all the EM subjects in the FLV coadministration group showed a similar decrease in CL/F after coadministration of FLV; no change in interindividual variability was found after coadministration of FLV. Similar results were obtained in the IM subjects (see Fig. 4).
It is assumed that most of the reduced CL/F due to coadministration of PRX is equivalent to the decrease in CL/F of CYP2D6, and the remainder represents the CL/F of CYP3A4 alone. Moreover, most of the reduced CL/F by coadministration of FLV is equivalent to the decrease in CL/F of CYP3A4.
These findings suggest that the relatively large interindividual variability in CL/F of APZ found in the EM subjects within the PRX and FLV coadministration groups was mainly attributable to the interindividual variability in CYP2D6 enzyme activity accompanied by the incidentally biased distribution of CYP2D6 diplotype in this study. Additionally, interindividual variability in CL/F of CYP3A4 in healthy subjects is presumed to be comparatively small.
However, since the decrease in CL/F of APZ by coadministration of FLV was comparable between CYP2D6 EM and IM subjects, the possibility that FLV may potentially inhibit CYP2D6 to some extent cannot be excluded.
The changes in the PK parameters of APZ by coadministration of PRX or FLV found in this study were similar to the following results previously reported in drug interaction studies: coadministration of APZ 10 mg with the CYP2D6 inhibitor quinidine 166 mg increased \( {\text{AU}}{{\text{C}}_{\infty }} \) of APZ by 112% and decreased \( {\text{AU}}{{\text{C}}_{\infty }} \) of DAPZ by 35% [2], coadministration of APZ 15 mg with the CYP3A4 inhibitor ketoconazole 200 mg increased \( {\text{AU}}{{\text{C}}_{\infty }} \) of APZ and DAPZ by 63 and 77%, respectively, in American subjects [2], and coadministration of APZ 3 mg with the CYP3A4 inhibitor itraconazole 100 mg decreased CL/F of APZ by 27 and 47% in Japanese EM and IM subjects, respectively [13].
Since the increase in blood exposure (APZ + DAPZ) after coadministration with SSRI was around three times at the maximum in both CYP2D6 EM and IM subjects, it was considered that APZ can be used safely in combination with SSRIs.
In conclusion, this study demonstrated that there were marked differences in the degree of influence on the pharmacokinetics of APZ by coadministration of PRX between CYP2D6 EM and IM genotypes, whereas these differences during FLV coadministration were small and almost negligible.
The percent contribution of enzymes CYP2D6 and CYP3A4 to the CL/F (i.e., metabolic activity) for APZ in each subject was calculated, assuming both the percent inhibition of CYP2D6 by coadministration of PRX and the percent inhibition of CYP3A4 by coadministration of FLV are constant.
Our findings suggest that the percent contribution of CYP enzymes to the metabolic activity for APZ can be estimated, and the pharmacokinetic behavior of each CYP genotype can definitely be presumed by inhibiting the metabolic enzymes of APZ with SSRIs.
References
Oshiro Y, Sato S, Kurahashi N, Tanaka T, Kikuchi T, Tottori K, Uwahodo Y, Nishi T (1998) Novel antipsychotic agents with dopamine autoreceptor agonist properties: synthesis and pharmacology of 7-[4-(4-phenyl-1-piperazinyl)butoxy]-3,4-dihydro-2(1H)-quinolinone derivatives. J Med Chem 41:658–667
Otsuka Pharmaceutical Co, Ltd (2009) Package insert of Abilify® (aripiprazole, U.S.), July 2009 revision. Otsuka, Tokyo
Perucca E, Gatti G, Spina E (1994) Clinical pharmacokinetics of fluvoxamine. Clin Pharmacokinet 27:175–190
Sproule BA, Naranjo CA, Brenmer KE, Hassan PC (1997) Selective serotonin reuptake inhibitors and CNS drug interactions. A critical review of the evidence. Clin Pharmacokinet 33:454–471
Jeppesen U, Gram LF, Vistisen K, Loft S, Poulsen HE, Brøsen K (1996) Dose-dependent inhibition of CYP1A2, CYP2C19 and CYP2D6 by citalopram, fluoxetine, fluvoxamine and paroxetine. Eur J Clin Pharmacol 51:73–78
Venkatakrishnan K, Obach RS (2005) In vitro-in vivo extrapolation of CYP2D6 inactivation by paroxetine: prediction of nonstationary pharmacokinetics and drug interaction magnitude. Drug Metab Dispos 33:845–852
van Harten J (1995) Overview of the pharmacokinetics of fluvoxamine. Clin Pharmacokinet 29(Suppl 1):1–9
Fleishaker JC, Hulst LK (1994) A pharmacokinetic and pharmacodynamic evaluation of the combined administration of alprazolam and fluvoxamine. Eur J Clin Pharmacol 46:35–39
Damkier P, Hansen LL, Brøsen K (1999) Effect of fluvoxamine on the pharmacokinetics of quinidine. Eur J Clin Pharmacol 55:451–456
Lam YW, Alfaro CL, Ereshefsky L, Miller M (2003) Pharmacokinetic and pharmacodynamic interactions of oral midazolam with ketoconazole, fluoxetine, fluvoxamine, and nefazodone. J Clin Pharmacol 43:1274–1282
Xu ZH, Xie HG, Zhou HH (1996) In vivo inhibition of CYP2C19 but not CYP2D6 by fluvoxamine. Br J Clin Pharmacol 42:518–521
Spina E, Pollicino AM, Avenoso A, Campo GM, Perucca E, Caputi AP (1993) Effect of fluvoxamine on the pharmacokinetics of imipramine and desipramine in healthy subjects. Ther Drug Monit 15:243–246
Kubo M, Koue T, Inaba A, Takeda H, Maune H, Fukuda T, Azuma J (2005) Influence of itraconazole co-administration and CYP2D6 genotype on the pharmacokinetics of the new antipsychotic aripiprazole. Drug Metab Pharmacokin 20:55–64
US FDA (2002) Drug approval package, Abilify (aripiprazole) tablets. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21-436_Abilify.cfm
Ozawa S, Soyama A, Saeki M, Fukushima-Uesaka H, Itoda M, Koyano S, Sai K, Ohno Y, Saito Y, Sawada J (2004) Ethnic differences in genetic polymorphisms of CYP2D6, CYP2C19, CYP3As, and MDR1/ABCB1. Drug Metab Pharmacokinet 19:83–95
Fukuda T, Azuma J (2000) Evaluation of in vivo and in vitro metabolic characteristics of CYP2D6*10. Drug Metab Pharmacokinet 15:165–170
Kubo M, Koue T, Maune H, Fukuda T, Azuma J (2007) Pharmacokinetics of aripiprazole, a new antipsychotic, following oral dosing in healthy adult Japanese volunteers: influence of CYP2D6 polymorphism. Drug Metab Pharmacokinet 22:358–366
Nakamura K, Goto F, Ray WA, McAllister CB, Jacqz E, Wilkinson GR, Branch RA (1985) Interethnic differences in genetic polymorphism of debrisoquin and mephenytoin hydroxylation between Japanese and Caucasian populations. Clin Pharmacol Ther 38:402–408
Ishizaki T, Eichelbaum M, Horai Y, Hashimoto K, Chiba K, Dengler HJ (1987) Evidence for polymorphic oxidation of sparteine in Japanese subjects. Br J Clin Pharmacol 23:482–485
Horai Y, Nakano M, Ishizaki T, Ishikawa K, Zhou HH, Zhou BI, Liao CL, Zhang LM (1989) Metoprolol and mephenytoin oxidation polymorphisms in Far Eastern Oriental subjects: Japanese versus mainland Chinese. Clin Pharmacol Ther 46:198–207
Kubo M, Mizooku Y, Hirao Y, Osumi T (2005) Development and validation of an LC-MS/MS method for the quantitative determination of aripiprazole and its main metabolite, OPC-14857, in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 822(1–2):294–299
Ebisawa A, Hiratsuka M, Sakuyama K, Konno Y, Sasaki T, Mizugaki M (2005) Two novel single nucleotide polymorphisms (SNPs) of the CYP2D6 gene in Japanese individuals. Drug Metabol Pharmacokinet 20:294–299
Sakuyama K, Sasaki T, Ujiie S, Obata K, Mizugaki M, Ishikawa M, Hiratsuka M (2008) Functional characterization of 17 CYP2D6 allelic variants (CYP2D6.2, 10, 14A–B, 18, 27, 36, 39, 47–51, 53–55, and 57). Drug Metab Dispos 36:2460–2467
Robertson GR, Field J, Goodwin B, Bierach S, Tran M, Lehnert A, Liddle C (2003) Transgenic mouse models of human CYP3A4 gene regulation. Mol Pharmacol 64:42–50
Kubo M, Koue T, Fukuda T, Azuma J (2007) Pharmacokinetics of aripiprazole, a new antipsychotic, following oral dosing in healthy adult Japanese volunteers: influence of CYP2D6 polymorphism. Drug Metab Pharmacokinet 22:358–366
Acknowledgments
This study was sponsored by Otsuka Pharmaceutical Co., Ltd, Japan. The authors gratefully acknowledge the volunteers and staff who participated in this study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Azuma, J., Hasunuma, T., Kubo, M. et al. The relationship between clinical pharmacokinetics of aripiprazole and CYP2D6 genetic polymorphism: effects of CYP enzyme inhibition by coadministration of paroxetine or fluvoxamine. Eur J Clin Pharmacol 68, 29–37 (2012). https://doi.org/10.1007/s00228-011-1094-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-011-1094-4