
Abstract
Atherosclerosis is a chronic inflammatory and metabolic disorder affecting large- and medium-sized arteries, and the leading cause of mortality worldwide. The pathogenesis of atherosclerosis involves accumulation of lipids and leukocytes in the intima of blood vessel walls creating plaque. How leukocytes accumulate in plaque remains poorly understood; however, chemokines acting at specific G protein-coupled receptors appear to be important. Studies using knockout mice suggest that chemokine receptor signaling may either promote or inhibit atherogenesis, depending on the receptor. These proof of concept studies have spurred efforts to develop drugs targeting the chemokine system in atherosclerosis, and several have shown beneficial effects in animal models. This study will review key discoveries in basic and translational research in this area.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis is the pathologic process underlying most strokes and heart attacks, which together are now the leading cause of death worldwide (Roger et al. 2012). Risk factors for atherosclerosis include age, gender, a high ratio of low-density lipoprotein (LDL) to high-density lipoprotein (HDL) in the blood, hypertension, diabetes, obesity, smoking and inheritance (Berger et al. 2010). It is generally accepted that atherosclerosis is a chronic metabolic and inflammatory disease (Hansson 2005). The pathologic hallmark is the atherosclerotic plaque, composed of lipids, collagen, platelets, fibroblasts, smooth muscle cells (SMCs) and leukocytes. Rupture of unstable plaques may result in thrombosis and ischemia in surrounding tissues (Weber et al. 2008; Weber and Noels 2011).
Both innate and adaptive immunity appear to be involved in the development of plaque (Packard et al. 2009). Innate immune cells, including macrophages, neutrophils, mast cells and platelets, accumulate early and express reactive oxygen species (ROS), proteinases, lipid mediators and various cytokines, leading to SMC proliferation, angiogenesis and additional inflammatory cell activation. Adaptive immune cells, including T and B lymphocytes, accumulate later and may accelerate disease progression through complex and poorly understood mechanisms (Galkina and Ley 2009; Weber et al. 2008). In particular, in mouse models, the B1 subset of B cells is atheroprotective, whereas the B2 subset is pro-atherogenic; Th1 cells are pro-atherogenic, and regulatory T cells (Tregs) are atheroprotective, whereas the roles of Th2 and Th17 cells are still controversial (Butcher and Galkina 2011; Kyaw et al. 2011; Robertson and Hansson 2006).
The classic mechanism of immune cell recruitment from the blood into tissues, including the blood vessel wall, is thought to involve sequential interactions between leukocytes and endothelial cells (ECs). These include selectin-mediated leukocyte rolling on endothelium, followed by leukocyte activation by chemokines, then integrin-mediated firm arrest of leukocytes to endothelium, culminating in leukocyte transendothelial migration (Ley et al. 2007). Chemokines are a large protein family of small (8–10 kDa) leukocyte chemoattractants that can be divided into four classes: C, CC, CXC and CX3C according to the number and spacing of conserved cysteines in the N-terminal domain of the molecule (Murphy et al. 2000). In human, approximately 46 chemokines and 20 chemokine receptors have been identified (Murphy 2002). Since the discovery in 1998 that the chemokine Ccl2 and its receptor Ccr2 are positive regulators of both the apolipoprotein E-deficient (ApoE −/−) and LDL receptor-deficient (Ldlr −/−) mouse models of atherosclerosis (Boring et al. 1998; Dawson et al. 1999; Gu et al. 1998), roles for many other chemokines and chemokine receptors have been identified using genetic and, in some cases, pharmacological criteria (Koenen and Weber 2011). In this review, we will discuss current concepts of the chemokine system as an immunoregulator and potential therapeutic target in atherosclerotic cardiovascular disease.
The Chemokine System as an Immunoregulator in Atherogenesis
Chemokines and chemokine receptors are able to regulate leukocyte trafficking in both homeostasis and inflammation (Murphy et al. 2000). Chemokine receptors identified in atherogenesis include inflammatory receptors (e.g., CCR2, CCR5, CXCR2 and CX3CR1), classical homeostatic receptors (e.g., CCR7) as well as receptors that have both inflammatory and homeostatic functions (e.g., CCR6) (Koenen and Weber 2011). We will discuss the chemokines and their receptors in three different categories: atherogenic, atheroprotective and controversial (Table 1).
Atherogenic Chemokines/Chemokine Receptors
CCL2–CCR2
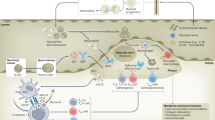
CCL2–CCR2 was the first chemokine/chemokine receptor pair to be implicated in the pathogenesis of atherosclerosis, and the one studied in the greatest detail. CCL2 (also known as monocyte chemotactic protein-1) is mainly produced by monocytes, macrophages, ECs and SMCs, and has been identified in both mouse and human atherosclerotic lesions (Nelken et al. 1991; Rayner et al. 2000). In human, single nucleotide polymorphisms (SNPs) in the promoter of CCL2, CCL2-2518G, and in the open reading frame of CCR2, CCR2-V64I, have been associated with increased risk of myocardial infarction (McDermott et al. 2005; Ortlepp et al. 2003; Szalai et al. 2001). CCL2-2518G appears to be functional since individuals with this allele have significantly higher serum CCL2 levels (McDermott et al. 2005). The biochemical effect of CCR2-V64I has not been clearly defined. In mice, genetic deletion of either Ccl2 or Ccr2 in both the ApoE −/− and Ldlr −/− models of atherosclerosis significantly reduced the size of lesions in the aorta (Boring et al. 1998; Dawson et al. 1999; Gu et al. 1998). In a third model, Ccl2 deficiency decreased atherosclerotic plaque burden in mice that overexpress human apolipoprotein B (Gosling et al. 1999). A pro-atherogenic role for Ccl2/Ccr2 was also demonstrated by blocking Ccr2 with 7ND, an N-terminally truncated mutant form of Ccl2, which attenuated the initiation and progression of atherosclerosis in ApoE −/− mice (Inoue et al. 2002; Ni et al. 2001). A study of radiation chimeric mice has attributed the pro-atherogenic effect of Ccr2 to expression on hematopoietic cells. In particular, Ccr2 −/− but not Ccr2 +/+ bone marrow transplantation into ApoE3-leiden mice reduced the size of atherosclerotic lesions; moreover, overexpression of Ccl2 on hematopoietic cells in ApoE −/− mice increased atherosclerotic lesion size (Aiello et al. 1999; Guo et al. 2003). However, blockade of Ccr2 by either 7ND treatment or Ccr2 −/− bone marrow transplantation did not affect the progression of established atherosclerotic lesion development (de Waard et al. 2010; Guo et al. 2005). Further studies in mice showed that Ccr2 deficiency abolished the egress of monocytes from the bone marrow and markedly reduced Ccl2-induced recruitment of monocytes from the blood into inflammatory sites (Boring et al. 1997; Tacke et al. 2007; Tsou et al. 2007). Considering that monocytes from the bone marrow and the spleen are critical for both the initiation and progression of atherosclerosis (Fig. 1) (Robbins et al. 2012; Swirski et al. 2009; Weber et al. 2008), this may partially explain why Ccl2 and Ccr2 deficiencies inhibit the development of atherosclerosis.

Schematic representation of chemokines/chemokine receptors and their target cells involved in the progress of atherosclerosis. CCL2–CCR2 and CCL20–CCR6 induce the egress of monocytes from the bone marrow and the spleen into the circulating blood (arrows), and then together with CCL5–CCR5 and CX3CL1–CX3CR1, they mediate the arrest of monocytes on the endothelium and their migration into the atherosclerotic plaque. In the plaque, monocytes differentiate into macrophages (foam cells) after digestion of oxLDL through CXCL16 and other scavenger receptors. CX3CL1–CX3CR1 is also important for the survival/retention of macrophages in the plaque and the formation of monocyte–platelet complex during atherogenesis. The egress of neutrophils from the bone marrow and their recruitment into the vessel wall are mediated by CXCL1/8–CXCR2 and CXCL12–CXCR4. The adhesion and migration of Th1 cells are controlled by CXCL10–CXCR3 and CCL5–CCR1/5, while the recruitment of Th2 and Th17 cells relies on CCR4 and CCL20–CCR6, respectively. The proliferation of Tregs in the plaque is controlled by CCR4 and its ligand CCL17, which is secreted by DCs. The recruitment of HSCs, B cells and T cells may depend on their specific chemokine receptors (dashed arrow). Macrophages, DCs and Tregs all express CCR7 and during atherosclerosis regression these cells may egress into the lymph node by signaling through CCL19 and CCL21. DCs Dendritic cells, EC endothelial cell, HSCs hematopoietic stem cells, Mono monocytes, oxLDL oxidized low-density lipoprotein, PMNs polymorphonuclear leukocytes (neutrophils), SMC smooth muscle cell, Tregs regulatory T cells
CCL5–CCR5
The inflammatory chemokine CCL5 [also known as regulated upon activation, normal T cell expressed and secreted (RANTES)] is expressed by monocytes, macrophages, T cells and SMCs in both mouse and human atherosclerotic lesions, and acts at the chemokine receptors CCR1, CCR3 and CCR5 (Krohn et al. 2007; Pattison et al. 1996). In human, the gain-of-function polymorphism CCL5-G403A affects basal CCL5 expression levels and is associated with a higher risk of coronary artery disease (CAD) (Boger et al. 2005; Simeoni et al. 2004), and loss-of-function polymorphism CCR5Δ32 may be associated with a lower risk of myocardial infarction (Gonzalez et al. 2001; Simeoni et al. 2004). Although genetic deletion of Ccr5 in ApoE −/− mice was not shown to reduce early spontaneous atherosclerosis in the initial study (Kuziel et al. 2003), later investigations suggested that it reduced both early- and late-stage atherosclerosis development in ApoE −/− mice fed either a chow or high-fat diet (Braunersreuther et al. 2007; Quinones et al. 2007). Also, Ldlr −/− mice reconstituted with Ccr5 −/− bone marrow showed reduced macrophage accumulation in atherosclerotic lesions and improved plaque stability (Potteaux et al. 2006). The mechanism of CCL5/CCR5 modulation of atherosclerosis has not been clearly defined. Possibilities include regulation of leukocyte trafficking to lesions and modulation of adaptive immunity. As indirect supportive evidence, CCR5 is important for the spreading and trans-endothelial migration of monocytes, neutrophils and Th1 cells, and the atheroprotective effects seen in Ccr5 −/− mice may be associated with up-regulation of interleukin (IL)-10 and reduction of Th1-type immune responses (Fig. 1) (Braunersreuther et al. 2007; Drechsler et al. 2010; Potteaux et al. 2006; Weber et al. 2001).
CCR6
Both CCR6 and its sole chemokine ligand CCL20 (also known as macrophage inflammatory protein 3α) are expressed at increased levels in human atherosclerotic plaques, and the circulating level of CCL20 is significantly increased in hypercholesterolemic subjects (Calvayrac et al. 2011; Yilmaz et al. 2007). In mice, Ccr6 and Ccl20 are both present constitutively in healthy aortas and in atherosclerotic plaques, and Ccr6 deletion in ApoE −/− mice fed a Western diet significantly reduced the atherosclerotic lesion size in both the whole aorta and the aortic root, accompanied by a reduction of macrophage content in the plaques (Wan et al. 2011). Bone marrow transplantation suggested that the lesion reduction seen in Ccr6 −/− ApoE −/− mice is caused by Ccr6 expression on hematopoietic cells. CCL20 is produced by epithelial cells, ECs and SMCs, whereas CCR6 is mainly expressed by immature dendritic cells (DCs), B cells, T cells, neutrophils and monocytes (Schutyser et al. 2003; Wan et al. 2011). Interestingly, Ccr6-deficient mice on the ApoE knockout background were relatively monocytopenic, whereas bone marrow monocyte content was slightly increased. Thus, modulation of atherosclerosis could be due to Ccr6 function on monocytes, mediating their egress from the bone marrow and their recruitment from the blood into the vessel wall (Fig. 1) (Koenen and Weber 2011; Wan et al. 2011). However, the effect of Ccr6 on atherogenesis may be cell-type specific since a second study suggested that Ccr6 might be involved in B-cell-mediated atheroprotection (Doran et al. 2012). Because mice globally deficient in Ccr6 have reduced atherosclerosis, the effect of Ccr6 deficiency on non-B cells appears to be dominant over the effect of B-cell Ccr6 deficiency (Wan and Murphy 2011). Additional work will be necessary to further clarify the underlying mechanism.
CCL17
CCL17 (also known as thymus and activation-regulated chemokine) is a DC-derived chemokine that induces chemotaxis of T cells through its receptor CCR4 (Imai et al. 1997a). CCL17 has been detected in both advanced mouse and human atherosclerotic lesions, and its expression is upregulated in human atherosclerotic plaques compared with healthy arteries (Greaves et al. 2004; Weber et al. 2011). Genetic targeting of Ccl17 in ApoE −/− mice resulted in reduced atherosclerotic lesion size, along with a decrease of macrophages and T cells in the plaques, which was confirmed by Ccl17 −/− bone marrow transplantation and Ccl17-specific antibody treatment (Weber et al. 2011). The beneficial effect may be dependent on Tregs that accumulate in the lymph nodes and aortas in the double knockout mice (Fig. 1). Moreover, co-culture of CD4+ T cells with Ccl17 −/− DCs resulted in less apoptosis and enhanced expansion of Tregs, suggesting that Ccl17 is a central regulator of Treg homeostasis (Weber et al. 2011). Taken together, this suggests that CCL17 expressed by DCs may drive atherosclerosis development through inhibiting expansion of Tregs.
CX3CL1–CX3CR1
CX3CL1 (Fractalkine), the sole ligand for CX3CR1, is a unique chemokine that exists in both membrane-tethered and soluble shed forms, thus mediating both cell adhesion and chemotaxis (Bazan et al. 1997). CX3CL1 is primarily produced by ECs and CX3CR1 is expressed on monocytes, lymphocytes, platelets, and DCs (Barlic and Murphy 2007a; Imai et al. 1997b). In addition, both molecules have been identified on foam cells and SMCs in mouse and human atherosclerotic lesions, and CX3CR1 is upregulated on monocytes from coronary artery disease (CAD) patients (Apostolakis et al. 2007; Lucas et al. 2003). In several retrospective cohort studies and the population-based prospective Framingham Heart Study Offspring Cohort, two SNPs of CX3CR1, V249I and T280M, were associated with a markedly reduced risk of CAD (Ghilardi et al. 2004; McDermott et al. 2001, 2003; Moatti et al. 2001; Norata et al. 2006). However, two other studies showed that these polymorphisms were not associated with peripheral arterial disease (522 patients) but were associated with increased risk of restenosis after coronary stenting (365 patients) (Gugl et al. 2003; Niessner et al. 2005). The biochemical effects of these polymorphisms on CX3CR1 function are also in dispute with two groups reporting that they affect function but one finding a gain of function and the other a loss of function (Daoudi et al. 2004; McDermott et al. 2003). In atherosclerosis mouse models (ApoE −/− mice and Ldlr −/− mice), genetic deletion of Cx3cl1 and Cx3cr1 significantly reduced the size of atherosclerotic lesions and inhibited the recruitment of macrophages/DCs into the vessel wall (Combadiere et al. 2003; Lesnik et al. 2003; Liu et al. 2008; Teupser et al. 2004). The role of Cx3cl1 and Cx3cr1 in atherogenesis appears to be independent of Ccl2, Ccr2 and Ccr5 since additive protection was observed in the ApoE knockout model in both Cx3cl1 −/−Ccr2−/− mice and Ccl2 −/− Cx3cr1 −/− mice treated with Met-RANTES, a pharmacological inhibitor of Ccr5 (Combadiere et al. 2008; Saederup et al. 2008). The proposed atherogenic mechanisms include Cx3cr1-dependent migration/retention of monocyte/macrophage in the vessel wall (Barlic and Murphy 2007b; Tacke et al. 2007); Cx3cr1-conferred monocyte/macrophage survival in atherosclerotic plaques (Landsman et al. 2009) and Cx3cr1 supported the formation of platelet–monocyte complexes in hyperlipidemic mice (Fig. 1) (Postea et al. 2012).
CXCL1–CXCR2
Both CXCR2 and its major ligands, CXCL1 (also known as growth-related oncogene α) and CXCL8 (also known as IL-8), have been identified in human atheromata, and mouse Cxcr2 and Cxcl1 have been found in plaque in mouse models of atherosclerosis (Apostolopoulos et al. 1996; Boisvert et al. 2000; Wang et al. 1996). Consistent with this, the plasma level of CXCL1 is significantly increased in patients with CAD (Breland et al. 2008), and expression of CXCL8 and CXCR2 are strongly upregulated on ECs and monocytes after oxidized LDL (oxLDL) stimulation (Lei et al. 2002; Yeh et al. 2001). A pro-atherogenic role for Cxcr2 was suggested by studies in which Cxcr2 −/− bone marrow was transplanted into irradiated Ldlr −/− mice, resulting in much smaller lesions with fewer lesional macrophages compared to controls (Boisvert et al. 1998). Also, genetic deletion of Cxcl1 significantly reduced atherosclerotic lesion size in both the whole aorta and the aortic root of Ldlr −/− mice (Boisvert et al. 2006). CXCL1 is mainly produced by macrophages, neutrophils and epithelial cells, whereas CXCR2 is found most prominently on neutrophils, with some expression on monocytes and mast cells (Murphy et al. 2000). The pro-atherogenic effect of the CXCL1–CXCR2 axis may be due to CXCL1-triggered monocyte arrest on early atherosclerotic endothelium and CXCR2-mediated macrophage accumulation in established lesions (Boisvert et al. 2006; Huo et al. 2001; Papadopoulou et al. 2008). In humans, CXCL8 levels were significantly increased in the setting of acute myocardial infarction (Neumann et al. 1995). Recent studies have suggested that neutrophils are also pro-atherogenic, and thus CXCR2 might be involved in the recruitment of neutrophils to the vessel wall during the initiation of atherosclerotic plaque formation (Fig. 1) (Drechsler et al. 2011).
CXCL10–CXCR3
CXCR3 and its ligand CXCL10 [also known as interferon (IFN)-γ-induced protein of 10 kDa or IP-10] are highly expressed in lesional T cells (Th1 cells) as well as ECs, SMCs and macrophages in human atheromata (Mach et al. 1999). The percentage of CXCR3+ lymphocytes has been reported to be significantly increased in the blood of CAD patients (Fernandes et al. 2004) and the plasma concentration of CXCL10 was also much higher in CAD patients compared to controls (Kawamura et al. 2003). In ApoE −/− mice, both Cxcr3 and Cxcl10 deficiencies resulted in a reduction of atherosclerotic lesion formation, accompanied by a decrease of CD4+ T-cell accumulation in the plaques (Heller et al. 2006; Veillard et al. 2005). This was associated with an elevated number of Treg cells and increased expression of the anti-inflammatory cytokine IL-10 within lesions, suggesting that the CXCL10–CXCR3 axis may promote atherogenesis by regulating the recruitment and balance of effector T cells and Tregs (Fig. 1) (Heller et al. 2006). The role of Cxcr3 in atherogenesis is not redundant since Cxcr3 −/− Ccr2 −/− ApoE −/− triple knockout mice show a further decrease of lesion formation compared with deletion of either Cxcr3 or Ccr2 alone in ApoE −/− mice (Veillard et al. 2005). Cxcl10 deficiency may also be harmful to the cardiovascular system in the ApoE −/− mouse model, since in addition to reduced atherosclerosis, increased aneurysm formation was identified in these mice (King et al. 2009).
CXCR6
CXCR6 is expressed on monocytes, macrophages, T cells and SMCs in both mouse and human atherosclerotic lesions (Wuttge et al. 2004). Genetic deletion of Cxcr6 in ApoE −/− mice significantly decreased the size of atherosclerotic plaques. This was accompanied by a reduced percentage of macrophages and Cxcr6+ T cells within the aortas (Fig. 1) (Galkina et al. 2007). The production of IFN-γ within the aortas of Cxcr6 −/− ApoE −/− mice was also diminished, consistent with the finding that Cxcr6+ T cells express high amounts of IFN-γ upon activation (Calabresi et al. 2002). These results suggested that Cxcr6 might accelerate the progress of atherosclerosis by regulating the migration of T cells into the aortic wall thus influencing the accumulation of macrophages indirectly. The ligand for CXCR6 is CXCL16 and its effect in atherogenesis will be discussed in the following section since an opposite phenotype has been suggested (Aslanian and Charo 2006).
Atheroprotective Chemokines and Chemokine Receptors
CCR1
CCR1 is a chemokine receptor for many pro-inflammatory CC chemokines, including CCL3 and CCL5, both of which have been identified in atherosclerotic plaques; it is mainly expressed on monocytes, macrophages and T cells in humans, but important functional roles have been identified for Ccr1 on mouse neutrophils (Murphy et al. 2000; Wilcox et al. 1994). In contrast to the pro-atherogenic role of Ccl5, Ccr1 −/− bone marrow reconstitution of Ldlr −/− mice resulted in markedly increased atherosclerotic lesion size in both the thoracic aorta and the aortic root (Potteaux et al. 2005). Similarly, Ccr1 −/− ApoE −/− mice showed increased atherosclerotic lesion formation compared with control mice and there is a significant increase of CD3+ T cells and IFN-γ production in the plaques, but the lesional macrophage content was not affected (Braunersreuther et al. 2007). Also, it has been reported that although both CCR1 and CCR5 support trans-endothelial chemotaxis toward CCL5, only CCR1 mediates CCL5-induced arrest of monocytes and Th1 cells on activated endothelium (Fig. 1) (Weber et al. 2001). These results suggest that CCR1 plays a protective role in atherogenesis, possibly by regulating T-cell activation and the trafficking of monocytes and T cells into the vessel wall.
CXCR4
CXCR4 is a receptor only for the chemokine CXCL12 (also known as stromal cell-derived factor-1α), which has been identified in ECs and SMCs of human atherosclerotic plaques (Abi-Younes et al. 2000). It is well known that the CXCL12–CXCR4 axis plays an important role in hematopoietic stem cell mobilization, organ development and angiogenesis, and recent data suggest that it may also be involved in atherosclerosis (Zernecke and Weber 2010). In particular, plasma levels of CXCL12 in CAD patients were significantly reduced compared to those in healthy controls (Damas et al. 2002) and aged ApoE −/− mice had much lower serum and bone marrow levels of Cxcl12 (Xu et al. 2011), indicating that the CXCL12–CXCR4 axis may exert a protective effect in atherogenesis. Consistent with this, chimeric ApoE −/− mice transplanted with Cxcr4 −/− bone marrow showed a marked increase of atherosclerotic lesions in both the whole aorta and the aortic root (Zernecke et al. 2008). This effect was confirmed by either blocking Cxcr4 through long-term administration of a specific pharmacologic antagonist, AMD3465, in ApoE −/− mice or by repopulating Ldlr −/− mice with bone marrow that had been transduced by a recombinant lentivirus encoding a Cxcr4-specific “degrakine”, which traps Cxcr4 in the endoplasmic reticulum (Zernecke et al. 2008). Blocking Cxcr4 with AMD3465 in ApoE −/− mice caused a pronounced leukocytosis and an expansion of circulating neutrophils, and correspondingly the recruitment of neutrophils into the plaques was significantly increased. There was also a significant reduction of SMCs and CD3+ T cells in aortic root plaques, implying that the plaques become more vulnerable (Zernecke et al. 2008). Taken together, these results suggest that the CXCL12–CXCR4 axis may play a protective role in atherogenesis possibly by controlling homeostasis of neutrophils and their recruitment to atherosclerotic lesions (Fig. 1).
Controversial Chemokines/Chemokine Receptors in Atherogenesis
CXCL16
CXCL16 (originally named scavenger receptor for phosphatidylserine and oxidized lipoprotein [SR-PSOX]) is the only other chemokine besides CX3CL1 that exists in both membrane-tethered and soluble shed forms, and it is expressed by macrophages, SMCs and T cells in mouse, rabbit and human atherosclerotic plaques (Hofnagel et al. 2011; Minami et al. 2001a, b; Wuttge et al. 2004). The role of CXCL16 in human atherosclerosis is controversial. One study showed that patients with stable angina pectoris and myocardial infarction have significantly lower plasma levels of CXCL16 (Sheikine et al. 2006), whereas several other groups found that patients with angina, stroke or acute coronary syndromes have elevated plasma CXCL16 levels compared with healthy controls (Lehrke et al. 2007; Smith et al. 2008; Sun et al. 2008; Ueland et al. 2012; Wang et al. 2010; Yi and Zeng 2008). In candidate gene analysis, two SNPs within the CXCL16 gene, rs3744700 and CXCL16-A181V, were reported to be independently associated with CAD development and the severity of coronary stenosis (Huang et al. 2010; Lundberg et al. 2005). In Ldlr −/− mice, genetic deletion of Cxcl16 significantly aggravated atherosclerosis and enhanced macrophage recruitment into the plaques (Aslanian and Charo 2006). However, this is in contrast to the deletion of its receptor Cxcr6 in ApoE −/− mice, which resulted in reduced atherosclerosis (Galkina et al. 2007). Conceptually, an atheroprotective role is plausible since CXCL16 is known to function not just as a chemokine but also as a scavenger receptor for the pro-atherogenic factors, phosphatidylserine and oxLDL (Fig. 1) (Fukumoto et al. 2004; Minami et al. 2001a). Consistent with this, CXCL16 promoted the internalization of oxLDL in human macrophages and Cxcl16 −/− mouse macrophages have a significant reduction in the capacity to bind and internalize oxLDL (Aslanian and Charo 2006; Barlic et al. 2009). However, a recent study showed that the plasma level of Cxcl16 was much higher in ApoE −/− mice receiving a high-fat diet (Yi et al. 2011). Moreover, overexpression of Cxcl16 in ApoE −/− mice did not affect the size of existing atherosclerotic lesions but instead promoted their evolution to vulnerable plaques, suggesting that CXCL16 may be an atherogenic marker in plasma (Yi et al. 2011). Considering the limited studies in atherosclerosis mouse models and the contradictory findings in human CAD patients, more work will be needed to clarify the role of CXCL16 in atherogenesis.
CCR7
CCR7 and its ligands, CCL19 and CCL21, have been identified in both mouse and human atherosclerotic lesions, especially in macrophages and T-cell rich areas (Damas et al. 2007). In humans, the plasma levels of CCL19 and CCL21 were found to be increased in patients with stable and unstable angina (Damas et al. 2007). In Ldlr −/− mice, genetic deletion of Ccr7 attenuated the size of atherosclerotic plaques and reduced the macrophage accumulation in those plaques (Luchtefeld et al. 2010). In contrast, Ccr7 deficiency did not affect the size of atherosclerotic plaques or the plaque macrophage content in ApoE −/− mice (Feig et al. 2010; Potteaux et al. 2011). In an atherosclerosis regression mouse model in which a segment of atherosclerotic ApoE −/− mouse aortic arch was transplanted into wild-type mice, it was found that Ccr7 was upregulated in foam cells during the regression of atherosclerotic lesions, accompanied by a significant reduction of macrophage content in the lesions (Trogan et al. 2006). Plaque regression and foam cell content reduction were both greatly inhibited by the blockade of Ccl19 and Ccl21 with specific antibodies, suggesting that Ccr7 may drive the egress of macrophages from the lesions and have a protective role in atherosclerosis development (Fig. 1) (Trogan et al. 2006). Consistent with this, atorvastatin and rosuvastatin treatment significantly increased the expression of Ccr7 in lesional macrophages and enhanced the emigration of macrophages from the plaques in ApoE −/− mice, while macrophages from Ccr7 −/− ApoE −/− mice failed to emigrate upon statin treatment (Feig et al. 2011a). Also, it was found that liver-X-receptor agonist- and HDL-induced atherosclerotic lesion regression in different atherogenic mouse models are associated with increased expression of Ccr7 on plaque macrophages (Feig et al. 2010, 2011b; Verschuren et al. 2009). However, in a non-surgical model, it was found that Ccr7 had no effect on atherosclerotic plaque regression in ApoE −/− mice, and the loss of plaque macrophages during disease regression was attributed to suppressed monocyte recruitment (Potteaux et al. 2011). In fact, although expression of CCR7 is increased in atherosclerotic plaques, its expression on circulating T cells was significantly decreased in angina patients, indicating that CCR7 may also be involved in infiltration and egress of T cells from the atherosclerotic vessel wall (Fig. 1) (Damas et al. 2007). Clearly, more studies are necessary to dissect the exact role of CCR7 in the development of atherosclerosis.
Chemokine/Chemokine Receptor Antagonists in Atherogenesis
Considering the above evidence from animal models that chemokines and chemokine receptors may modulate atherogenesis (Fig. 1), it is reasonable to consider them as potential drug targets for the treatment of cardiovascular disease. In the USA, more than 30 % of drugs on the market target G protein-coupled receptors and numerous antagonists of chemokine receptors have been developed (Horuk 2009). We will focus on the antagonists that have been tested in atherosclerosis (Table 2). The difficulty in demonstrating efficacy for these agents is the endpoint that must be considered: clinical events or inflammation of the blood vessel wall. The former is difficult to assess in a short trial, and the latter is difficult to quantitate with precision.
Drugs targeting CCR2 and its ligand CCL2 have been the most extensively evaluated in preclinical and clinical studies. In a phase II clinical trial of patients at risk for atherosclerotic cardiovascular disease, MLN1202 (Millennium), a specific humanized monoclonal antibody directed against CCR2, was found to significantly reduce the serum levels of C-reactive protein, a surrogate marker of inflammation in cardiovascular disease (Gilbert et al. 2011). CCX140 (Chemocentryx) is a small molecule inhibitor of CCR2 that has successfully completed a phase II clinical trial in type 2 diabetes, but has not yet been tested in cardiovascular disease (Koenen and Weber 2011). The CCR2 small molecule antagonists, INCB-3344 and GSK1344386B (Glaxo-Smith-Kline), did not affect the size of atherosclerotic lesions in ApoE −/− mice, even though they markedly reduced the number of circulating inflammatory monocytes and macrophage content in plaques (Aiello et al. 2010; Olzinski et al. 2010). As for CCL2 antagonists, 11K2 (an inhibitory antibody against CCL2 and CCL12; Biogen Idec Inc.) treatment of ApoE −/− mice significantly reduced both atherosclerotic plaque formation and lesional macrophage content (Lutgens et al. 2005). Treatment of ApoE −/− mice with PA508, a modified CCL2 form with increased glycosaminoglycan binding activity but reduced affinity for CCR2, markedly reduced neointimal plaque formation after arterial injury, due to reduced inflammatory monocyte recruitment to the lesions (Liehn et al. 2010). A humanized monoclonal antibody against CCL2, ABN-912 (Novartis), has been tested in patients with rheumatoid arthritis, but the treatment did not show any benefit and whether it may affect atherosclerosis still remains unknown (Haringman et al. 2006).
CCR5 and its ligand CCL5 is another chemokine/chemokine receptor pair that has been targeted in atherogenesis. TAK-779 is a small molecule antagonist for both CCR5 and CXCR3 that was originally developed as an HIV entry inhibitor (Baba et al. 1999; Gao et al. 2003). TAK-779 treatment of Ldlr −/− mice significantly reduced atherosclerotic plaque formation and Th1 cell infiltration into the lesions (van Wanrooij et al. 2005). Maraviroc (Pfizer) is a small molecule CCR5 antagonist approved by the FDA for HIV treatment and HGS004/HGS101 (antibodies raised against CCR5; Human Genome Sciences) has also been used to treat HIV in clinical trials (Latinovic et al. 2011), but whether they can affect atherosclerosis is still unclear. Treatment of Ldlr −/− mice with Met-RANTES, an N-methionylated variant of CCL5 with potent CCR5 antagonist activity (Proudfoot et al. 1996), reduced the size of atherosclerotic lesions in both the whole aorta and the aortic root, accompanied by increased plaque stability (Veillard et al. 2004). Met-RANTES treatment of ApoE −/− Ccl2 −/− Cx3cr1 −/− triple knockout mice induced a further decrease of atherosclerotic plaque size, suggesting that it might also be considered alone or in combination with other chemokine receptor antagonists to treat atherosclerosis (Combadiere et al. 2008). Another antagonist for CCL5 is [ 44 AANA 47 ]-CCL5, a CCL5 variant with specific mutations in the principal RANTES/GAG binding site, that was shown to prevent the progression of established atherosclerotic lesions in Ldlr −/− mice (Braunersreuther et al. 2008). MKEY (Carolus Therapeutics Inc.) is a synthetic peptide designed to disrupt the heteromerization of CCL5 and CXCL4, and administration of MKEY into ApoE −/− mice markedly reduced the formation of atherosclerotic lesions as well as macrophage accumulation in plaque (Koenen et al. 2009).
Antagonists against CXCR2, CXCR3 and CXCR4 have also been developed and tested in atherosclerosis models. SB-517785-M (Glaxo-Smith-Kline) is an antagonist that selectively inhibits CXCR2. This agent significantly reduced angiotensin II-induced infiltration of mononuclear cells and neutrophils into rat arterioles, suggesting that it may inhibit atherogenesis by controlling recruitment of neutrophils into the vessel wall (Drechsler et al. 2011; Nabah et al. 2007). Treatment of Ldlr −/− mice with NBI-74330, a CXCR3 antagonist, markedly increased the content of Tregs in atherosclerotic plaques but reduced effector T cells in lymph nodes draining the aortic arch, thus attenuating atherosclerotic lesion formation in these mice (van Wanrooij et al. 2008). As discussed in the previous section, CXCR4 blockade by its antagonist AMD3465 led to aggravated atherosclerotic lesion formation in ApoE −/− mice, probably due to increased neutrophil expansion in the blood and their recruitment into plaque (Zernecke et al. 2008). AMD3100 (Plerixafor; Genzyme) is a CXCR4 antagonist that has been approved by the FDA for stem cell mobilization for autologous transplantation in the setting of ablative chemotherapy for multiple myeloma and non-Hodgkin’s lymphoma, but its role in atherogenesis is still unknown (Keating 2011).
Human CX3CL1 has been modified at the N-terminus to produce a CX3CR1 antagonist named F1 (Dorgham et al. 2009). F1 did not induce signaling through CX3CR1 but potently inhibited CX3CL1-induced chemotaxis, calcium flux and cell adhesion. In a peritonitis mouse model, F1 was found to strongly inhibit macrophage accumulation, indicating that it has anti-inflammatory activity (Dorgham et al. 2009). However, it is still not clear whether this antagonist may affect the progress of atherosclerosis.
There are also some naturally occurring chemokine-blocking agents that target more than one chemokine. For example, Evasins are a group of multipotential chemokine binding proteins found in tick saliva. Evasin-1 binds to CCL3, CCL4, and CCL18; Evasin-3 binds to CXCL1 and CXCL8; and Evasin-4 binds to CCL5 and CCL11 (Deruaz et al. 2008). A single administration of Evasin-3 significantly reduced post-ischemic infarct size during myocardial ischemia in C57BL/6 mice, associated with reduced infiltration of neutrophils into the site of injury and decreased production of ROS (Montecucco et al. 2010). These results suggest that Evasins have anti-inflammatory functions that may be useful for the treatment of atherosclerosis. M-T7 is a 37-kDa glycoprotein that also binds to a broad range of C, CC and CXC chemokines through the conserved C-terminal GAG binding domain of the chemokines (Lalani et al. 1997). Administration of purified M-T7 into rats caused a significant reduction of intimal hyperplasia after angioplasty injury (Liu et al. 2000) and inhibited aortic allograft vasculopathy associated with reduced inflammatory cell invasion (Dai et al. 2010; Liu et al. 2004), indicating that it may prevent recurrent atherosclerotic plaque growth. NR58-3.14.3 is a broad-spectrum chemokine-blocking peptide that effectively inhibits the activities of CCL2, CCL3, CXCL8 and CXCL12, and treatment of ApoE −/− mice with this peptide significantly reduced macrophage accumulation in vascular lesions and increased the content of collagen and SMCs, although the vascular lipid lesion area was not changed (Reckless et al. 2005). Also, tail-vein injection of a recombinant adenovirus encoding soluble protein 35K, a broad-spectrum CC-chemokine blocking agent encoded by Vaccinia virus, into ApoE −/− mice markedly reduced atherosclerotic plaque formation and atherosclerosis in carotid–caval vein grafts, accompanied by decreased macrophage recruitment into the lesions (Ali et al. 2005; Bursill et al. 2004). These results have suggested that broad-spectrum chemokine-blocking peptides might be useful to inhibit the progress of atherosclerosis and promote the stabilization of atherosclerotic plaques.
Recent studies suggest that statins (HMG-CoA reductase inhibitors), a class of drugs used for the primary and secondary prevention of CAD, may also affect the expression of chemokines and chemokine receptors during atherosclerosis (Feig et al. 2011a; Waehre et al. 2003). Statins are known to lower cholesterol levels in hypercholesterolemic patients and it has been shown that normalization of plasma lipid levels may cause atherosclerosis regression in both humans and atherosclerosis-prone mouse models (Nissen et al. 2006; Trogan et al. 2006). The regression of atherosclerosis is accompanied by reduced plaque inflammatory cell content (e.g., macrophages), which may be due to increased efflux of leukocytes from plaques (Trogan et al. 2006) and decreased influx of inflammatory cells into plaques (Potteaux et al. 2011). The increased efflux and reduced influx of leukocytes are associated with increased expression of Ccr7 and reduced expression of Ccl2, respectively (Lieu et al. 2003; Trogan et al. 2006). In support of this, statin treatment significantly increased the expression of Ccr7 in lesional macrophages and enhanced their emigration from the plaques of ApoE −/− mice (Feig et al. 2011a). In vitro statin treatment significantly inhibited the expression of CCL2 in peripheral blood mononuclear cell (PBMC), ECs and monocytes/macrophages (Jougasaki et al. 2010; Morikawa et al. 2002; Romano et al. 2000; Veillard et al. 2006). In vivo simvastatin treatment reduced expression of CCL2 and CCR2 on circulating monocytes in both hypercholesterolemic patients and healthy people (Han et al. 2005; Rezaie-Majd et al. 2002); both atorvastatin and simvastatin significantly inhibited the expression of CCR2 and CX3CR1 on PBMCs in CAD patients (Damas et al. 2005; Waehre et al. 2003). In addition, statin treatment has been reported to downregulate the expression of CCR1, CCL3, CCL4, CCL19, CCL21, CXCL8, CXCL16 and CX3CL1 in different studies (Damas et al. 2005, 2007; Smith et al. 2008; Waehre et al. 2003), suggesting that statins may have a broad effect on the expression of atherogenic chemokines during the reversal of hypercholesterolemia in cardiovascular patients. However, not all patients can benefit from using statins and as many as 20 % of the patients who use statins develop adverse effects such as muscle fatigue and weakness and impaired cognition (Maningat and Breslow 2011). New drugs are needed to treat those statin-intolerant patients and chemokine and chemokine receptor antagonists are worth considering in this setting, either as prevention or in combination with statins in treatment of atherosclerosis.
Conclusions
Atherosclerosis is the leading cause of mortality worldwide. Although many drugs in several classes are approved for the treatment of cardiovascular disease, so far only statins target atherosclerosis by lowering the cholesterol level. However, not all patients benefit from statins, and new therapeutics are needed. In the past decade, numerous studies in mice and humans have suggested that chemokines and their corresponding G protein-coupled chemokine receptors play important roles in the pathophysiology of atherosclerosis development. For example, CCL2–CCR2, CCL5–CCR5 and CX3CL1–CX3CR1 are found to be pathogenic whereas CCR1 and CXCR4 are protective, indicating that antagonists for CCR2, CCR5, CX3CR1 and agonists for CCR1 and CXCR4 may be beneficial for therapeutic intervention. However, although more than 30 % of the drugs on the USA market are targeted against G protein-coupled receptors and many chemokine receptor antagonists have been tested in different mouse models and clinical trials, only two small molecule antagonists of chemokine receptors have been approved by the FDA: the CCR5 antagonist Maraviroc for HIV treatment and the CXCR4 antagonist AMD3100 (Plerixafor) for multiple myeloma and non-Hodgkin’s lymphoma. Despite substantial progress in identifying chemokine system targets and developing lead compounds, much more research will be necessary to define the cellular mechanisms by which they operate as well as to evaluate current drug candidates and identify new ones. Also, it would be necessary to evaluate the effect of these new drugs on the immune system and antimicrobial host defense during long-term use.
References
Abi-Younes S, Sauty A, Mach F et al (2000) The stromal cell-derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ Res 86:131–138
Aiello RJ, Bourassa PA, Lindsey S et al (1999) Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 19:1518–1525
Aiello RJ, Perry BD, Bourassa PA et al (2010) CCR2 receptor blockade alters blood monocyte subpopulations but does not affect atherosclerotic lesions in apoE(−/−) mice. Atherosclerosis 208:370–375
Ali ZA, Bursill CA, Hu Y et al (2005) Gene transfer of a broad spectrum CC-chemokine inhibitor reduces vein graft atherosclerosis in apolipoprotein E-knockout mice. Circulation 112:I235–I1241
Apostolakis S, Krambovitis E, Vlata Z et al (2007) CX3CR1 receptor is up-regulated in monocytes of coronary artery diseased patients: impact of pre-inflammatory stimuli and renin–angiotensin system modulators. Thromb Res 121:387–395
Apostolopoulos J, Davenport P, Tipping PG (1996) Interleukin-8 production by macrophages from atheromatous plaques. Arterioscler Thromb Vasc Biol 16:1007–1012
Aslanian AM, Charo IF (2006) Targeted disruption of the scavenger receptor and chemokine CXCL16 accelerates atherosclerosis. Circulation 114:583–590
Baba M, Nishimura O, Kanzaki N et al (1999) A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci USA 96:5698–5703
Barlic J, Murphy PM (2007a) Chemokine regulation of atherosclerosis. J Leukoc Biol 82:226–236
Barlic J, Murphy PM (2007b) An oxidized lipid-peroxisome proliferator-activated receptor gamma-chemokine pathway in the regulation of macrophage-vascular smooth muscle cell adhesion. Trends Cardiovasc Med 17:269–274
Barlic J, Zhu W, Murphy PM (2009) Atherogenic lipids induce high-density lipoprotein uptake and cholesterol efflux in human macrophages by up-regulating transmembrane chemokine CXCL16 without engaging CXCL16-dependent cell adhesion. J Immunol 182:7928–7936
Bazan JF, Bacon KB, Hardiman G et al (1997) A new class of membrane-bound chemokine with a CX3C motif. Nature 385:640–644
Berger JS, Jordan CO, Lloyd-Jones D et al (2010) Screening for cardiovascular risk in asymptomatic patients. J Am Coll Cardiol 55:1169–1177
Boger CA, Fischereder M, Deinzer M et al (2005) RANTES gene polymorphisms predict all-cause and cardiac mortality in type 2 diabetes mellitus hemodialysis patients. Atherosclerosis 183:121–129
Boisvert WA, Santiago R, Curtiss LK et al (1998) A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor-deficient mice. J Clin Invest 101:353–363
Boisvert WA, Curtiss LK, Terkeltaub RA (2000) Interleukin-8 and its receptor CXCR2 in atherosclerosis. Immunol Res 21:129–137
Boisvert WA, Rose DM, Johnson KA et al (2006) Up-regulated expression of the CXCR2 ligand KC/GRO-alpha in atherosclerotic lesions plays a central role in macrophage accumulation and lesion progression. Am J Pathol 168:1385–1395
Boring L, Gosling J, Chensue SW et al (1997) Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C–C chemokine receptor 2 knockout mice. J Clin Invest 100:2552–2561
Boring L, Gosling J, Cleary M et al (1998) Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 394:894–897
Braunersreuther V, Zernecke A, Steffens S et al (2007) Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler Thromb Vasc Biol 27:373–379
Braunersreuther V, Steffens S, Arnaud C et al (2008) A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arterioscler Thromb Vasc Biol 28:1090–1096
Breland UM, Halvorsen B, Hol J et al (2008) A potential role of the CXC chemokine GROalpha in atherosclerosis and plaque destabilization: downregulatory effects of statins. Arterioscler Thromb Vasc Biol 28:1005–1011
Bursill CA, Choudhury RP, Ali Z et al (2004) Broad-spectrum CC-chemokine blockade by gene transfer inhibits macrophage recruitment and atherosclerotic plaque formation in apolipoprotein E-knockout mice. Circulation 110:2460–2466
Butcher M, Galkina E (2011) Current views on the functions of interleukin-17A-producing cells in atherosclerosis. Thromb Haemost 106:787–795
Calabresi PA, Yun SH, Allie R et al (2002) Chemokine receptor expression on MBP reactive T cells: CXCR6 is a marker of IFNgamma-producing effector cells. J Neuroimmunol 127:96–105
Calvayrac O, Rodríguez-Calvo R, Alonso J et al (2011) CCL20 is increased in hypercholesterolemic subjects and is upregulated by LDL in vascular smooth muscle cells: role of NF-κB. Arterioscler Thromb Vasc Biol 31:2733–2741
Combadiere C, Potteaux S, Gao JL et al (2003) Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation 107:1009–1016
Combadiere C, Potteaux S, Rodero M et al (2008) Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117:1649–1657
Dai E, Liu LY, Wang H et al (2010) Inhibition of chemokine-glycosaminoglycan interactions in donor tissue reduces mouse allograft vasculopathy and transplant rejection. PLoS One 5:e10510
Damas JK, Waehre T, Yndestad A et al (2002) SDF-1α in unstable angina: potential antiinflammatory and matrix-stabilizing effects. Circulation 106:36–42
Damas JK, Boullier A, Waehre T et al (2005) Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, is elevated in coronary artery disease and is reduced during statin therapy. Arterioscler Thromb Vasc Biol 25:2567–2572
Damas JK, Smith C, Oie E et al (2007) Enhanced expression of the homeostatic chemokines CCL19 and CCL21 in clinical and experimental atherosclerosis: possible pathogenic role in plaque destabilization. Arterioscler Thromb Vasc Biol 27:614–620
Daoudi M, Lavergne E, Garin A et al (2004) Enhanced adhesive capacities of the naturally occurring Ile249-Met280 variant of the chemokine receptor CX3CR1. J Biol Chem 279:19649–19657
Dawson TC, Kuziel WA, Osahar TA et al (1999) Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 143:205–211
de Waard V, Bot I, de Jager SC et al (2010) Systemic MCP1/CCR2 blockade and leukocyte specific MCP1/CCR2 inhibition affect aortic aneurysm formation differently. Atherosclerosis 211:84–89
Deruaz M, Frauenschuh A, Alessandri AL et al (2008) Ticks produce highly selective chemokine binding proteins with antiinflammatory activity. J Exp Med 205:2019–2031
Doran AC, Lipinski MJ, Oldham SN et al (2012) B-cell aortic homing and atheroprotection depend on Id3. Circ Res 110:e1–e12
Dorgham K, Ghadiri A, Hermand P et al (2009) An engineered CX3CR1 antagonist endowed with anti-inflammatory activity. J Leukoc Biol 86:903–911
Drechsler M, Megens RT, van Zandvoort M et al (2010) Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 122:1837–1845
Drechsler M, Döring Y, Megens RT et al (2011) Neutrophilic granulocytes—promiscuous accelerators of atherosclerosis. Thromb Haemost 106:839–848
Feig JE, Pineda-Torra I, Sanson M et al (2010) LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J Clin Invest 120:4415–4424
Feig JE, Shang Y, Rotllan N et al (2011a) Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS One 6:e28534
Feig JE, Rong JX, Shamir R et al (2011b) HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci USA 108:7166–7171
Fernandes JL, Mamoni RL, Orford JL et al (2004) Increased Th1 activity in patients with coronary artery disease. Cytokine 26:131–137
Fukumoto N, Shimaoka T, Fujimura H et al (2004) Critical roles of CXC chemokine ligand 16/scavenger receptor that binds phosphatidylserine and oxidized lipoprotein in the pathogenesis of both acute and adoptive transfer experimental autoimmune encephalomyelitis. J Immunol 173:1620–1627
Galkina E, Ley K (2009) Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol 27:165–197
Galkina E, Harry BL, Ludwig A et al (2007) CXCR6 promotes atherosclerosis by supporting T-cell homing, interferon-gamma production, and macrophage accumulation in the aortic wall. Circulation 116:1801–1811
Gao P, Zhou XY, Yashiro-Ohtani Y et al (2003) The unique target specificity of a nonpeptide chemokine receptor antagonist: selective blockade of two Th1 chemokine receptors CCR5 and CXCR3. J Leukoc Biol 73:273–280
Ghilardi G, Biondi ML, Turri O et al (2004) Internal carotid artery occlusive disease and polymorphisms of fractalkine receptor CX3CR1: a genetic risk factor. Stroke 35:1276–1279
Gilbert J, Lekstrom-Himes J, Donaldson D et al (2011) Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am J Cardiol 107:906–911
Gonzalez P, Alvarez R, Batalla A et al (2001) Genetic variation at the chemokine receptors CCR5/CCR2 in myocardial infarction. Genes Immun 2:191–195
Gosling J, Slaymaker S, Gu L et al (1999) MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J Clin Invest 103:773–778
Greaves DR, Hakkinen T, Lucas AD et al (2004) Linked chromosome 16q13 chemokines, macrophage-derived chemokine, fractalkine, and thymus- and activation-regulated chemokine, are expressed in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol 21:923–929
Gu L, Okada Y, Clinton SK et al (1998) Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell 2:275–281
Gugl A, Renner W, Seinost G et al (2003) Two polymorphisms in the fractalkine receptor CX3CR1 are not associated with peripheral arterial disease. Atherosclerosis 166:339–343
Guo J, Van Eck M, Twisk J et al (2003) Transplantation of monocyte CC-chemokine receptor 2-deficient bone marrow into ApoE3-Leiden mice inhibits atherogenesis. Arterioscler Thromb Vasc Biol 23:447–453
Guo J, de Waard V, Van Eck M et al (2005) Repopulation of apolipoprotein E knockout mice with CCR2-deficient bone marrow progenitor cells does not inhibit ongoing atherosclerotic lesion development. Arterioscler Thromb Vasc Biol 25:1014–1019
Han KH, Ryu J, Hong KH et al (2005) HMG-CoA reductase inhibition reduces monocyte CC chemokine receptor 2 expression and monocyte chemoattractant protein-1-mediated monocyte recruitment in vivo. Circulation 111:1439–1447
Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352:1685–1695
Haringman JJ, Gerlag DM, Smeets TJ et al (2006) A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum 54:2387–2392
Heller EA, Liu E, Tager AM et al (2006) Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation 113:2301–2312
Hofnagel O, Engel T, Severs NJ et al (2011) SR-PSOX at sites predisposed to atherosclerotic lesion formation mediates monocyte–endothelial cell adhesion. Atherosclerosis 217:371–378
Horuk R (2009) Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov 8:23–33
Huang M, Han Y, Zhang X et al (2010) An intron polymorphism in the CXCL16 gene is associated with increased risk of coronary artery disease in Chinese Han population: a large angiography-based study. Atherosclerosis 210:160–165
Huo Y, Weber C, Forlow SB et al (2001) The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J Clin Invest 108:1307–1314
Imai T, Baba M, Nishimura M et al (1997a) The T cell-directed CC chemokine TARC is a highly specific biological ligand for CC chemokine receptor 4. J Biol Chem 272:15036–15042
Imai T, Hieshima K, Haskell C et al (1997b) Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91:521–530
Inoue S, Egashira K, Ni W et al (2002) Anti-monocyte chemoattractant protein-1 gene therapy limits progression and destabilization of established atherosclerosis in apolipoprotein E-knockout mice. Circulation 106:2700–2706
Jougasaki M, Ichiki T, Takenoshita Y et al (2010) Statins suppress interleukin-6-induced monocyte chemo-attractant protein-1 by inhibiting Janus kinase/signal transducers and activators of transcription pathways in human vascular endothelial cells. Br J Pharmacol 159:1294–1303
Kawamura A, Miura S, Fujino M et al (2003) CXCR3 chemokine receptor–plasma IP10 interaction in patients with coronary artery disease. Circ J 67:851–854
Keating GM (2011) Plerixafor: a review of its use in stem-cell mobilization in patients with lymphoma or multiple myeloma. Drugs 71:1623–1647
King VL, Lin AY, Kristo F et al (2009) Interferon-gamma and the interferon-inducible chemokine CXCL10 protect against aneurysm formation and rupture. Circulation 119:426–435
Koenen RR, Weber C (2011) Chemokines: established and novel targets in atherosclerosis. EMBO Mol Med 3:713–725
Koenen RR, von Hundelshausen P, Nesmelova IV et al (2009) Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med 15:97–103
Krohn R, Raffetseder U, Bot I et al (2007) Y-box binding protein-1 controls CC chemokine ligand-5 (CCL5) expression in smooth muscle cells and contributes to neointima formation in atherosclerosis-prone mice. Circulation 116:1812–1820
Kuziel WA, Dawson TC, Quinones M et al (2003) CCR5 deficiency is not protective in the early stages of atherogenesis in apoE knockout mice. Atherosclerosis 167:25–32
Kyaw T, Tipping P, Toh BH et al (2011) Current understanding of the role of B cell subsets and intimal and adventitial B cells in atherosclerosis. Curr Opin Lipidol 22:373–379
Lalani AS, Graham K, Mossman K et al (1997) The purified myxoma virus gamma interferon receptor homolog M-T7 interacts with the heparin-binding domains of chemokines. J Virol 71:4356–4363
Landsman L, Bar-On L, Zernecke A et al (2009) CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood 113:963–972
Latinovic O, Reitz M, Le NM et al (2011) CCR5 antibodies HGS004 and HGS101 preferentially inhibit drug-bound CCR5 infection and restore drug sensitivity of Maraviroc-resistant HIV-1 in primary cells. Virology 411:32–40
Lehrke M, Millington SC, Lefterova M et al (2007) CXCL16 is a marker of inflammation, atherosclerosis, and acute coronary syndromes in humans. J Am Coll Cardiol 49:442–449
Lei ZB, Zhang Z, Jing Q et al (2002) OxLDL upregulates CXCR2 expression in monocytes via scavenger receptors and activation of p38 mitogen-activated protein kinase. Cardiovasc Res 53:524–532
Lesnik P, Haskell CA, Charo IF (2003) Decreased atherosclerosis in CX3CR1−/− mice reveals a role for fractalkine in atherogenesis. J Clin Invest 111:333–340
Ley K, Laudanna C, Cybulsky MI et al (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7:678–689
Liehn EA, Piccinini AM, Koenen RR et al (2010) A new monocyte chemotactic protein-1/chemokine CC motif ligand-2 competitor limiting neointima formation and myocardial ischemia/reperfusion injury in mice. J Am Coll Cardiol 56:1847–1857
Lieu HD, Withycombe SK, Walker Q et al (2003) Eliminating atherogenesis in mice by switching off hepatic lipoprotein secretion. Circulation 107:1315–1321
Liu L, Lalani A, Dai E et al (2000) The viral anti-inflammatory chemokine-binding protein M-T7 reduces intimal hyperplasia after vascular injury. J Clin Invest 105:1613–1621
Liu L, Dai E, Miller L et al (2004) Viral chemokine-binding proteins inhibit inflammatory responses and aortic allograft transplant vasculopathy in rat models. Transplantation 77:1652–1660
Liu P, Yu YR, Spencer JA et al (2008) CX3CR1 deficiency impairs dendritic cell accumulation in arterial intima and reduces atherosclerotic burden. Arterioscler Thromb Vasc Biol 28:243–250
Lucas AD, Bursill C, Guzik TJ et al (2003) Smooth muscle cells in human atherosclerotic plaques express the fractalkine receptor CX3CR1 and undergo chemotaxis to the CX3C chemokine fractalkine (CX3CL1). Circulation 108:2498–2504
Luchtefeld M, Grothusen C, Gagalick A et al (2010) Chemokine receptor 7 knockout attenuates atherosclerotic plaque development. Circulation 122:1621–1628
Lundberg GA, Kellin A, Samnegard A et al (2005) Severity of coronary artery stenosis is associated with a polymorphism in the CXCL16/SR-PSOX gene. J Intern Med 257:415–422
Lutgens E, Faber B, Schapira K et al (2005) Gene profiling in atherosclerosis reveals a key role for small inducible cytokines: validation using a novel monocyte chemoattractant protein monoclonal antibody. Circulation 111:3443–3452
Mach F, Sauty A, Iarossi AS et al (1999) Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest 104:1041–1050
Maningat P, Breslow JL (2011) Needed: pragmatic clinical trials for statin-intolerant patients. N Engl J Med 365:2250–2251
McDermott DH, Halcox JPJ, Schenke WH et al (2001) Association between polymorphism in the chemokine receptor CX3CR1 and coronary vascular endothelial dysfunction and atherosclerosis. Circ Res 89:401–407
McDermott DH, Fong AM, Yang Q et al (2003) Chemokine receptor mutant CX3CR1–M280 has impaired adhesive function and correlates with protection from cardiovascular disease in humans. J Clin Invest 111:1241–1250
McDermott DH, Yang Q, Kathiresan S et al (2005) CCL2 polymorphisms are associated with serum monocyte chemoattractant protein-1 levels and myocardial infarction in the Framingham Heart Study. Circulation 112:1113–1120
Minami M, Kume N, Shimaoka T et al (2001a) Expression of SR-PSOX, a novel cell-surface scavenger receptor for phosphatidylserine and oxidized LDL in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol 21:1796–1800
Minami M, Kume N, Shimaoka T et al (2001b) Expression of scavenger receptor for phosphatidylserine and oxidized lipoprotein (SR-PSOX) in human atheroma. Ann N Y Acad Sci 947:373–376
Moatti D, Faure S, Fumeron F et al (2001) Polymorphism in the fractalkine receptor CX3CR1 as a genetic risk factor for coronary artery disease. Blood 97:1925–1928
Montecucco F, Lenglet S, Braunersreuther V et al (2010) Single administration of the CXC chemokine-binding protein Evasin-3 during ischemia prevents myocardial reperfusion injury in mice. Arterioscler Thromb Vasc Biol 30:1371–1377
Morikawa S, Takabe W, Mataki C et al (2002) The effect of statins on mRNA levels of genes related to inflammation, coagulation, and vascular constriction in HUVEC. J Atheroscler Thromb 9:178–183
Murphy PM (2002) International Union of Pharmacology. XXX. Update on chemokine receptor nomenclature. Pharmacol Rev 54:227–229
Murphy PM, Baggiolini M, Charo IF et al (2000) International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev 52:145–176
Nabah YNA, Losada M, Estelles R et al (2007) CXCR2 blockade impairs angiotensin II induced CC chemokine synthesis and mononuclear leukocyte infiltration. Arterioscler Thromb Vasc Biol 27:2370–2376
Nelken NA, Coughlin SR, Gordon D et al (1991) Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest 88:1121–1127
Neumann FJ, Ott I, Gawaz M et al (1995) Cardiac release of cytokines and inflammatory responses in acute myocardial infarction. Circulation 92:748–755
Ni W, Egashira K, Kitamoto S et al (2001) New anti-monocyte chemoattractant protein-1 gene therapy attenuates atherosclerosis in apolipoprotein E-knockout mice. Circulation 103:2096–2101
Niessner A, Marculescu R, Kvakan H et al (2005) Fractalkine receptor polymorphisms V2491 and T280M as genetic risk factors for restenosis. Thromb Haemost 94:1251–1256
Nissen SE, Nicholls SJ, Sipahi I et al (2006) Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 295:1556–1565
Norata GD, Garlaschelli K, Ongari M et al (2006) Effects of fractalkine receptor variants on common carotid artery intima-media thickness. Stroke 37:1558–1561
Olzinski AR, Turner GH, Bernard RE et al (2010) Pharmacological inhibition of C–C chemokine receptor 2 decreases macrophage infiltration in the aortic root of the human C–C chemokine receptor 2/apolipoprotein E−/− mouse: magnetic resonance imaging assessment. Arterioscler Thromb Vasc Biol 30:253–259
Ortlepp JR, Vesper K, Mevissen V et al (2003) Chemokine receptor (CCR2) genotype is associated with myocardial infarction and heart failure in patients under 65 years of age. J Mol Med 81:363–367
Packard RR, Lichtman AH, Libby P (2009) Innate and adaptive immunity in atherosclerosis. Semin Immunopathol 31:5–22
Papadopoulou C, Corrigall V, Taylor PR et al (2008) The role of the chemokines MCP-1, GRO-alpha, IL-8 and their receptors in the adhesion of monocytic cells to human atherosclerotic plaques. Cytokine 43:181–186
Pattison JM, Nelson PJ, Huie P et al (1996) RANTES chemokine expression in transplant-associated accelerated atherosclerosis. J Heart Lung Transplant 15:1194–1199
Postea O, Vasina EM, Cauwenberghs S et al (2012) Contribution of platelet CX3CR1 to platelet–monocyte complex formation and vascular recruitment during hyperlipidemia. Arterioscler Thromb Vasc Biol 32:1186–1193
Potteaux S, Combadiere C, Esposito B et al (2005) Chemokine receptor CCR1 disruption in bone marrow cells enhances atherosclerotic lesion development and inflammation in mice. Mol Med 11:16–20
Potteaux S, Combadiere C, Esposito B et al (2006) Role of bone marrow-derived CC-chemokine receptor 5 in the development of atherosclerosis of low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol 26:1858–1863
Potteaux S, Gautier EL, Hutchison SB et al (2011) Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest 121:2025–2036
Proudfoot AE, Power CA, Hoogewerf AJ et al (1996) Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem 271:2599–2603
Quinones MP, Martinez HG, Jimenez F et al (2007) CC chemokine receptor 5 influences late-stage atherosclerosis. Atherosclerosis 195:e92–e103
Rayner K, Van ES, Groot PH et al (2000) Localization of mRNA for JE/MCP-1 and its receptor CCR2 in atherosclerotic lesions of the ApoE knockout mouse. J Vasc Res 37:93–102
Reckless J, Tatalick L, Wilbert S et al (2005) Broad-spectrum chemokine inhibition reduces vascular macrophage accumulation and collagenolysis consistent with plaque stabilization in mice. J Vasc Res 42:492–502
Rezaie-Majd A, Maca T, Bucek RA et al (2002) Simvastatin reduces expression of cytokines interleukin-6, interleukin-8, and monocyte chemoattractant protein-1 in circulating monocytes from hypercholesterolemic patients. Arterioscler Thromb Vasc Biol 22:1194–1199
Robbins CS, Chudnovskiy A, Rauch PJ et al (2012) Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 125:364–374
Robertson AK, Hansson GK (2006) T cells in atherogenesis: for better or for worse? Arterioscler Thromb Vasc Biol 26:2421–2432
Roger VL, Go AS, Lloyd-Jones DM et al (2012) Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 125:e2–e220
Romano M, Diomede L, Sironi M et al (2000) Inhibition of monocyte chemotactic protein-1 synthesis by statins. Lab Invest 80:1095–1100
Saederup N, Chan L, Lira SA et al (2008) Fractalkine deficiency markedly reduces macrophage accumulation and atherosclerotic lesion formation in CCR2−/− mice: evidence for independent chemokine functions in atherogenesis. Circulation 117:1642–1648
Schutyser E, Struyf S, Van Damme J (2003) The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev 14:409–426
Sheikine Y, Bang CS, Nilsson L et al (2006) Decreased plasma CXCL16/SR-PSOX concentration is associated with coronary artery disease. Atherosclerosis 188:462–466
Simeoni E, Winkelmann BR, Hoffmann MM et al (2004) Association of RANTES G-403A gene polymorphism with increased risk of coronary arteriosclerosis. Eur Heart J 25:1438–1446
Smith C, Halvorsen B, Otterdal K et al (2008) High levels and inflammatory effects of soluble CXC ligand 16 (CXCL16) in coronary artery disease: down-regulatory effects of statins. Cardiovasc Res 79:195–203
Sun Y, Chang Z, Zhang S (2008) Increased serum CXCL16 level is a marker for acute coronary syndromes. Arch Med Res 39:332–337
Swirski FK, Nahrendorf M, Etzrodt M et al (2009) Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325:612–616
Szalai C, Duba J, Prohaszka Z et al (2001) Involvement of polymorphisms in the chemokine system in the susceptibility for coronary artery disease (CAD). Coincidence of elevated Lp(a) and MCP-1 -2518 G/G genotype in CAD patients. Atherosclerosis 158:233–239
Tacke F, Alvarez D, Kaplan TJ et al (2007) Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 117:185–194
Teupser D, Pavlides S, Tan M et al (2004) Major reduction of atherosclerosis in fractalkine (CX3CL1)-deficient mice is at the brachiocephalic artery, not the aortic root. Proc Natl Acad Sci USA 101:17795–17800
Trogan E, Feig JE, Dogan S et al (2006) Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci USA 103:3781–3786
Tsou CL, Peters W, Si Y et al (2007) Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117:902–909
Ueland T, Smedbakken LM, Hallén J et al (2012) Soluble CXCL16 and long-term outcome in acute ischemic stroke. Atherosclerosis 220:244–249
van Wanrooij EJ, Happe H, Hauer AD et al (2005) HIV entry inhibitor TAK-779 attenuates atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 25:2642–2647
van Wanrooij EJ, de Jager SC, van Es T et al (2008) CXCR3 antagonist NBI-74330 attenuates atherosclerotic plaque formation in LDL receptor deficient mice. Arterioscler Thromb Vasc Biol 28:251–257
Veillard NR, Kwak B, Pelli G et al (2004) Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res 94:253–261
Veillard NR, Steffens S, Pelli G et al (2005) Differential influence of chemokine receptors CCR2 and CXCR3 in development of atherosclerosis in vivo. Circulation 112:870–878
Veillard NR, Braunersreuther V, Arnaud C et al (2006) Simvastatin modulates chemokine and chemokine receptor expression by geranylgeranyl isoprenoid pathway in human endothelial cells and macrophages. Atherosclerosis 188:51–58
Verschuren L, de Vries-van der Weij J, Zadelaar S et al (2009) LXR agonist suppresses atherosclerotic lesion growth and promotes lesion regression in apoE*3Leiden mice: time course and mechanisms. J Lipid Res 50:301–311
Waehre T, Damås JK, Gullestad L et al (2003) Hydroxymethylglutaryl coenzyme a reductase inhibitors down-regulate chemokines and chemokine receptors in patients with coronary artery disease. J Am Coll Cardiol 41:1460–1467
Wan W, Murphy PM (2011) Regulation of atherogenesis by chemokine receptor CCR6. Trends Cardiovasc Med 21:140–144
Wan W, Lim JK, Lionakis MS et al (2011) Genetic deletion of chemokine receptor Ccr6 decreases atherogenesis in ApoE-deficient mice. Circ Res 109:374–381
Wang N, Tabas I, Winchester R et al (1996) Interleukin 8 is induced by cholesterol loading of macrophages and expressed by macrophage foam cells in human atheroma. J Biol Chem 271:8837–8842
Wang KD, Liu ZZ, Wang RM et al (2010) Chemokine CXC Ligand 16 serum concentration but not A181V genotype is associated with atherosclerotic stroke. Clin Chim Acta 411:1447–1451
Weber C, Noels H (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17:1410–1422
Weber C, Weber KS, Klier C et al (2001) Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and T(H)1-like/CD45RO(+) T cells. Blood 97:1144–1146
Weber C, Zernecke A, Libby P (2008) The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol 8:802–815
Weber C, Meiler S, Döring Y et al (2011) CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest 121:2898–2910
Wilcox JN, Nelken NA, Coughlin SR et al (1994) Local expression of inflammatory cytokines in human atherosclerotic plaques. J Atheroscler Thromb 1:S10–S13
Wuttge DM, Zhou X, Sheikine Y et al (2004) CXCL16/SR-PSOX is an interferon-gamma-regulated chemokine and scavenger receptor expressed in atherosclerotic lesions. Arterioscler Thromb Vasc Biol 24:750–755
Xu Q, Wang J, He J et al (2011) Impaired CXCR4 expression and cell engraftment of bone marrow-derived cells from aged atherogenic mice. Atherosclerosis 219:92–99
Yeh M, Leitinger N, de Martin R et al (2001) Increased transcription of IL-8 in endothelial cells is differentially regulated by TNF-alpha and oxidized phospholipids. Arterioscler Thromb Vasc Biol 21:1585–1591
Yi GW, Zeng QT (2008) Circulating CXCL16 is related to the severity of coronary artery stenosis. Arch Med Res 39:531–535
Yi GW, Zeng QT, Mao XB et al (2011) Overexpression of CXCL16 promotes a vulnerable plaque phenotype in apolipoprotein E-knockout mice. Cytokine 53:320–326
Yilmaz A, Lipfert B, Cicha I et al (2007) Accumulation of immune cells and high expression of chemokines/chemokine receptors in the upstream shoulder of atherosclerotic carotid plaques. Exp Mol Pathol 82:245–255
Zernecke A, Weber C (2010) Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc Res 86:192–201
Zernecke A, Bot I, Djalali-Talab Y et al (2008) Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res 102:209–217
Acknowledgments
This work was supported by the Intramural Research Program of the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (Bethesda, MD, USA).
Conflict of interest
None.
Note added in proof
We have recently reported that Ccr7-deficient ApoE knockout mice fed a Western diet have markedly increased atherosclerotic plaque. This effect was due to Ccr7 expression on a bone marrow-derived cell and was associated with increased lesional and blood T cells. (Reference: Wan W, Lionakis MS, Liu Q et al (2012) Genetic deletion of chemokine receptor Ccr7 exacerbates atherogenesis in ApoE-deficient mice. Cardiovasc Res PMID: 23180724).
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Wan, W., Murphy, P.M. Regulation of Atherogenesis by Chemokines and Chemokine Receptors. Arch. Immunol. Ther. Exp. 61, 1–14 (2013). https://doi.org/10.1007/s00005-012-0202-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-012-0202-1