
Abstract
The immune system is the central regulator of tissue homeostasis, ensuring tissue regeneration and protection against both pathogens and the neoformation of cancer cells. Its proper functioning requires homeostatic properties, which are maintained by an adequate balance of myeloid and lymphoid responses. Aging progressively undermines this ability and compromises the correct activation of immune responses, as well as the resolution of the inflammatory response. A subclinical syndrome of “homeostatic frailty” appears as a distinctive trait of the elderly, which predisposes to immune debilitation and chronic low-grade inflammation (inflammaging), causing the uncontrolled development of chronic and degenerative diseases. The innate immune compartment, in particular, undergoes to a sequela of age-dependent functional alterations, encompassing steps of myeloid progenitor differentiation and altered responses to endogenous and exogenous threats. Here, we will review the age-dependent evolution of myeloid populations, as well as their impact on frailty and diseases of the elderly.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Life expectancy is increasing worldwide and by 2040, it is expected to further improve and exceed 80 years in most countries. This implicates a potential raise of incidence and prevalence of diseases associated with aging, including cardiovascular, neurological, musculoskeletal, and oncological diseases [1]. Therefore, medicine has to make an effort to face changes in the epidemiology of diseases and to improve the management of complex and frail patients. In parallel, scientific research has to deepen our knowledge in the mechanisms involved in the onset of age-related diseases.
Aging is considered as the progressive decline of functional performance leading to an increased risk of mortality [2]. Nonetheless, it is still debated whether aging is a programmed and regulated process, or a consequence of casual accumulation of stressor events ending in a reduced performance state. In fact, a central role is played by cellular senescence, i.e., the progressive and irreversible cellular loss of proliferative capacity. Therefore, the relative contribution of the genetic background individual and the environmental stresses that it encounters, in determining aging and cellular senescence, as well as in undermining our adaptive capacities against life events, is still far to be elucidated. [3]. Notably, cellular loss of proliferative capacity might be beneficial in some contexts, as embryonic development, tissue repair, and tumor suppression, but it can also become detrimental, as senescent cells in old tissues acquire an altered phenotype, with the production and secretion of pro-inflammatory molecules [3]. In this context, the concept of aging had been introduced, as the result of the imbalance between stressors and compensatory mechanisms, leading to the accumulation of damage, reduced functional reserve, and impaired healing capacity [4].
The immune system has been shown to play a primary role in these processes, as a common feature of tissue aging and age-related diseases is the presence of “inflammaging,” a sterile chronic low-grade inflammation that correlates with increased morbidity and mortality [5, 6]. The etiopathogenesis of inflammaging and the precise mechanisms by which it induces poor health outcomes is still far to be completely elucidated. However, mechanical trauma, ischemia, stress, or environmental conditions such as ultra-violet radiation may induce pathogen-free inflammation, through the secretion of damage-associated molecular patterns (DAMPs), that interact with innate immune receptors [6]. A further understanding of the underlying pathogenic pathways is an essential requirement to identify targetable therapeutic targets in age-related diseases. Both arms of the immune response, innate and adaptive, appear to functionally evolve with aging. The adaptive immune response is known to decline in the elderly, due to thymus involution and impairment of lymphocyte subsets [7]. Therefore, infections and external stressors may not be adequately contained, and antigenic stimulation may persist over time [8]. In turn, unresolved infections or tissue damage may lead to chronic stimulation of innate immune cells, since aged innate immune cells retain the capacity to recognize pathogen-associated molecular patterns (PAMPs) and DAMPs, promoting inflammaging [6, 8]. Given its role in both activation and resolution of the immune response, innate immunity is a key player in aging, and its malfunction may affect aging and senescence. Indeed, innate immune dysfunctions can predispose to frailty, morbidity [9], and more severe infections, as extensively discussed below.
In this review, we discuss the role of the innate immune system in inflammaging, senescence, and aging and provide an up-to-date overview of the putative pathways involved in the development of age-related diseases.
Immunosenescence and Inflammaging: the Role of Myeloid Cells
Aging is a highly complex physio(patho)logical process characterized by progressive loss of integrity at the organ, tissue, cellular, and molecular levels, leading to impaired systemic homeostasis and increased vulnerability to disease and, eventually, death [10]. The complex biological processes of aging are far from being elucidated. However, an attempt to describe the common features of aging was recently summarized in nine schematic albeit interconnected hallmarks: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, stem cell exhaustion, altered intercellular communication, and cellular senescence [10]. This latter is considered a key and central element of aging process and is characterized by morphological, genetic, and epigenetic remodeling, as well as metabolic reprogramming which influence the capacity of cells to renew themselves and their tissue environment, as the cell cycle comes to an exhaustion [11]. Naturally, as aging affects the entire organism, also the immune system undergoes to substantial changes which, in turn, as recently emerged, act itself as effector and regulator of aging process [12, 13]. Closely connected to cellular senescence, the alterations affecting the immune system during aging are termed “immunosenescence” and represent two interconnected processes: the intrinsic senescence process of immune cells, and the immune cell-extrinsic stimulation by senescent tissue and cells, which are sensed by the immune system [14]. Indeed, while senescent cells arrest their replicative cycle, they maintain an active metabolic, although altered, status which induces a senescence-associated secretory phenotype (SASP) (Fig. 1) [10]. The SASP results in the secretion of a wide range of soluble and insoluble factors such as cytokines, chemokines, and growth factors. Among these, interleukin (IL)-6, IL-8, IL-1β, transforming growth factor beta (TGFβ), and granulocyte–macrophage colony stimulating factor (GM-CSF) represent important mediators for the further recruitment of immune cells with potential additive and deleterious inflammatory effects [15].

Schematic network connecting senescence/immunosenescence, inflammaging, frailty, and age-related diseases. The physiological cellular and tissue senescence, which occurs during aging, contributes to inflammaging through the disruption of tissue barriers. This event increases the release of PAMPs mediated by the entry of pathogens, via the production of SASP factors, and the release of cellular debris (DAMPs and/or GARBage). PAMPs, DAMPs, and GARBage elements are recognized by myeloid cells (neutrophils, macrophages, dendritic cells) via their PRRs, thus enhancing the production of pro-inflammatory mediators (i.e., IL-6, TNFα, IL-1β). These last exacerbate the inflammaging process. In addition, senescence of HSCs leads to their reduced renewal and to a biased differentiation of hematopoietic progenitors toward myelopoiesis, rather than lymphopoiesis. Inflammaging appears increasingly responsible for the onset and progression of the most common diseases in the elderly. Among them, atherosclerosis, obesity, diabetes, neurodegenerative and musculoskeletal (e.g., sarcopenia) diseases, cancer, and infections (e.g., COVID-19) are well characterized for their inflammatory facet and/or etiology. These pathologies meet the definition of age-related disease, also in terms of clinical management among frail elderly subjects. PAMP, pathogen-associated molecular patterns, SASP, senescence-associated secretory phenotype, DAMP, damage-associated molecular patterns, PRR, pattern recognition receptor, HSC, hematopoietic stem cell. For details, see the text
The immune response is composed by two distinct but closely interrelated parts: the innate and the adaptive immunity. While it is clearly recognized since 1980s, the concept of immunosenescence as the immune changes occurring during aging in the adaptive immune response [16, 17], in the last two decades, it has become clear that even the innate immune compartment acquires age-related alterations [17,18,19,20]. In adaptive immunity, these changes include increased proportions of antigen-experienced B and T cells at the cost of naïve cell populations. These changes reflect diminished thymic and bone marrow lymphopoiesis, reduced antigen-receptors repertoire, altered developmental and signal transduction, resulting in impaired generation of protective humoral (B lymphocytes) and cell-mediated (CD4+ and CD8+ T lymphocytes) immunity [19, 21]. Aging of the innate immune system in humans, instead, demonstrated a paradoxical increase in levels of pro-inflammatory mediators such as IL-6, tumor necrosis factor alpha (TNFα), C-reactive protein (CRP), and blood clotting factors [19]. This state of low-grade, chronic inflammation has been termed “inflammaging” [22, 23].
Hematopoietic Stem Cell Senescence
Both adaptive and innate immune cells rise from hematopoietic stem cell (HSC) progenitors in the bone marrow, which were shown to undergo themselves the age-related changes and to have a limited replicative potency [24]. In 1978, Harrison et al. demonstrated that bone marrow cells from aged mice, transplanted into recipients, showed loss of stem cell repopulating ability, suggesting that aging promotes HSC exhaustion or replicative senescence [25]. More recently, several evidences supported the substantial difference between young and aged HSCs [26]. Geiger et al. suggested a set of criteria to define the phenotype of aged HSCs, although in certain cases, they can appear ambiguous since the aging process is a gradual and highly heterogenous process [24]. These criteria include (a) increased HSCs number and decreased regenerative potential: unexpectedly, aged HSCs appear increased in their number in the bone marrow niche, probably due to an enhanced short-term self-renewal activity. However, aged HSCs under conditions of stress, as in serial transplantation assays, exhibit several functional defects, including a reduced long-term self-renewal capacity. The increase in the number of HSCs does not compensate for their loss in function, resulting in an overall reduction in the regenerative capacity of the pool of aged HSCs; (b) altered homing to, and mobilization from, the bone marrow: aged HSCs localize outside the endosteal stem cell niche following their transplantation. This implies that aged HSCs select distinct niches from young HSCs. Moreover, transfer of aged HSCs into recipient mice showed less efficiency in their homing in the bone marrow, as well as higher responsiveness to inflammatory stimuli which promotes their mobilization from the bone marrow niche; (c) finally, a central hallmark of aged HSCs, and of relevance for this review, is their skewed differentiative potential toward myeloid cell lineage, thus providing more myeloid and fewer lymphoid progenitor cells compared to young HSCs (Fig. 1) [24, 26, 27].
HSCs committed to the myeloid lineage outnumber lymphoid cells in both mice [28] and humans [29]. This imbalanced ratio of myeloid/lymphoid cell number in human is corroborated by the upregulation of genes associated with myeloid lineage commitment (e.g., GM-CSF signaling) and malignancies (e.g., AURKA, FOS, MYC), and by the down-modulation of genes mediating lymphoid specificity and function (e.g., FLT3, SOX4) [29]. More recently, comparison between aged and young HSCs from humans or mice has been characterized for several other changes, including DNA damage, impaired DNA repair, altered cell polarity, increased production of reactive oxygen species, and a higher number of differences in gene expression profile in total HSCs as well as in myeloid- or lymphoid-committed HSCs [30]. Some of the changes in gene expression of old HSCs may be due to loss of epigenetic regulations [31].
Beyond the alterations in the number, frequency, or lineage commitment of aged immune cells, substantial changes were underlined in their functionality. In the following sections, we will describe the myeloid immune cell–mediated inflammaging and its centrality between frailty syndrome and age-related diseases. The main alterations of myeloid cells during aging are summarized in Table 1. T and B cell aging is finely described elsewhere [32, 33].
Myeloid Cells and Inflammaging
As briefly described above, the innate immune cells remain relatively well functioning in elderly and represent the main source of inflammatory mediators in response to challenging agents, thus contributing to the age-related increase of low-grade, chronic, and systemic production of pro-inflammatory mediators (i.e., inflammaging), feeding the immunosenescence process [17, 34, 35]. This process is caused by the chronic, repetitive, lifelong antigenic stimulation by challenging self and non-self agents, recently named “immunobiography” [36]. This phenomenon shapes the immune system throughout the entire life, generating different health conditions in old age, from healthy status (successful aging) to multiple age-related diseases (pathological aging) [36]. These immunological stresses can be provided either by several species of pathogens, such as cytomegalovirus (CMV) and herpes simplex virus-1, or by metabolic products or cellular debris from damaged or senescent cells and from damage caused by reactive oxygen species (ROS), by advanced glycation and by nitrosylation. The accumulation of this ‘garbage’ products in aging cells which actively contribute to inflammaging was recently dubbed “garbaging” [37].
The innate immune system professionally recognizes the exogenous and endogenous challenges and ‘garbage’ and reacts to them exerting heterogenous inflammatory activities [37, 38]. Indeed, the innate cells rely on pattern recognition receptors (PRR) (i.e., Toll-like receptors (TLR), NOD-like receptors (NLR), and the RIG-like receptors (RLR)) to identify and respond to exogenous microbial PAMPs (e.g., LPS, viral RNA), as well as to endogenous cellular debris and DAMPs, including heat-shock proteins, cytokines, and damaged genomic and mitochondrial DNA fragments [37]. It is intuitive that senescent cells can directly promote the release of DAMPs which directly contribute to SASP and, eventually, to inflammaging (Fig. 1). In addition to the intrinsic ‘sterile’ inflammation induced by DAMPs, aging hallmarks can be associated with dysfunctional anatomical barriers to exogenous insults, thus increasing the exposure to microbes and the circulation of PAMPs [10, 39,40,41]. Associated with increased PAMPs and DAMPs, as well as SASP, the aging process results in altered number and function of innate immune cells, mainly myeloid, such as neutrophils, monocytes/macrophages, myeloid-derived suppressor cells (MDSCs), and dendritic cells. The intrinsic immunosenescence of these cells, as well as their chronic abnormal activity in response to the aging-associated perturbations of organismal homeostasis, represent the key regulator and effector of the inflammaging process [14, 17, 20]. More in detail, inflammaging is characterized by a 2–fourfold increased levels of pro-inflammatory cytokines, in comparison with healthy young subjects, in particular IL-1β, IL-6, and TNFα, as well as CRP, and by reduced serum level of anti-inflammatory cytokines, including IL-10 and TGFβ (Fig. 1) [7, 42]. Myeloid cells are highly heterogeneous and contribute to inflammaging at different levels, within different pathophysiological conditions [20, 43].
Neutrophils
Neutrophils are the most abundant leukocytes in the blood and represent the first line of defense against bacteria, fungi, and yeast challenges. They are the first immune cells to reach sites of infection, where they recognize and phagocytose pathogens. While in absence of specific stimuli their lifespan is relatively short, the presence of pro-inflammatory stimuli (e.g., LPS) can significantly increase it [20]. Even though ageing showed to have no significant effect in the numbers of circulating neutrophils in both healthy [44] and hospitalized with bacterial infections [45] old adults, as compared to young people, relevant age-related alterations in neutrophil function were described [20].
Neutrophils expand from HSCs and migrate from blood to the site of infection or tissue damage in response to chemoattractants. Major evidences from in vivo and in vitro studies reported an impaired chemotactic capability of neutrophils from elderly subjects. Indeed, cells isolated from old subjects exhibit markedly reduced migration in response to chemotactic agents, including the bacterial products formyl-methionine-leucine-phenylalanine (fMLP), and altered responses to LPS and to myelopoietic GM-CSF [46]. This effect was recently associated to a constitutive activation of the gamma and delta isoforms of phosphoinositide 3-kinase (PI3K) [47]. Noteworthy, the impaired in vitro migrative capability of neutrophils from old adults with occurring infections was directly correlated to the reduced survival rate of these patients [48]. Accordingly, the aberrant neutrophil migration with age was demonstrated in healthy aged volunteers [49] and in a mouse model of cutaneous wound infection [50], where it was associated to a reduced expression of the adhesion molecule ICAM-1 on the activated endothelium of aged mice. Conversely, other studies suggested an unaffected neutrophil chemotaxis in both murine and human models of inflammation [51].
Once arrived at the site of infection, neutrophils engulf the invading agents to expose them to a microbicidal action. A marked impairment of complement- and immunoglobulin (Ig)-opsonized pathogens was described in aged-neutrophils, attributable to a decline in the surface expression of the Fc receptor CD16 [20]. One of the main mechanisms of neutrophil cytotoxic activity is exerted by a huge production of ROS once the pathogen is engulfed. Controversial evidences regarding ROS production by neutrophils were associated to specific stimuli triggering their release. Indeed, while C. albicans, E. coli, and zymosan did not show significant difference in the induction of superoxide release by aged neutrophils in comparison to young controls, the exposure to fMLP, GM-CSF, or S. aureus was less efficient to prime the superoxide production by older cells [52]. Neutrophil extracellular traps (NETs), consisting of a DNA backbone and granule-derived enzymes, can entrap and neutralize pathogens. It has been demonstrated that neutrophils of aged mice challenged with S. aureus produced significantly lower amount of NETs [53].
The accredited explanations of dysfunctional neutrophils converge in the alteration of their plasma membrane composition. Indeed, aged neutrophils from mice showed an increased membrane fluidity as consequence of reduced cholesterol presence in favor of phospholipid enrichment [54]. This could represent the reason why the recruitment of receptors into lipid rafts, the plasma membrane microdomains rich in phospholipids and cholesterol, has been shown markedly reduced [46, 55].
In a very recent work, it has been highlighted that CXCR2+ neutrophils from aged mice are recruited and activated by CXCL1-producing senescent mast cells, into inflamed aged tissues. Once neutrophils have been activated, they are able to reverse transmigrate through the endothelium into the blood circulation and, subsequently, to reach distant organs where they are capable of inducing tissue damage [56].
Macrophages
At the first conceptualization of inflammaging, Franceschi et al. proposed that the main responsible of the sustained production of pro-inflammatory mediators were the chronically activated macrophages (hence ‘macrophaging’) [22]. The recent advances in immunosenescence suggest that macrophages are not the unique players in inflammaging; however, it is widely accepted that monocytes and macrophages are the central components in initiating this process [57].
Macrophages constitute important innate immune cells, comprising a heterogeneous population that may differ in phenotype and behavior according to the site and to the pathophysiological conditions which they encounter. These cells sense microenvironmental and senescence signals [58]. Activated macrophages are usually divided into two subsets: M1-like macrophages mainly involved in pro-inflammatory responses and M2-like macrophages driving anti-inflammatory and resolving responses. However, this dichotomy represents extremes of an intricate continuum [58] influenced by environmental stimulation, metabolic status, and cellular stress [59].
In elderly, the number of macrophages showed contradictory indications, despite major evidences show increased output of myeloid progenitor cells [24] and an elevated number of macrophages in the spleen and bone marrow of aged mice, as compared to young controls [57]. Moreover, these tissue-resident macrophages from old mice demonstrated a more anti-inflammatory, pro-angiogenic phenotype as compared to the young counterpart [60]. Conversely, human studies underlined a reduction of mono/macrophage cells in the blood and in the bone marrow from older adults [61].
One of the main mechanisms proposed to explain the increased basal inflammatory rate in elderly is the higher permeability of gut to microbiota due to senescence and deterioration of tissues, resulting in higher level of circulating LPS, responsible of the systemic, low-grade increase of pro-inflammatory cytokines (e.g., TNFα) [41, 62]. Indeed, elevated pro-inflammatory mediators impaired the phagocytosis of bacteria by peritoneal and bone marrow–derived macrophages, thus perpetuating an inflammatory phenotype. Of note, germ-free mice did not develop age-dependent inflammation and had preserved macrophage function [62].
It was recently shown that intestinal alkaline phosphatase activity declines in aged mice and humans. In mice, this decline promoted the increase of hepatic TNFα levels and of circulating pro-inflammatory cytokines produced by bone marrow–derived macrophages, as well as the liver dysfunction [63]. In the murine lung, the resident alveolar macrophage population changes dramatically with age, impairing the expression of macrophage scavenger receptor 1 (MSR1 or CD204) and macrophage receptor with collagenous structure (MARCO) genes, which have a critical role in the bacterial phagocytosis and in the efferocytosis of apoptotic neutrophils; this critically perpetuates the pro-inflammatory responses [64, 65]. Classically activated macrophages are professional antigen-presenting cells (APCs) that can stimulate T-cell activation. Aged macrophages from both human and mice showed a reduced expression of MHC class II molecules that can limit T cell response in elderly [66].
The production of pro-inflammatory cytokines by aged macrophages showed discrepant results. Although the major concept that macrophages produce more IL-1β, IL-6, and TNFα during aging, other studies demonstrated that splenic and thioglycolate-elicited peritoneal macrophage from aged mice, stimulated with LPS, produced less pro-inflammatory cytokines, as a result of reduced expression of TLRs [57]. It is, however, accepted that aged macrophages secrete more prostaglandin E2 (PGE2) than younger counterparts, as a result of increased expression of cyclooxygenase 2 (COX2) [67]. This leads to increased expression of inflammatory cytokines such as TNFα and IL-6 and decreased MHC class II molecules on macrophages, as well as increased production of IL-10 which suppresses T-cell activation, thus impairing pathogen’s elimination [57]. A possible explanation of these contradictory evidences is the difference in the tissue sources and activation states of the macrophages tested, as well as the health state of population tested.
Of note, the observed reduction of TLRs expression in elderly may be responsible, at least in part, for the higher susceptibility of old people to bacterial, mycotic, and viral infections [57]. Aged murine peritoneal macrophages showed also a reduced phagocytic capacity in comparison with younger controls [68], as a result of diminished production of macrophage inflammatory protein (MIP), eotaxin [57], and nitrous oxide and superoxide [69]. In addition, old mice showed reduced wound healing compared to younger mice, which was correlated with reduced phagocytic capacity of macrophages [57]. Recently, the impaired phagocytosis of macrophage has also been observed in elderly humans, and associated with chronic inflammatory response, regulated by an elevation p38 mitogen-activated protein kinase (MAPK) activity [70]. Conversely, aged alveolar macrophages from rats challenged with K. pneumoniae showed an enhanced phagocytic activity as compared to young, without differences in the production of pro-inflammatory lipid mediator leukotrienes and prostaglandins [71].
As mentioned, aged macrophages have impaired activity in the wound healing process. Several studies showed poor cutaneous wound healing in aging, characterized by enhanced platelet aggregation, delayed re-epithelialization, angiogenesis, and collagen deposition, in association with reduced infiltration of macrophages. Moreover, the transplantation of young macrophages into aged mice partially restored the rate of wound repair [72]. The reduced angiogenesis in elderly was proposed to be associated to the decreased TLR expression on macrophages, which are also involved in VEGF signaling [73], or to the decrease in adhesion molecules VLA-4 on monocytes that may impair the rate of macrophage infiltration to the wound [74].
The metabolic pathways utilized by the macrophages have important implications for their phenotype and showed several influences in the aging process [75]. Notably, NAD+ has been suggested to be a therapeutic target for ageing, since its levels decrease significantly with age [76]. In macrophages, NAD+ synthesis decreases with age and more prominently during the immune responses, affecting macrophage effector activity and resulting in increased pro-inflammatory functions [57, 77].
Myeloid-Derived Suppressor Cells
MDSCs are a group of highly heterogenous immature myeloid cells that rise during prolonged pathological state. Indeed, under conditions of immunological stress, including cancer, infections, and autoimmune disorders, the signals arriving from damaged tissues alter the magnitude and quality of the hematopoietic output which guarantees an adequate supply of lymphoid and myeloid cells. This “emergency” state perpetually reprograms myeloid cells toward suppressive functions of T cell-specific immune response. MDSCs are conventionally divided into two major subpopulations: polymorphonuclear (PMN)-MDSCs and monocytic (M)-MDSCs [78,79,80].
Both human and murine studies underlined that during aging, there is an increased frequency of circulating MDSCs [81, 82]. Indeed, PMN-MDSCs were increased in the circulation of very old people (> 80 years), especially with a frailty profile, in comparison with younger subjects; conversely, the monocytic subset was found unchanged [82, 83]. In murine models, several studies showed that MDSCs frequency is significantly increased with aging in the bone marrow, spleen, and peripheral lymph nodes [84]. Of note, the progeroid mouse models Ercc1 and BubR1 mutants showed higher number of MDSCs in the bone marrow, as compared to wild-type controls [85]. However, the exact mechanism by which aging influences the output and differentiation of MDSCs is far to be clarified.
Nonetheless, many aspects of inflammaging may influence the MDSCs output. As mentioned above, it is known that senescent cells and tissues secrete a plethora of pro-inflammatory cytokines (e.g., IL-1β, IL-6, IL-18, TNFα), as well as different alarmins, such as HMGB1, S100A8/9, and PGE2 [37, 86]. Interestingly, these pro-inflammatory factors are potent inducers of both proliferation and activation of MDSCs [78]. MDSC activity induces an immunosuppressive microenvironment which inhibits both innate and adaptive immune responses [78]. Of note, MDSCs secrete the anti-inflammatory cytokine TGFβ which is a potent inducer of cellular senescence in inflamed tissues [87].
Dendritic Cells
Dendritic cells (DCs) are a primary interface between innate and adaptive immune responses, as they are the predominant antigen-presenting cells for naïve T cells. They are generally classified in conventional or plasmacytoid dendritic cells [88]. Their impact in the aging process is quite elusive. However, some studies showed that the numbers of both plasmacytoid and conventional circulating DCs is reduced in elderly, more markedly in frailty conditions, as compared to young subjects [89]. In contrast, another study underlined no differences in DC distribution, in different tissues [90]. Noteworthy, plasmacytoid DCs from old donors have reduced TLR expression, decreased production of interferons class I and II, and damped antiviral response [89]. In addition, aged DCs showed increased production of IL-6 and TNFα, suggesting their contribution to inflammaging [14, 91]. Conversely, conventional DCs from old mice revealed an impaired capability to induce CD8+ T cell cytotoxic responses [14].
Inflammaging and Frailty
Frailty is widely recognized as an important and common geriatric state associated with increased vulnerability to internal and external stressors, resulting from a significant loss of homeostatic reserve involving multiple interrelated physiological systems [92, 93]. In 2013, a group of six major international societies in the field of aging and geriatric medicine conceptualize frailty as “a medical syndrome with multiple causes and contributors, that is characterized by diminished strength, endurance, and reduced physiologic functions that increase individual’s vulnerability and dependency, and/or death” [94].
In frailty, accumulation of cell damage accompanied by impairment of repair mechanisms (i.e., DNA repair, synthesis and control, detection and clearance of damaged proteins and lipids, clearance of abnormal organelles and cells) and defense against injury lead to loss of the homeostasis and physiological decline [95]. There are several methods to assess the presence and entity of frailty [96, 97]. Among others, the two main systems are (1) frailty phenotype (FP), which diagnoses frailty when at least three of five criteria (weight loss, exhaustion, weakness, slow walking speed and low levels of physical activity) are present; one or two criteria identify a pre-frail person [98]; and (2) Frailty Index (FI), which measures the number of age-related deficits that an individual accumulates over time [99]. The latter method considers a more complex scenario reflecting dysfunction of physiological systems, including age-related diseases, as well as cognitive deficits. Indeed, evaluating different pathophysiological factors, such as cellular senescence, oxidative stress, mitochondrial dysfunction, and dysregulation of inflammatory processes (immunosenescence and inflammaging), the Frailty Index can describe several heterogenous conditions at different stages of the aging process [95, 97].
Although the concept of frailty has been clearly discerned from that of multimorbidity [94], multimorbidity is emerging as a frequent parallel condition mutually connected to frailty [97, 100]. The term multimorbidity refers to the co-occurrence of two or more chronic diseases in the same person [100, 101]. Of relevance, multimorbidity was not conceptualized as a sum of single diseases, but rather as synergy of different diseases associated with worse health outcomes and more complex clinical management [100]. Multimorbidity prevalence increase consistently with elderly (up to 98% in subject older than 65 years) [100, 102]. Subjects affected by cardiovascular disorders exhibit a greater multimorbidity than individuals not affected [34]. Diseases most often associated with multimorbidity are diabetes, chronic kidney disease, anemia, chronic pulmonary disease, depression, and dementia, which all involve inflammaging as an important risk factor [34, 75]. It becomes intuitive that frailty and multimorbidity are closely related (Fig. 1); both can be considered as a cause and a consequence of each other, as well as indicators of healthy status and predictors of disability and mortality [97, 99, 100, 102]. Multimorbidity can cause an accumulation of deficits participating in frailty onset and evaluation. Within this scenario, inflammaging was directly correlated to an increased risk of age-related chronic diseases and frailty [95].
It was recently introduced the concept of “anti-inflammation” which underlines the production of anti-inflammatory cytokines in aging, to neutralize the dangerous inflammatory processes. The balance between inflammation and anti-inflammation has been suggested to determine the onset and severity of age-associated disorders and the individual’s ability to achieve extreme longevity [7, 103]. Several studies underlined that the major pro-inflammatory factors directly correlated with frailty, independently of the chronic disease states, are IL-6, TNFα, CRP, fibrinogen, and factor VIII [95, 104]. In particular, recent meta-analysis of more than 20,000 older adults highlighted that frailty and pre-frailty conditions directly correlate with indicators of inflammation, particularly CRP and IL-6 levels [105, 106].
Furthermore, frail subjects displayed higher expression of CXCL-10, a potent pro-inflammatory chemokine in monocyte and macrophage, which directly correlated with IL-6 serum levels [107]. In line, the levels of circulating IL-10 were reduced in aged humans [103]. In agreement, IL-10-deficient mice develops increased typical signs of frailty with age, including muscle weakness and decreased strength, and in direct correlation with increased levels of IL-6 [108].
Inflammaging-Related Diseases as Multimorbidity of Frailty
While it is clear that chronic low-grade inflammation develops with aging and immunosenescence, it is also evident that age-related inflammatory conditions worsen the aging process. This creates a negative loop that influences the outcome of a successful or unsuccessful aging, and thus determining longevity.
The principal age-related inflammatory diseases also represent the most common multimorbidity of frail old people [97, 100]. These includes cardiovascular diseases (CVDs), the metabolic syndrome (i.e., diabetes and obesity), neurodegenerative diseases, musculoskeletal disorders (e.g., osteoarthritis, rheumatoid arthritis, sarcopenia) (Table 2), and cancer (Table 3) and infective diseases [34, 109,110,111]. In each of these conditions, alteration of various molecules or macromolecules can act as DAMPs detected by the innate immune cells, and thus initiating and maintaining the chronic inflammatory response. In turn, inflammation contributes to the formation of further DAMPs thereby accelerating the disease process.
Cardiovascular Diseases
Cardiovascular diseases (CVDs), such as heart failure, myocardial infarction, atrial fibrillation, and stroke, are classified as a group of heart and blood vessel disorders which are directly associated with features of organismal aging and loss of homeostasis, and increased risk of frailty [34, 112].
Both cardiovascular comorbidities and frailty are influenced by the same risk factors, including low physical activity, smoking, dietary pattern, obesity, and diabetes. Additionally, many biomarkers reflecting multisystem dysfunction are common for the two [34]. Of concern, numerous studies have shown that inflammaging is a major risk factor for CVDs [34, 75, 110].
Atherosclerosis and insulin resistance are the leading mechanisms in the development of CVDs [113]. Atherosclerosis is a typical inflammatory disease [114] and is the main pathological basis of cardiovascular dysfunctions, which are extremely frequent in the elderly [115]. This pathological condition is characterized by mutual relationship between the age-related increase of pro-inflammatory mediators and tissue and cellular senescence; these impact on the pathogenic process, as well as on the intrinsic production of antigens by atherosclerotic plaques, which trigger and sustain an inflammatory response. Therefore, a vicious cycle is generated, influencing and amplifying the role of inflammation in atherogenesis [34].
Atherosclerosis is a process that continues throughout human life. It usually begins at an early age and become clinically evident at more advanced age in the form of coronary heart or cerebrovascular or peripheral arterial disease [111]. Therefore, it appears that aging of the immune system contributes to the development of clinically manifested atherosclerosis such as coronary heart disease [34, 112, 116]. This inflammatory condition may be initiated by auto-antigens released by damaged cells, such as high-mobility group box 1 (HMGB1) and heat shock protein 60 (HSP60), by the accumulation of cholesterol-containing LDL particles, or by infective agents such as C. pneumoniae [117]. These agents are recognized by pattern recognition receptors (PRRs) and several scavenger receptors expressed mainly by innate immune cells, stimulating the production of large amount of pro-inflammatory cytokines and the subsequent engagement of immune cells in the intima of the arterial wall [116]. Classical mediators of inflammation by the innate immune response, including CRP, IL-1β, IL-6, and TNFα, correlate with the occurrence of myocardial infarction and stroke in healthy and individuals with known coronary disease [118].
Monocytes that migrate into the intima of the arterial wall differentiate into macrophages and then transform into foam cells in the lipid necrotic core of the atheroma. The macrophage content of the plaque represents the kinetic balance between the recruitment of blood monocytes, their differentiation into tissue macrophages and proliferation in situ, and their emigration or death. Their activation state changes during both the progression and regression of atherosclerosis. The cholesterol-engulfed macrophage (foam cell) promotes innate immune responses and inflammation by both increasing the sensitivity of TLRs to their ligands and activating the NLRP3 inflammasome. These result in the release of IL-1β, IL-18, and other pro-inflammatory cytokines [114, 119]. While in early phase of the atherosclerosis process the clearance of senescent myofibroblasts through recruitment of inflammatory phagocytic cells may be beneficial, as they can limit tissue fibrosis, late atherosclerosis is characterized by massive cell apoptosis and accumulation of cells with senescent features, which support an exacerbated pro-inflammatory status and lead to the formation of a necrotic core that ultimately causes fragility and rupture of the plaque, formation of a thrombus, and acute vascular occlusion [34, 110].
Elevated secretion of TNFα, IL-6, and IL-1β from cardiomyocytes and peripheral tissues has been shown to play an important role in the pathogenesis and progression of myocardial dysfunction; the plasma levels of these pro-inflammatory cytokines can predict the short- or long-term survival in patients with cardiac heart failure [110, 118]. In agreement, in elderly individuals, genetic gain-of-function modifications of IL-6 receptor (IL6R) and CRP genes were associated with increased risk of CVDs, as well as other inflammatory diseases, regardless of other cardiovascular risk factors [34]. Of note, a genome-wide study on Chinese centenarians highlighted that a single nucleotide polymorphism (SNP) of IL6 gene was significantly associated with extreme longevity confirming the role of IL-6 in conditioning morbidity and mortality, especially in old age [120]. The gold standard therapies for the treatment of CVDs rely mainly in the metabolic regulation of hyperlipidemia through life style changes and/or hypolipidemic drugs (e.g., statins), and blood pressure medications [121]. However, recent indications from several mouse model studies as well as from two big clinical trials showed that pro-inflammatory cytokine-regulating drugs reduces major cardiovascular events in subjects with previous myocardial infarction and with residual inflammation, independently of lowered lipid levels. These studies include CANTOs trial using canakinumab, an anti-IL1β monoclonal antibody, and MEASURE trial using tocilizumab, an IL-6 receptor blocker monoclonal antibody [111].
Metabolic Syndrome
In the last decades, the prevalence of metabolic syndrome has dramatically increased worldwide [122]. This condition is characterized by concurring and high prevalent disorders and diseases including obesity, type 2 diabetes mellitus (T2DM), hypercholesterolemia, non-alcoholic fatty liver disease, and their multiple complications [123]. In recent years, it has become evident that systemic low-grade inflammation in the liver, muscle, and adipose tissue is a major contributor to the development of obesity and insulin resistance [124].
Obesity
Obesity represents a serious health condition strongly associated with inflammaging and constitutes a model of accelerated aging [125]. Indeed, adipose tissue dysfunction is a central etiological mechanism of obesity; beyond the altered metabolic homeostasis, its high immune cell infiltration is a key contributor to pathological metabolic alterations. This immunometabolic circuit is emerging for its role in human aging process [126]. Thus, the inflamed adipose tissue makes obesity a condition strongly associated to pro-inflammatory state [127]. The seminal discovery by Hotamisligil et al. showed a TNFα overexpression in obesity, whereas the TNFα blockade restored insulin sensitivity [128]. Moreover, hypertrophic and hyperplastic adipocytes in abdominal, intramuscular, liver, and pericardial fat can produce pro-inflammatory and chemotactic compounds, such as IL-6, IL-1β, TNFα, and CCL2, as well as hormones that modulate inflammation, such as adiponectin and leptin [34]. These lead to an increased infiltration of immune cells, in particular macrophages and T lymphocytes, which further amplify the production of pro-inflammatory mediators, generating a vicious cycle of chronic inflammatory state named “metaflammation.” This metabolic-driven inflammation resembles the critical inflammatory process of aging [129]. Additionally, in vivo administration of IL-10 prevents the IL-6-induced insulin resistance [130]. Thus, the balance between pro- and anti-inflammatory cytokines seems likely to impact metabolic homeostasis. Concordantly, obesity in humans has been associated with increased risk for autoimmune diseases [131]. Weight loss combined with exercise improves functional status, reduces some of the features of frailty in obese older individuals, and improves the cardiovascular risk profile. However, the connection of these beneficial effects and inflammation is still undefined [132].
Neutrophils showed contradictory evidence for their enrichment in adipose tissue from obese individuals; however, while some evidence suggests that their increase in response to high fat feeding is transient, others show a perpetuating and chronic supply of adipose tissue neutrophils (ATNs) [133]. Adipose tissue macrophages (ATMs) are the most abundant immune cell in adipose tissue, representing more than half of leucocytes in fat depots, in both lean and obese animals [133]. Emerging evidences have highlighted that insulin resistance and lipotoxicity increase neutrophil infiltration, macrophage proliferation, and the release of inflammatory mediators that induce smooth muscle and endothelial cell activation, thus accelerating the atherogenesis [34].
Type 2 Diabetes
T2DM alone or associated with metabolic syndrome is related to chronic inflammation [111]. T2DM is very closely related to obesity, which is not a specific disease of the elderly, and in some cases may be paradoxically a protective factor against many adverse events related to aging [134]. However, following the new classification of diabetes into five different clusters, Cluster 5, which represents the most common form of diabetes (about 40%), called mild age-related diabetes, is linked to physiological maladaptation during aging which includes the early onset of inflammaging [34, 135]. T2DM is also a strong risk and prognostic factor for CVDs; in agreement, the effect of diabetes on the risk of CVDs and the CVD mortality increase dramatically with elderly and frailty conditions [136]. Inflammation also plays a role in the pathogenesis of diabetes or insulin resistance. The basic underlying mechanism in T2DM is the reduced insulin sensitivity of the target organs, due to changes in the intracellular signaling pathways. Insulin resistance can be also a consequence of obesity condition [137].
Since T2DM is commonly present in the elderly, it is expected that its immunological profile overlaps immunosenescence. However, inflammation-driven T2DM is observed at early ages, suggesting that T2DM could represent a model of premature immunosenescence [110]. Other studies indicated that T2DM and hyperglycemia increased some inflammatory markers such as IL-6, TNFα, and CRP, which were associated with increased oxidative stress and represent typical features of aging process [138, 139].
A classical feature of immunosenescence presented in T2DM patients is the diminished phagocytic activity and TLR responsiveness of peripheral blood monocytes, as well as their higher susceptibility to apoptosis in culture [140]. In addition, elevated levels of glycated hemoglobin (HbA1c), the major marker for diabetes diagnosis, correlate with a decreased phagocytic activity of circulating monocytes and neutrophils; this is restored in patients treated with the insulin-sensitizing drug, metformin [141].
Neurodegenerative Diseases
As mentioned above, the Frailty Index includes the evaluation of cognitive status of old people [99], and epidemiological evidence suggests that physical frailty may be associated with cognitive impairment and decline in late life. However, the causal relationship between physical decline and neurocognitive disorders is far to be clarified [142]; nevertheless, when considering immunosenescence and inflammaging as causal network, a bidirectional interconnection emerges [111, 143]. Inflammaging is considered one of the most important processes involved in neurodegenerative disorders [143, 144], which in turn are also considered as systemic inflammatory conditions and, thus, influencing or contributing to inflammaging and, eventually, to other age-related inflammatory diseases [145, 146]. Immunosenescence may accelerate brain aging and memory loss, while aging of the brain either directly, by the innate cell recirculation, or indirectly, by the neuro-endocrine–immune network, may influence systemic aging of the immune system [111]. Of note, the most important risk factor for neurodegenerative diseases is aging itself [147]. In this context, inflammaging and peripheral immunosenescence can modulate neuronal immune cell activity and reactivity, leading to a chronic low-grade inflammation in the central nervous system (CNS), called “neuroinflammaging” [148].
The most relevant and frequent forms of neurodegenerative disease are Alzheimer’s disease (AD) and Parkinson’s disease (PD). As for most of the pathological processes involving inflammation, immune reactions can be both beneficial, at early stages, and detrimental when the pathological challenges are chronically sustained [149]. The triggering events of neurodegeneration according to our present knowledge may be infections, metabolic alterations, traumas, and vascular dysfunctions. These result in the activation of resident innate immune system which start a process favoring the recruitment of further immune cells from the periphery and in the production of amyloid-beta (Aβ) peptide by CNS cells [111].
Microglia are tissue-resident macrophages in the CNS, derived from the yolk sac during embryogenesis, which colonize the developing brain where they stay during the individual’s lifetime, acting as the first line of defense. They detect molecules of damaged CNS cells or infiltrated pathogens throughout PRRs expressed on their surface and on the surface of infiltrating monocytes. Senescent microglia are characterized by a higher proliferative rate which in turn can culminate in the shortening of telomeres and replicative senescence [150]. Aged microglia have an increased production of pro-inflammatory cytokines that are accompanied by reduced capacity to phagocyte Αβ fibrils. Further, senescent microglia are characterized by morphological changes, by an accumulation of mitochondrial DNA damages that induce an overproduction of ROS, and of increased expression of TLRs [151,152,153]. Accumulation of misfolded proteins [154] and chronic exposure to TGFβ that impairs the production of anti-inflammatory cytokine by microglia [153] were proposed as mechanisms contributing to the aged phenotype of microglia.
Indeed, in AD, neuronal senescence and degeneration activates microglia that are directly involved in the generation of Aβ senile plaques; this mechanism triggers a chronic inflammation inducing a further infiltration and activation of peripheral monocytes and macrophages in the CNS, thus contributing to the release of pro-inflammatory cytokines and the upregulation of immune receptor such as CD14, TLRs, and MHC-II, which promote brain tissue damage [151]. The Aβ-rich areas were shown to induce an M2 to M1 functional switch of macrophages, with increased production of IL-1β and TNFα, promoting oxidative stress, apoptosis, and neuronal loss [155]. Of note, AD patients have elevated levels of IL-6, TNFα, and IFNγ in the circulation and in cerebrospinal fluid; these are associated with increased risk for AD cognitive decline [151]. PD is characterized by neuroinflammatory process akin to AD and to other neurodegenerative process; increased MHC-II and TLR expressions, as well as the production of TNFα and IL-6, were described for their involvement in PD pathological process [156].
Musculoskeletal Disorders
Musculoskeletal health and frailty are in close and interconnected relationship. While musculoskeletal functioning is a central parameter for the definition of frailty, frailty is commonly associated with the main age-related musculoskeletal disorders such as rheumatoid arthritis (RA), osteoarthritis (OA), osteoporosis, and sarcopenia [96, 104, 109, 157]. The direct link between the pathogenesis of RA, OA, osteoporosis, and the processes of inflammaging and immunosenescence is widely characterized and described elsewhere [9, 158]. Conversely, the immune-related events involved in sarcopenia are less defined and are emerging as new tools to understand and treat sarcopenic patients [104].
Sarcopenia
Sarcopenia is a very common geriatric disease defined as a syndrome characterized by progressive and generalized loss of skeletal muscle mass and strength, with a risk of adverse outcomes such as physical disability and poor quality of life and death. Progressive loss of muscle mass begins as early as 40 years of age, and its rate has been estimated at about 8.0% per decade until the age of 70 years [157, 159]. Similar to the frailty phenotype, the European Working Group on Sarcopenia in Older People (EWGSOP) recommends categorizing sarcopenia into pre-sarcopenia, Sarcopenia, and severe sarcopenia, depending on the presence of specific criteria. Severe sarcopenia as defined by EWGSOP is pre-frailty by the Frailty Phenotype. Sarcopenia is often considered a precursor syndrome or the physical component to frailty [160]. Moreover, sarcopenia is reported to be twice as common as frailty in the general population [161].
One of the main links between inflammation and sarcopenia comes from the evidence that patients with sepsis (the majority of them are in elderly [20]) are subject to both chronic pro-inflammatory state and periods of extreme inactivity. These were suggested as a model of accelerated aging, as in conditions of critical care, patients reported prolonged weakness as result of muscle loss and changes in muscle cell functionality [162]. Another clinical condition intersecting inflammation and muscle deterioration is the so-called sarcopenic obesity where fat accumulation and sarcopenia, which both increase in elderly, co-occur [163]. The chronic inflammatory alterations present in old obese and diabetic subjects (see above) are shown to contribute to the development and progression of sarcopenia [109], as well as to affect insulin sensitivity. Since insulin is a central anabolic signal, and insulin resistance is considered the main factor controlling the development of T2DM, these observations connect obesity-associated metabolic syndrome, immunity, and sarcopenia [164]. Moreover, RA, OA, and osteoporosis, which, as mentioned, are critical chronic inflammatory diseases, are closely associated with sarcopenia [157].
Of note, inflammaging impairs many physiological functions such as insulin sensitivity, hormonal secretion, endothelial functionality, and vascular structures which are directly correlated with muscular strength, cellular metabolism, and energy regulation, thus impacting on the sarcopenia development [165]. Moreover, the homeostatic balance between protein synthesis and catabolism has been described as central players in the relationship between pro-inflammatory state and muscle degeneration. Increased protein degradation (via altered mitochondrial function), cell death or apoptosis, increase of insulin resistance–dependent muscle adiposity, and impaired muscle repair capacity are the main mechanisms underlying inflammation-induced muscle degeneration [166].
In particular, the inflammaging markers IL-6, TNFα, and CRP have been suggested to chronically induce further inflammatory signals in muscle tissue, to increase muscle adiposity that interferes with the insulin-like growth factor 1 (IGF-1)-dependent protein synthesis homeostasis, thus promoting the overall pathogenesis of sarcopenia [165]. Furthermore, immunosenescence and SASP factors have been shown to directly affect the regenerative capacity of muscle stem cells (satellite cells) [165, 167]. Notably, in human, several epidemiological studies (among them the InCHIANTI and the Framingham Heart and Health ABC studies) have found that high circulating levels of IL-6, TNFα, IL1ra, and CRP significantly associated with poor overall physical performance and reduced muscle strength, indicating reduced muscle mass and increased fat mass, status [167]. At tissue levels, IL-6, TNFα, and CRP were associated with decreased mitochondrial oxidative capacity, increased p65 NF-κB activation (essential signal in the production of pro-inflammatory cytokines [168]), and decreased lean mass and muscle strength [167].
Myeloid Cells in Skeletal Muscle Homeostasis
Myeloid cells, in particular macrophages, are essential regulator of muscle regeneration following an acute injury [169]. Higher neutrophil counts are associated with low levels of physical activity and frailty [104]. Neutrophils are central to tissue repair, and studies have demonstrated that they are recruited to the site of injury within a couple of hours. However, migrating neutrophils also cause secondary damage to healthy muscle, and few studies reported that muscle damage is reduced when functioning neutrophils recruitment to the damaged tissue is prevented [104].
Macrophages contribute to the repair of damaged skeletal muscle by clearing tissue debris and releasing cytokines and growth factors that stimulate the proliferation and differentiation of satellite cells, as well as tissue revascularization. They are prominently in an M1 phenotype (producing TNFα and IL-1β) during the early, proliferative stages of muscle regeneration, and in an M2 phenotype (producing IL-10 and TGFβ) during the differentiation and regeneration phase [170]. The functional change of macrophage activation during muscle aging has shown controversial results. Some evidences underlined that pro-inflammatory TNFα produced by macrophage favors sarcopenia [171]; others indicated that aging of skeletal muscle correlated with and increased M2 phenotype, which contributes to the fibrosis of muscular tissue in association with increased intermuscular adipose tissue deposition [170]. Ultimately, the mechanisms and the inflammatory phenotype of macrophages need to be further elucidated, as their role in muscle regeneration and their immunometabolic pathways become increasingly relevant [169, 170, 172].
Immunosenescence, Inflammaging, and Cancer
The probability to develop a progressive and invasive cancer is more than double in subjects over 60 as compared to younger individuals, which defines cancer as both a disease of aging and as the leading cause of death in both males and females between the ages of 60 and 79 [173,174,175]. Accumulation of DNA damage and epigenetic dysregulations, which favor cellular senescence and oncogenesis, as well as persistency of chronic low-grade inflammation in elderly patients (inflammaging), has been pointed as the links between aging and cancer development (Table 3) [109, 176]. As described above, inflammaging is characterized by increased plasma levels of pro-inflammatory cytokines such as IL-1β, IL-6, and TNFα. The prolonged cell exposure to IL-1β, IL-6, and TNFα has been associated with high risk of cancer onset and progression, since these cytokines support tumor growth and metastasis spread [177,178,179]. Furthermore, SASP phenotype of senescent cells has been identified as an important promoter of tumorigenesis during aging [180].
In this scenario, a double role is attributed to the immune system, since it can provide antitumor activities, in spite of its tight association with the disease onset and progression [181]. In young and healthy tissues, effector cells, such as T cells, neutrophils, macrophages, and dendritic cells, work together to prevent malignant tissue growth and to clear damaged cells. Contrariwise, the progressive decline in overall immune function that characterizes immunosenescence plays as direct promoter of SASP, inflammaging, and tumor development [182, 183]. This physiopathological network affects both adaptive and innate immune functions, increasing recruitment of immune cells and their orientation toward immunosuppressive functions, which facilitate the greater susceptibility toward cancer development [184].
Accumulation of MDSCs has been observed in bone marrow, spleen, and blood of both tumor-bearing and aged mice [81, 185]. While the signals that regulate expansion (e.g., CSFs), infiltration (e.g., CCL2, CXCL2, IL-8), and activation (e.g., IL-1β, IL-6, TNFα) of MDSCs are significantly upregulated in elderly [186, 187], a clear association with the increased cancer susceptibility is still missing. However, the interplay between immunosuppressive populations and the secretomes associated with SASP may support MDSC infiltration of the tumor site. In agreement, the age-related expansion of MDSCs decreased the cytotoxicity of T cells, impairing senescent and cancerous cell clearance and leading to tumor immune escape [185]. Similar results were observed with an increased number of immunosuppressive M2 macrophages in the spleen and bone marrow of aged tumor-bearing mice [188, 189]. In general, aged monocytes and tumor-associated macrophages (TAMs), which are potentially important for the recognition and clearance of cancer cells, are defective in antitumor functions, including cytokine production and antigen presentation, thus facilitating tumor progression [190, 191].
Previous studies have described two functional states of neutrophil activation, the N1 (cytotoxic and anti-tumoral) and N2 (immunosuppressive and pro-angiogenic) [192]. While these two subsets were identified and characterized in young mice, their role in cancer development has not been defined in the elderly. Of note, in young mice, it was reported that the anti-inflammatory N2 tumor-associated neutrophils (TANs) play a relevant role, in many cancers [187]. These immunosuppressive N2 TANs are characterized by a decreased chemotactic and phagocytic activity, impaired production of free radicals and altered apoptosis [193, 194], features that resemble senescent neutrophils [52]. Interestingly, N2 TANs are increased systemically in aged patients and exhibit a similar function to MDSCs [195, 196].
Noteworthy, loss of extracellular matrix (ECM) integrity is a hallmark of cancers associated with tumor progression and metastasis [197]. The ECM acts as a physical barrier, an anchorage site, or a “trail” for cell migration. It has been established that ECM integrity decreases substantially as we age. Age-related changes in ECM physical properties include decreased collagen density [198], reduced ECM fiber area and thickness, and altered mechanical features, such as stiffness [198, 199]. Of relevance, recent studies highlighted that many of the molecules characterizing the SASP are ECM-degrading enzymes (e.g., MMPs). These can sustain cancer development by creating a permissive environment for invasion, dissemination of cancer cells, and immune cell trafficking through ECM remodeling [199, 200]. Of note, immunosuppressive myeloid cells play a pro-tumoral role also through alteration of ECM composition [201]. It appears therefore that inflammaging, SASP factors, and ECM remodeling bridge immunosuppressive myeloid cells with age-related tumorigenesis, suggesting the need for further studies on this front.
Inflammaging and Infections: the Paradigm of COVID-19
It is established that frail elder population is more susceptible to experience more frequently infections than younger population [202, 203]. Moreover, it is known that immunosenescence is associated to increased susceptibility to infectious diseases and decreased vaccine efficacy [204]. On the other side, the infections can support the chronic inflammaging during aging, through the release of PAMPs which stimulate innate myeloid cells to produce pro-inflammatory cytokines [203]. Such an immune profile is a serious challenge for the limited capacity of the aged immune system to resolve infections. Onen et al. reported that frail HIV-infected patients show an increased number of comorbidities as well as an increased risk of developing malignancies [205]. In addition, while CMV infection is commonly asymptomatic, frail patients tend to be symptomatic. Indeed, the high levels of circulating IL-6, found in frail patients, activate latent CMV infection and positively feed back on its replication, and thus inducing the production of further pro-inflammatory mediators such as TNFα, IL-1β, and IL-6, perpetuating viral infection [206].
In this scenario, the recent coronavirus disease 2019 (COVID-19) pandemic is leading a rapid, although dramatic, comprehension of aging, frailty, and inflammatory dysregulation. Indeed, COVID-19, which is caused by the SARS-CoV2 virus infection, suddenly emerged as a disease of aging [207]. Subjects over 60 years of age with pre-existing pathological conditions, most of them described as age-related diseases (e.g., hypertension, diabetes, obesity, cancer), are more prevalent among hospitalized COVID-19 patients [208] and have a mortality rate about three-fold higher as compared to patients below 60 years old [209].
Despite the mechanisms leading SARS-CoV2-positive elderly immunocompromised patients toward a bad prognosis are still unclear, there is a common consensus that the viral entry triggers a potent inflammatory phenomenon leading to an acute respiratory distress syndrome (ARDS), which resembles the cytokine release syndrome (CRS) or cytokine storm phenomenon observed in leukemia patients after chimeric antigen receptor (CAR) T-cell therapy [210]. Specifically, the release of pro-inflammatory cytokines involved in the acute phase response (TNFα and IL-1β), as well as chemoattractant factors like IL-8 and CCL2, orchestrates a progressive increase in circulating IL-6, which in turns feeds back a potent hyperinflammatory status [211]. Furthermore, it was seen that ACE2-mediated SARS-CoV2 endocytosis induces the expression of angiotensin II (Ang-II), which by binding AT1R (Ang-II type I receptor) leads to the activation of the metalloprotease ADAM17 and cleavage of (IL-6R), switching it to its soluble form (sIL-6R). This latter binds circulating IL-6, and the resulting complex is able to signal through gp130 expressed by non-immune cells, activating that JAK/STAT3 signaling pathway, thus perpetuating the inflammatory state [212].
Of note, several studies have underlined the central role of innate myeloid cells in triggering the inflammatory burden and in promoting the pathological process of COVID-19 [213,214,215,216,217,218,219]. Indeed, two separated studies by Silvin et al. [214] and Schulte-Schrepping et al. [213] observed in the peripheral blood and in the lungs of COVID-19 patients an increase of classical inflammatory monocytes, at the expenses of non-classical subset, correlating with the severity of the disease progression. The over-represented monocytic subsets showed a marked pro-inflammatory interferon-stimulated signature and a massive production of calprotectin (S100A8/S100A9), a potent inflammatory and myelopoietic stimulator. In addition, it was described that bronchoalveolar fluid (BALF) from patients with severe COVID-19 is enriched in CCL2 and CCL7, which drive the recruitment of CCR2+ monocytes [215, 220]. A significant expansion of populations of intermediate CD14+CD16+ monocytes producing IL-6 was observed in the peripheral blood of patients with COVID-19 requiring intensive care unit (ICU) hospitalization [221]. In such a scenario, it is not surprising that a combined treatment with corticosteroid and the monoclonal antibody targeting the IL-6 receptor (tocilizumab) reduces the mortality rate in hospitalized COVID-19 patients by one third [222].
Emerging evidence has also shown that microRNAs (miRNAs) are involved in the inflammaging process and that changes in the expression of these miRNAs have been linked to increased production of IL-6, TNFα, and IL-1β [223]. Among these, the expression of miRNA-146a-5p, which in physiological conditions, is able to dampen NF-κB activation and the consequent IL-6 production has been shown to be reduced during inflammaging [224]. Of note, miRNA-146a-5p levels in COVID-19 patients treated with tocilizumab were found to be significantly increased in subjects who responded to therapy, suggesting a direct link and a role of the pre-existing inflammaging in the COVID-19 progression and outcome [224].
Research and Clinical Perspective
Despite the increasing understanding of the mechanisms underlying innate immunity modifications inducing inflammaging and age-related diseases, whether these processes can be targeted and modified is still far to be elucidated. Different approaches have been proposed, with contradictory results.
As mentioned above, targeting specific inflammatory molecules has led to conflicting outcomes. Administration of canakinumab, a selective inhibitor of IL-1β, to patients with previous myocardial infarction and high CRP levels resulted in a significantly lower rate of recurrence of cardiovascular events compared with placebo, independent of lipid-level lowering. Nonetheless, the incidence of fatal infections was higher than in the placebo group [225]. Similarly, patients receiving canakinumab were also analyzed for lung cancer. Those diagnosed with cancer had higher levels CRP and IL-6. Treatment with canakinumab significantly reduced the levels of inflammatory molecules and mortality rates, but fatal infections or sepsis were significantly more common than in the placebo group [225]. Tocilizumab showed mixed results. In fact, while in RA patients the MEASURE trial showed that it modulates lipids and proteins toward an anti-inflammatory composition compared with placebo [226], the net cardiovascular risk of patients treated with tocilizumab seems to be comparable or slightly increased than those in patients receiving etanercept, a TNFα antagonist. In fact, in a recent randomized controlled trial, individuals affected by RA and treated with tocilizumab showed significantly increased serum levels of cholesterol and triglyceride compared with RA patients treated with etanercept, and the estimated hazard ratio for major adverse cardiovascular events in the tocilizumab group relative to the etanercept group was 1.05. Serious infection occurred more frequently in patients treated with tocilizumab [227]. Tocilizumab was also studied in a double-blind, placebo-controlled trial on patients with ST-segment elevation myocardial infarction. The myocardial salvage index as measured by magnetic resonance imaging was larger in the tocilizumab group than in the placebo group, and adverse events were similar among groups [228]. On the contrary, IL-6 blockade did not affect coronary flow reserve during hospitalization or after 6 months in patients with non-ST-elevation myocardial infarction [229]. Tocilizumab may also have a role in cancer, as demonstrated in pre-clinical studies, where it seems to suppress metastasis formation to enhance the cytotoxic effect of cisplatin, and to overcome resistance to chemotherapy [230, 231].
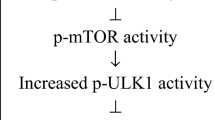
Another approach to target inflammaging could be addressing the upstream pathways responsible for inflammatory molecule production. Immunometabolic sensors able to modulate gene programming have been identified, as sirtuins, insulin-like signals, and mammalian target of rapamycin (mTOR) [75, 232]. Targeting them could be a promising way to reach longevity and reduce age-related diseases [233]. In fact, sirtuins play a role in metabolism, oxidative stress, cell survival, and autophagy. They act as deacetylase and depend on NAD + for their activity; therefore, reduced NAD + levels with aging impair their activity [234]. Restoring an adequate sirtuin function could be an important step in improving lifespan. Moreover, mTOR is a metabolic sensor for amino acids, oxygen, and hormones and modulates ribosome function and mitochondrial metabolism by tuning gene expression. Its dysregulation has been implicated in age-related diseases, and studies in human cellular models demonstrated that mTOR inhibition suppresses the SASP [235]. Other immunometabolic sensors with a described role in aging are forkhead box O transcription factor (FOXO) and AMP-activated protein kinase (AMPK), and they all act in an interrelated network linking nutrient sensing and gene expression. Among the genes modulated by these sensors are those related to metabolism, oxidative stress, cell fate decisions, and inflammation [234]. They also modulate NF-κB, a pleiotropic transcription factor and a major regulator of the SASP, therefore a promising target to address inflammaging [3].
Dietary restriction appears to effectively regulate innate immunity and to extend lifespan, by its impact on the immunometabolic sensors, leading to gene reprogramming, as demonstrated in pre-clinical models [236, 237]. In particular, caloric restriction increases sirtuin expression and modulates mTOR and other immunometabolic sensor pathways, thus reducing pro-inflammatory molecule levels [238, 239].
Further studies in this direction and the standardization of the caloric restriction approach could allow metabolic and anti-inflammatory effects, inducing both delayed aging and genetic reprogramming with an impact on lifespan. It would be a low-cost, high-impact lifestyle modification that would overcome the potential side effects of pharmacologic targeting of specific inflammatory molecules, as cytokines or NF-κB, or the oncologic risk associated with interventions of epigenetic reprogramming of senescent cells [3]. Another recent and intriguing research field concerns the activity of transposable elements (transposons), repetitive sequences contained in the genome of all organisms that can move among different genomic locations, potentially amplifying their copy number. Evidence shows that retrotransposons, a subgroup of transposons, can promote aging and age-related diseases. In young cells, transcriptional regulators control retrotransposons in the genome, promoting the formation of heterochromatin. In adult somatic cells, regulators are reduced and retrotransposons can activate, with detrimental consequences. Genetic and epigenetic modification occurs, DNA can be damaged, and retrotransposon sequences can accumulate in the cytoplasm, triggering the host innate immune response. This can become a vicious circle, as DNA damage induces inflammation, but in turn, inflammation can enhance DNA damage. Retrotransposons have been implicated in age-related diseases, mostly in neurodegenerative diseases, and a role has been proposed in cancer, cardiovascular diseases, and diabetes, due to their potential pro-inflammatory activity which can drive inflammaging. The understanding of the peculiar activity and regulation of retrotransposon in aging and in the stimulation of the immune response can lead to the development of targeted therapies. Interestingly, nucleoside reverse transcriptase inhibitors, currently approved for the treatment of HIV infection, were demonstrated to reduce cytoplasmic retrotransposons and to decrease IFN activation [240].
Of note, chronic inflammation can also have a beneficial role, as occurs in tissue remodeling or in the response against tumors. Moreover, in centenarians, high levels of pro-inflammatory molecules have been found, but they did not present signs of age-related diseases, suggesting that inflammaging can co-exist with healthy aging. Probably, the inflammatory state can be both detrimental or pro-adaptive, depending on other conditions as its magnitude, gene variants conferring reduced sensitivity to inflammation, or the presence of an anti-inflammatory response, which modulate the final effect [5].
Conclusion
Aging can induce modifications in cellular metabolism, function, and gene expression, leading to secretion of pro-inflammatory molecules and to a chronic sterile inflammatory state, named inflammaging. The innate arm of the immune system plays a key role in this process, undergoing alterations which ultimately result in an impairment of multiple systems, as cardiovascular, neurologic, and musculoskeletal systems. An increased risk of age-related diseases has been associated with inflammaging and immunosenescence, including cancer. A deeper understanding of the mechanisms underlying the detrimental effects of inflammaging can allow the identification of potential targets, in order to promote healthy aging and increase longevity.
References
Foreman KJ, Marquez N, Dolgert A et al (2018) Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016–40 for 195 countries and territories. Lancet. https://doi.org/10.1016/S0140-6736(18)31694-5
da Costa JP, Vitorino R, Silva GM et al (2016) A synopsis on aging—theories, mechanisms and future prospects. Ageing Res Rev
Schmeer C, Kretz A, Wengerodt D et al (2019) Dissecting aging and senescence-current concepts and open lessons. Cells. https://doi.org/10.3390/cells8111446
Bektas A, Schurman SH, Sen R, Ferrucci L (2018) Aging, inflammation and the environment. Exp Gerontol
Franceschi C, Campisi J (2014) Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. Journals Gerontol. - Ser A Biol Sci Med Sci
Feldman N, Rotter-Maskowitz A, Okun E (2015) DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res Rev
Fülöp T, Larbi A, Witkowski JM (2019) Human inflammaging. Gerontology. https://doi.org/10.1159/000497375
Sadighi Akha AA (2018) Aging and the immune system: an overview. J Immunol Methods
Motta F, Sica A, Selmi C (2020) Frailty in rheumatic diseases. Front Immunol
López-Otín C, Blasco MA, Partridge L et al (2013) The hallmarks of aging. Cell
Herranz N, Gil J (2018) Mechanisms and functions of cellular senescence. J Clin Invest
Vicente R, Mausset-Bonnefont AL, Jorgensen C et al (2016) Cellular senescence impact on immune cell fate and function. Aging Cell
Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K (2013) Causes, consequences, and reversal of immune system aging. J Clin Invest
Rodrigues LP, Teixeira VR, Alencar-Silva T et al (2021) Hallmarks of aging and immunosenescence: connecting the dots. Cytokine Growth Factor Rev
Basisty N, Kale A, Jeon OH et al (2020) A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. https://doi.org/10.1371/journal.pbio.3000599
Goronzy JJ, Fang F, Cavanagh MM et al (2015) Naive T cell maintenance and function in human aginG. J Immunol. https://doi.org/10.4049/jimmunol.1500046
Fulop T, Larbi A, Dupuis G et al (2018) Immunosenescence and inflamm-aging as two sides of the same coin: Friends or Foes? Front Immunol
Solana R, Tarazona R, Gayoso I et al (2012) Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol
Montgomery RR, Shaw AC (2015) Paradoxical changes in innate immunity in aging: recent progress and new directions. J Leukoc Biol. https://doi.org/10.1189/jlb.5mr0315-104r
Hazeldine J, Lord JM (2015) Innate immunesenescence: underlying mechanisms and clinical relevance. Biogerontology
Goronzy JJ, Weyand CM (2013) Understanding immunosenescence to improve responses to vaccines. Nat Immunol
Franceschi C, Bonafè M, Valensin S et al (2000) Inflamm-aging. An evolutionary perspective on immunosenescence. In: Annals of the New York Academy of Sciences
Kirkwood KL (2018) Inflammaging. Immunol Invest
Geiger H, De Haan G, Carolina Florian M (2013) The ageing haematopoietic stem cell compartment. Nat Rev Immunol
Harrison DE, Astle CM, Delaittre JA (1978) Loss of proliferative capacity in immunohemopoietic stem cells caused by serial transplantation rather than aging*. J Exp Med. https://doi.org/10.1084/jem.147.5.1526
Dorshkind K, Höfer T, Montecino-Rodriguez E et al (2020) Do haematopoietic stem cells age? Nat Rev Immunol. https://doi.org/10.1038/s41577-019-0236-2
Beerman I, Maloney WJ, Weissmann IL, Rossi DJ (2010) Stem cells and the aging hematopoietic system. Curr Opin Immunol
Rossi DJ, Bryder D, Zahn JM et al (2005) Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.0503280102
Pang WW, Price EA, Sahoo D et al (2011) Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1116110108
Montecino-Rodriguez E, Kong Y, Casero D et al (2019) Lymphoid-biased hematopoietic stem cells are maintained with age and efficiently generate lymphoid progeny. Stem Cell Reports. https://doi.org/10.1016/j.stemcr.2019.01.016
Chambers SM, Shaw CA, Gatza C et al (2007) Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. https://doi.org/10.1371/journal.pbio.0050201
Mittelbrunn M, Kroemer G (2021) Hallmarks of T cell aging. Nat Immunol
Hagen M, Derudder E (2020) Inflammation and the Alteration of B-Cell Physiology in Aging. Gerontology
Ferrucci L, Fabbri E (2018) Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol
Franceschi C, Santoro A, Capri M (2020) The complex relationship between Immunosenescence and Inflammaging: special issue on the New Biomedical Perspectives. Semin Immunopathol
Franceschi C, Salvioli S, Garagnani P et al (2017a) Immunobiography and the heterogeneity of immune responses in the elderly: a focus on inflammaging and trained immunity. Front Immunol
Franceschi C, Garagnani P, Vitale G et al (2017b) Inflammaging and garb-aging. Trends Endocrinol. Metab
Rivera A, Siracusa MC, Yap GS, Gause WC (2016) Innate cell communication kick-starts pathogen-specific immunity. Nat Immunol
Parrish AR (2017) The impact of aging on epithelial barriers. Tissue Barriers
Man AL, Gicheva N, Nicoletti C (2014) The impact of ageing on the intestinal epithelial barrier and immune system. Cell Immunol
Conway J, Duggal NA (2021) Ageing of the gut microbiome: potential influences on immune senescence and inflammageing. Ageing Res Rev
Baylis D, Bartlett DB, Patel HP, Roberts HC (2013) Understanding how we age: insights into inflammaging. Longev Heal. https://doi.org/10.1186/2046-2395-2-8
Mantovani A, Marchesi F, Jaillon S et al (2021) Tumor-associated myeloid cells: diversity and therapeutic targeting. Cell Mol Immunol
Born J, Uthgenannt D, Dodt C et al (1995) Cytokine production and lymphocyte subpopulations in aged humans. An assessment during nocturnal sleep. Mech Ageing Dev. https://doi.org/10.1016/0047-6374(95)01638-4
Butcher SK, Killampalli V, Lascelles D et al (2005) Raised cortisol: DHEAS ratios in the elderly after injury: potential impact upon neutrophil function and immunity. Aging Cell. https://doi.org/10.1111/j.1474-9726.2005.00178.x
Fulop T, Larbi A, Douziech N et al (2004) Signal transduction and functional changes in neutrophils with aging. Aging Cell
Sapey E, Greenwood H, Walton G et al (2014) Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. https://doi.org/10.1182/blood-2013-08-519520
Niwa Y, Kasama T, Miyachi Y, Kanoh T (1989) Neutrophil chemotaxis, phagocytosis and parameters of reactive oxygen species in human aging: cross-sectional and longitudinal studies. Life Sci. https://doi.org/10.1016/0024-3205(89)90482-7
MacGregor RR, Shalit M (1990) Neutrophil function in healthy elderly subjects. Journals Gerontol. https://doi.org/10.1093/geronj/45.2.M55
Brubaker AL, Rendon JL, Ramirez L et al (2013) Reduced neutrophil chemotaxis and infiltration contributes to delayed resolution of cutaneous wound infection with advanced age. J Immunol. https://doi.org/10.4049/jimmunol.1201213
Nomellini V, Faunce DE, Gomez CR, Kovacs EJ (2008) An age-associated increase in pulmonary inflammation after burn injury is abrogated by CXCR2 inhibition. J Leukoc Biol. https://doi.org/10.1189/jlb.1007672
Wenisch C, Patruta S, Daxböck F et al (2000) Effect of age on human neutrophil function. J Leukoc Biol. https://doi.org/10.1002/jlb.67.1.40
Tseng CW, Kyme PA, Arruda A et al (2012) Innate immune dysfunctions in aged mice facilitate the systemic dissemination of methicillin-resistant S. aureus. PLoS One. https://doi.org/10.1371/journal.pone.0041454
Alvarez E, Santa-María C, Ruiz-Gutiérrez V, Sobrino F (2001) Age-related changes in membrane lipid composition, fluidity and respiratory burst in rat peritoneal neutrophils. Clin Exp Immunol. https://doi.org/10.1046/j.1365-2249.2001.01490.x
Fortin CF, Lesur O, Fulop T (2007) Effects of aging on triggering receptor expressed on myeloid cells (TREM)-1-induced PMN functions. FEBS Lett. https://doi.org/10.1016/j.febslet.2007.02.029
Barkaway A, Rolas L, Joulia R et al (2021) Age-related changes in the local milieu of inflamed tissues cause aberrant neutrophil trafficking and subsequent remote organ damage. Immunity
De Maeyer RPH, Chambers ES (2021) The impact of ageing on monocytes and macrophages. Immunol Lett
Hall BM, Balan V, Gleiberman AS et al (2016) Aging of mice is associated with p16(Ink4a)- and β-galactosidasepositive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY). https://doi.org/10.18632/aging.100991
van Beek AA, Van den Bossche J, Mastroberardino PG et al (2019) Metabolic alterations in aging macrophages: ingredients for inflammaging? Trends Immunol
Kelly J, Khan AA, Yin J et al (2007) Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest. https://doi.org/10.1172/JCI32430
Ogawa T, Kitagawa M, Hirokawa K (2000) Age-related changes of human bone marrow: a histometric estimation of proliferative cells, apoptotic cells, T cells, B cells and macrophages. Mech Ageing Dev. https://doi.org/10.1016/S0047-6374(00)00137-8
Thevaranjan N, Puchta A, Schulz C et al (2017) Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe. https://doi.org/10.1016/j.chom.2017.03.002
Kühn F, Adiliaghdam F, Cavallaro PM et al (2020) Intestinal alkaline phosphatase targets the gut barrier to prevent aging. JCI Insight. https://doi.org/10.1172/jci.insight.134049
Wong CK, Smith CA, Sakamoto K et al (2017) Aging impairs alveolar macrophage phagocytosis and increases influenza-induced mortality in mice. J Immunol. https://doi.org/10.4049/jimmunol.1700397
Li Z, Jiao Y, Fan EK et al (2017) Aging-impaired filamentous actin polymerization signaling reduces alveolar macrophage phagocytosis of bacteria. J Immunol. https://doi.org/10.4049/jimmunol.1700140
Herrero C, Sebastiaán C, Marqueés L et al (2002) Immunosenescence of macrophages: Reduced MHC class II gene expression. Exp Gerontol. https://doi.org/10.1016/S0531-5565(01)00205-4
Chen H, Ma F, Hu X et al (2013) Elevated COX2 expression and PGE2 production by downregulation of RXRα in senescent macrophages. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2013.09.047
Linehan E, Dombrowski Y, Snoddy R et al (2014) Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell. https://doi.org/10.1111/acel.12223
Alvarez E, Machado A, Sobrino F, Maria CS (1996) Nitric oxide and superoxide anion production decrease with age in resident and activated rat peritoneal macrophages. Cell Immunol. https://doi.org/10.1006/cimm.1996.0103
De Maeyer RPH, van de Merwe RC, Louie R et al (2020) Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat Immunol. https://doi.org/10.1038/s41590-020-0646-0
Mancuso P, McNish RW, Peters-Golden M, Brock TG (2001) Evaluation of phagocytosis and arachidonate metabolism by alveolar macrophages and recruited neutrophils from F344xBN rats of different ages. Mech Ageing Dev. https://doi.org/10.1016/S0047-6374(01)00322-0
Stahl EC, Brown BN (2015) Cell therapy strategies to combat immunosenescence. Organogenesis
Pinhal-Enfield G, Ramanathan M, Hasko G et al (2003) An angiogenic switch in macrophages involving synergy between toll-like receptors 2, 4, 7, and 9 and adenosine A2A receptors. Am J Pathol. https://doi.org/10.1016/S0002-9440(10)63698-X
Ashcroft GS, Horan MA, Ferguson MWJ (1998) Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab Investig
Franceschi C, Garagnani P, Parini P et al (2018) Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat Rev Endocrinol
Potente M, Ghaeni L, Baldessari D et al (2007) SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. https://doi.org/10.1101/gad.435107
Minhas PS, Liu L, Moon PK et al (2019) Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat Immunol. https://doi.org/10.1038/s41590-018-0255-3
Consonni FM, Porta C, Marino A et al (2019) Myeloid-derived suppressor cells: ductile targets in disease. Front Immunol 10
Bronte V, Brandau S, Chen SH et al (2016) Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 7
Hegde S, Leader AM, Merad M (2021) MDSC: Markers, development, states, and unaddressed complexity. Immunity
Enioutina EY, Bareyan D, Daynes RA (2011) A role for immature myeloid cells in immune senescence. J Immunol. https://doi.org/10.4049/jimmunol.1002987
Alves AS, Ishimura ME, de Duarte YA, O, Bueno V, (2018) Parameters of the immune system and vitamin D levels in old individuals. Front Immunol. https://doi.org/10.3389/fimmu.2018.01122
Verschoor CP, Johnstone J, Millar J et al (2013) Blood CD33(+)HLA-DR(−) myeloid-derived suppressor cells are increased with age and a history of cancer. J Leukoc Biol. https://doi.org/10.1189/jlb.0912461
Salminen A, Kaarniranta K, Kauppinen A (2019) Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci
Flores RR, Clauson CL, Cho J et al (2017) Expansion of myeloid-derived suppressor cells with aging in the bone marrow of mice through a NF-κB-dependent mechanism. Aging Cell. https://doi.org/10.1111/acel.12571
Wiley CD, Velarde MC, Lecot P et al (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. https://doi.org/10.1016/j.cmet.2015.11.011
Acosta JC, Banito A, Wuestefeld T et al (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. https://doi.org/10.1038/ncb2784
Worbs T, Hammerschmidt SI, Förster R (2017) Dendritic cell migration in health and disease. Nat Rev Immunol
Jing Y, Shaheen E, Drake RR et al (2009) Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum Immunol. https://doi.org/10.1016/j.humimm.2009.07.005
Granot T, Senda T, Carpenter DJ et al (2017) Dendritic cells display subset and tissue-specific maturation dynamics over human life. Immunity. https://doi.org/10.1016/j.immuni.2017.02.019
Agrawal A, Gupta S (2011) Impact of aging on dendritic cell functions in humans. Ageing Res Rev
Hoogendijk EO, Afilalo J, Ensrud KE et al (2019) Frailty: implications for clinical practice and public health. Lancet
Proietti M, Cesari M (2020) Frailty: what is it? In: Advances in Experimental Medicine and Biology
Morley JE, Vellas B, Abellan van Kan G et al (2013) Frailty consensus: a call to action. J Am Med Dir Assoc. https://doi.org/10.1016/j.jamda.2013.03.022
Pansarasa O, Pistono C, Davin A et al (2019) Altered immune system in frailty: genetics and diet may influence inflammation. Ageing Res Rev
Cesari M, Nobili A, Vitale G (2016) Frailty and sarcopenia: from theory to clinical implementation and public health relevance. Eur J Intern Med
Cesari M, Pérez-Zepeda MU, Marzetti E (2017) Frailty and multimorbidity: different ways of thinking about geriatrics. J Am Med Dir Assoc. https://doi.org/10.1016/j.jamda.2016.12.086
Fried LP, Tangen CM, Walston J et al (2001) Frailty in older adults: evidence for a phenotype. Journals Gerontol - Ser A Biol Sci Med Sci. https://doi.org/10.1093/gerona/56.3.m146
Rockwood K, Mitnitski A (2007) Frailty in relation to the accumulation of deficits. Journals Gerontol. - Ser A Biol Sci Med Sci
Villacampa-Fernández P, Navarro-Pardo E, Tarín JJ, Cano A (2017) Frailty and multimorbidity: two related yet different concepts. Maturitas
Salisbury C (2012) Multimorbidity: redesigning health care for people who use it. Lancet
Barnett K, Mercer SW, Norbury M et al (2012) Epidemiology of multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet. https://doi.org/10.1016/S0140-6736(12)60240-2
Minciullo PL, Catalano A, Mandraffino G et al (2016) Inflammaging and anti-inflammaging: the role of cytokines in extreme longevity. Arch. Immunol. Ther Exp (Warsz)
Wilson D, Jackson T, Sapey E, Lord JM (2017) Frailty and sarcopenia: the potential role of an aged immune system. Ageing Res Rev
Piggott DA, Varadhan R, Mehta SH et al (2015) Frailty, inflammation, and mortality among persons aging with HIV infection and injection drug use. Journals Gerontol - Ser A Biol Sci Med Sci. https://doi.org/10.1093/gerona/glv107
Soysal P, Stubbs B, Lucato P et al (2016) Inflammation and frailty in the elderly: a systematic review and meta-analysis. Ageing Res Rev
Qu T, Yang H, Walston JD et al (2009) Upregulated monocytic expression of CXC chemokine ligand 10 (CXCL-10) and its relationship with serum interleukin-6 levels in the syndrome of frailty. Cytokine. https://doi.org/10.1016/j.cyto.2009.02.015
Walston J, Fedarko N, Yang H et al (2008) The physical and biological characterization of a frail mouse model. Journals Gerontol - Ser A Biol Sci Med Sci. https://doi.org/10.1093/gerona/63.4.391
Fougère B, Boulanger E, Nourhashémi F et al (2017) Chronic inflammation: accelerator of biological aging. Journals Gerontol. - Ser A Biol Sci Med Sci
Barbé-Tuana F, Funchal G, Schmitz CRR et al (2020) The interplay between immunosenescence and age-related diseases. Semin Immunopathol
Fulop T, Witkowski JM, Olivieri F, Larbi A (2018) The integration of inflammaging in age-related diseases. Semin Immunol
Veronese N (2020) Frailty as cardiovascular risk factor (and vice versa). In: Advances in Experimental Medicine and Biology
Ormazabal V, Nair S, Elfeky O et al (2018) Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol
Libby P (2012) Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. https://doi.org/10.1161/ATVBAHA.108.179705
Fülöp T, Dupuis G, Witkowski JM, Larbi A (2016) The role of immunosenescence in the development of age-related diseases. Rev Investig Clin
Soysal P, Arik F, Smith L et al (2020) Inflammation, frailty and cardiovascular disease. In: Advances in Experimental Medicine and Biology
Wick G, Perschinka H, Millonig G (2001) Atherosclerosis as an autoimmune disease: an update. Trends Immunol
Biasucci LM, La Rosa G, Pedicino D et al (2017) Where does inflammation fit? Curr Cardiol Rep
Moore KJ, Sheedy FJ, Fisher EA (2013) Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13:709–721
Zeng Y, Nie C, Min J et al (2016) Novel loci and pathways significantly associated with longevity. Sci Rep. https://doi.org/10.1038/srep21243
Schmidt AM (2019) Diabetes mellitus and cardiovascular disease. Arterioscler Thromb Vasc Biol
Saklayen MG (2018) The global epidemic of the metabolic syndrome. Curr Hypertens Rep
Eckel RH, Grundy SM, Zimmet PZ (2005) The metabolic syndrome. In: Lancet
Zmora N, Bashiardes S, Levy M, Elinav E (2017) The role of the immune system in metabolic health and disease. Cell Metab
Salvestrini V, Sell C, Lorenzini A (2019) Obesity may accelerate the aging process. Front Endocrinol (Lausanne)
Trim W, Turner JE, Thompson D (2018) Parallels in immunometabolic adipose tissue dysfunction with ageing and obesity. Front Immunol
Rocha VZ, Libby P (2009) Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. https://doi.org/10.1038/nrcardio.2009.55
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science (80- ). https://doi.org/10.1126/science.7678183
Hotamisligil GS (2017) Inflammation, metaflammation and immunometabolic disorders. Nature 542:177–185
Kim HJ, Higashimori T, Park SY et al (2004) Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes. https://doi.org/10.2337/diabetes.53.4.1060
Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y (2014) Obesity in autoimmune diseases: Not a passive bystander. Autoimmun Rev
Villareal DT, Aguirre L, Gurney AB et al (2017) Aerobic or resistance exercise, or both, in dieting obese older adults. N Engl J Med. https://doi.org/10.1056/nejmoa1616338
Ferrante AW (2013) The immune cells in adipose tissue. Diabetes Obes Metab
Hainer V, Aldhoon-Hainerová I (2013) Obesity paradox does exist. Diabetes Care. https://doi.org/10.2337/dcS13-2023
Ahlqvist E, Storm P, Käräjämäki A et al (2018) Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. https://doi.org/10.1016/S2213-8587(18)30051-2
Halter JB, Musi N, Horne FMF et al (2014) Diabetes and cardiovascular disease in older adults: current status and future directions. Diabetes
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature
Guarner V, Rubio-Ruiz ME (2014) Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip Top Gerontol. https://doi.org/10.1159/000364934
Esposito K, Nappo F, Marfella R et al (2002) Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: Role of oxidative stress. Circulation. https://doi.org/10.1161/01.CIR.0000034509.14906.AE
Komura T, Sakai Y, Honda M et al (2010) CD14+ monocytes are vulnerable and functionally impaired under endoplasmic reticulum stress in patients with type 2 diabetes. Diabetes. https://doi.org/10.2337/db09-0659
Lecube A, Pachón G, Petriz J et al (2011) Phagocytic activity is impaired in type 2 diabetes mellitus and increases after metabolic improvement. PLoS One. https://doi.org/10.1371/journal.pone.0023366
Panza F, Solfrizzi V, Frisardi V et al (2011) Different models of frailty in predementia and dementia syndromes. J Nutr Heal Aging. https://doi.org/10.1007/s12603-011-0126-1
Santoro A, Spinelli CC, Martucciello S et al (2018) Innate immunity and cellular senescence: the good and the bad in the developmental and aged brain. J Leukoc Biol
Salter MW, Stevens B (2017) Microglia emerge as central players in brain disease. Nat Med
Labzin LI, Heneka MT, Latz E (2018) Innate immunity and neurodegeneration. Annu Rev Med. https://doi.org/10.1146/annurev-med-050715-104343
Verdile G, Keane KN, Cruzat VF et al (2015) Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediators Inflamm
Rizzi L, Rosset I, Roriz-Cruz M (2014) Global epidemiology of dementia: Alzheimer’s and vascular types. Biomed Res Int. https://doi.org/10.1155/2014/908915
Pizza V, Agresta A, W D’Acunto C et al (2012) Neuroinflamm-aging and neurodegenerative diseases: an overview. CNS Neurol Disord - Drug Targets. https://doi.org/10.2174/187152711796235014
Raposo C, Graubardt N, Cohen M et al (2014) CNS repair requires both effector and regulatory T cells with distinct temporal and spatial profiles. J Neurosci. https://doi.org/10.1523/JNEUROSCI.0076-14.2014
Niraula A, Sheridan JF, Godbout JP (2017) Microglia priming with aging and stress. Neuropsychopharmacology
Costantini E, D’Angelo C, Reale M (2018) The role of immunosenescence in neurodegenerative diseases. Mediators Inflamm
Heneka MT, Carson MJ, Khoury JL et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol
Letiembre M, Hao W, Liu Y et al (2007) Innate immune receptor expression in normal brain aging. Neuroscience. https://doi.org/10.1016/j.neuroscience.2007.01.004
Flanary BE, Sammons NW, Nguyen C et al (2007) Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. https://doi.org/10.1089/rej.2006.9096
Thériault P, Elali A, Rivest S (2015) The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimer’s Res Ther
Calabrese V, Santoro A, Monti D et al (2018) Aging and Parkinson’s disease: inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic Biol Med
Salaffi F, Farah S, Carlo MD (2020) Frailty syndrome in musculoskeletal disorders: an emerging concept in rheumatology. Acta Biomed
Rezuș E, Cardoneanu A, Burlui A et al (2019) The link between inflammaging and degenerative joint diseases. Int J Mol Sci
Faulkner JA, Larkin LM, Claflin DR, Brooks SV (2007) Age-related changes in the structure and function of skeletal muscles. Clin Exp Pharmacol Physiol
Cruz-Jentoft AJ, Baeyens JP, Bauer JM et al (2010) Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing. https://doi.org/10.1093/ageing/afq034
von Haehling S, Morley JE, Anker SD (2010) An overview of sarcopenia: facts and numbers on prevalence and clinical impact. J Cachexia Sarcopenia Muscle
Singer M, Deutschman CS, Seymour C et al (2016) The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA - J Am Med Assoc
Roh E, Choi KM (2020) Health consequences of sarcopenic obesity: a narrative review. Front Endocrinol (Lausanne)
Kalinkovich A, Livshits G (2017) Sarcopenic obesity or obese sarcopenia: a cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing Res Rev
Chhetri JK, de Souto Barreto P, Fougère B et al (2018) Chronic inflammation and sarcopenia: a regenerative cell therapy perspective. Exp Gerontol
Jo E, Lee SR, Park BS, Kim JS (2012) Potential mechanisms underlying the role of chronic inflammation in age-related muscle wasting. Aging Clin Exp Res
Beyer I, Mets T, Bautmans I (2012) Chronic low-grade inflammation and age-related sarcopenia. Curr Opin Clin Nutr Metab Care
Taniguchi K, Karin M (2018) NF-B, inflammation, immunity and cancer: coming of age. Nat Rev Immunol
Patsalosa A, Tzerposb P, Weia X, Nagy L (2021) Myeloid cell diversification during regenerative inflammation: lessons from skeletal muscle. Semin Cell Dev Biol S1084–9521:00114–00122
Tidball JG (2017) Regulation of muscle growth and regeneration by the immune system. Nat Rev Immunol
Wang Y, Welc SS, Wehling-Henricks M, Tidball JG (2018) Myeloid cell-derived tumor necrosis factor-alpha promotes sarcopenia and regulates muscle cell fusion with aging muscle fibers. Aging Cell. https://doi.org/10.1111/acel.12828
Shang M, Cappellesso F, Amorim R et al (2020) Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature. https://doi.org/10.1038/s41586-020-2857-9
Laconi E, Marongiu F, DeGregori J (2020) Cancer as a disease of old age: changing mutational and microenvironmental landscapes. Br J Cancer
Siegel RL, Miller KD, Jemal A (2018) Cancer statistics, 2018. CA Cancer J Clin. https://doi.org/10.3322/caac.21442
Hsu T (2016) Educational initiatives in geriatric oncology – who, why, and how? J Geriatr Oncol
Rea IM, Gibson DS, McGilligan V et al (2018) Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol
Trinchieri G (2012) Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell
Cuollo L, Antonangeli F, Santoni A, Soriani A (2020) The senescence-associated secretory phenotype (Sasp) in the challenging future of cancer therapy and age-related diseases. Biology (Basel)
Yang L, Li A, Lei Q, Zhang Y (2019) Tumor-intrinsic signaling pathways: key roles in the regulation of the immunosuppressive tumor microenvironment. J Hematol Oncol
Lian J, Yue Y, Yu W, Zhang Y (2020) Immunosenescence: a key player in cancer development. J Hematol Oncol
DeSantis CE, Miller KD, Dale W et al (2019) Cancer statistics for adults aged 85 years and older, 2019. CA Cancer J Clin. https://doi.org/10.3322/caac.21577
Hurez V, Padrón S, Svatek RS, Curiel TJ (2017) Considerations for successful cancer immunotherapy in aged hosts. Clin Exp Immunol
Grizzle WE, Xu X, Zhang S et al (2007) Age-related increase of tumor susceptibility is associated with myeloid-derived suppressor cell mediated suppression of T cell cytotoxicity in recombinant inbred BXD12 mice. Mech Ageing Dev. https://doi.org/10.1016/j.mad.2007.10.003
Veglia F, Perego M, Gabrilovich D (2018) Myeloid-derived suppressor cells coming of age review-article. Nat Immunol
Ostrand-Rosenberg S, Fenselau C (2018) Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. https://doi.org/10.4049/jimmunol.1701019
Aras S, Raza Zaidi M (2017) TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer
Jackaman C, Radley-Crabb HG, Soffe Z et al (2013) Targeting macrophages rescues age-related immune deficiencies in C57BL/6J geriatric mice. Aging Cell. https://doi.org/10.1111/acel.12062
Dace DS, Apte RS (2008) Effect of senescence on macrophage polarization and angiogenesis. Rejuvenation Res
van Duin D, Mohanty S, Thomas V et al (2007) Age-associated defect in human TLR-1/2 function. J Immunol. https://doi.org/10.4049/jimmunol.178.2.970
Fridlender Z, Sun J, Kim S et al (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: N1 versus N2 TAN. Cancer Cell
Butcher SK, Chahal H, Nayak L et al (2001) Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol. https://doi.org/10.1189/jlb.70.6.881
Fülöp T, Fouquet C, Allaire P et al (1997) Changes in apoptosis of human polymorphonuclear granulocytes with aging. Mech Ageing Dev. https://doi.org/10.1016/S0047-6374(96)01881-7
Schröder AK, Rink L (2003) Neutrophil immunity of the elderly. In: Mechanisms of Ageing and Development
Fridlender ZG, Albelda SM (2012) Tumor-associated neutrophils: friend or foe? Carcinogenesis
Walker C, Mojares E, Del Río Hernández A (2018) Role of extracellular matrix in development and cancer progression. Int J Mol Sci
Panwar P, Lamour G, Mackenzie NCW et al (2015) Changes in structural-mechanical properties and degradability of collagen during aging-associated modifications. J Biol Chem. https://doi.org/10.1074/jbc.M115.644310
Mavrogonatou E, Pratsinis H, Papadopoulou A et al (2019) Extracellular matrix alterations in senescent cells and their significance in tissue homeostasis. Matrix Biol
Zhu Y, Herndon JM, Sojka DK et al (2017) Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47:323-338.e6. https://doi.org/10.1016/j.immuni.2017.07.014
Jiang D, Lim SY (2016) Influence of immune myeloid cells on the extracellular matrix during cancer metastasis. Cancer Microenviron
McElhaney JE, Effros RB (2009) Immunosenescence: what does it mean to health outcomes in older adults? Curr Opin Immunol
Liang SY (2016) Sepsis and other infectious disease emergencies in the elderly. Emerg Med Clin North Am
Crooke SN, Ovsyannikova IG, Poland GA, Kennedy RB (2019) Immunosenescence and human vaccine immune responses. Immun Ageing. https://doi.org/10.1186/s12979-019-0164-9
Önen NF, Agbebi A, Shacham E et al (2009) Frailty among HIV-infected persons in an urban outpatient care setting. J Infect. https://doi.org/10.1016/j.jinf.2009.08.008
Schmaltz HN, Fried LP, Xue QL et al (2005) Chronic cytomegalovirus infection and inflammation are associated with prevalent frailty in community-dwelling older women. J Am Geriatr Soc. https://doi.org/10.1111/j.1532-5415.2005.53250.x
Santesmasses D, Castro JP, Zenin AA et al (2020) COVID-19 is an emergent disease of aging. Aging Cell. https://doi.org/10.1111/acel.13230
Richardson S, Hirsch JS, Narasimhan M et al (2020) Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA - J Am Med Assoc. https://doi.org/10.1001/jama.2020.6775
Verity R, Okell LC, Dorigatti I et al (2020) Estimates of the severity of coronavirus disease 2019: a model-based analysis. Lancet Infect Dis. https://doi.org/10.1016/S1473-3099(20)30243-7
Fajgenbaum DC, June CH (2020) Cytokine storm. N Engl J Med. https://doi.org/10.1056/nejmra2026131
Mangalmurti N, Hunter CA (2020) Cytokine storms: understanding COVID-19. Immunity
Hojyo S, Uchida M, Tanaka K et al (2020) How COVID-19 induces cytokine storm with high mortality. Inflamm Regen
Schulte-Schrepping J, Reusch N, Paclik D et al (2020) Severe COVID-19 is markeD by a dysregulated myeloid cell compartment. Cell. https://doi.org/10.1016/j.cell.2020.08.001
Silvin A, Chapuis N, Dunsmore G et al (2020) Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from milD COVID-19. Cell. https://doi.org/10.1016/j.cell.2020.08.002
Szabo PA, Dogra P, Gray JI et al (2021) Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity. https://doi.org/10.1016/j.immuni.2021.03.005
Lu Q, Liu J, Zhao S et al (2021) SARS-CoV-2 exacerbates proinflammatory responses in myeloid cells through C-type lectin receptors and Tweety family member 2. Immunity. https://doi.org/10.1016/j.immuni.2021.05.006
Cavalcante-Silva LHA, Carvalho DCM, Lima É de A et al (2021) Neutrophils and COVID-19: the road so far. Int Immunopharmacol
D’Alessio FR, Heller NM (2020) COVID-19 and myeloid cells: complex interplay correlates with lung severity. J Clin Invest
Merad M, Martin JC (2020) Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol
Zhou Z, Ren L, Zhang L et al (2020) Overly exuberant innate immune response to SARS-CoV-2 infection. SSRN Electron J. https://doi.org/10.2139/ssrn.3551623
Zhang D, Guo R, Lei L et al (2021) Frontline science: COVID-19 infection induces readily detectable morphologic and inflammation-related phenotypic changes in peripheral blood monocytes. J Leukoc Biol. https://doi.org/10.1002/JLB.4HI0720-470R
Abani O, Abbas A, Abbas F et al (2021) Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. https://doi.org/10.1016/S0140-6736(21)00676-0
Grants JM, Wegrzyn J, Hui T et al (2020) Altered microRNA expression links IL6 and TNF-induced inflammaging with myeloid malignancy in humans and mice. Blood. https://doi.org/10.1182/blood.2019003105
Sabbatinelli J, Giuliani A, Matacchione G et al (2021) Decreased serum levels of the inflammaging marker miR-146a are associated with non-clinical response to tocilizumab in COVID-19 patients. Mech Ageing Dev. https://doi.org/10.1016/j.mad.2020.111413
Ridker PM, Everett BM, Thuren T et al (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. https://doi.org/10.1056/nejmoa1707914
McInnes IB, Thompson L, Giles JT et al (2015) Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann Rheum Dis. https://doi.org/10.1136/annrheumdis-2013-204345
Giles JT, Sattar N, Gabriel S et al (2020) Cardiovascular safety of tocilizumab versus etanercept in rheumatoid arthritis: a randomized controlled trial. Arthritis Rheumatol. https://doi.org/10.1002/art.41095
Broch K, Anstensrud AK, Woxholt S et al (2021) Randomized trial of interleukin-6 receptor inhibition in patients with acute ST-segment elevation myocardial infarction. J Am Coll Cardiol. https://doi.org/10.1016/j.jacc.2021.02.049
Holte E, Kleveland O, Ueland T et al (2017) Effect of interleukin-6 inhibition on coronary microvascular and endothelial function in myocardial infarction. Heart. https://doi.org/10.1136/heartjnl-2016-310875
Alraouji NN, Al-Mohanna FH, Ghebeh H et al (2020) Tocilizumab potentiates cisplatin cytotoxicity and targets cancer stem cells in triple-negative breast cancer. Mol Carcinog. https://doi.org/10.1002/mc.23234
Ham IH, Oh HJ, Jin H et al (2019) Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Mol Cancer. https://doi.org/10.1186/s12943-019-0972-8
Guarente L (2011) Sirtuins, aging, and metabolism. Cold Spring Harb Symp Quant Biol. https://doi.org/10.1101/sqb.2011.76.010629
Weichhart T (2018) mTOR as regulator and lifespan, aging and cellular senescence. Gerontology
Pignatti C, D’adamo S, Stefanelli C et al (2020) Nutrients and pathways that regulate health span and life span. Geriatr
Bjedov I, Rallis C (2020) The target of rapamycin signalling pathway in ageing and lifespan regulation. Genes (Basel)
Wu Z, Isik M, Moroz N et al (2019) Dietary restriction extends lifespan through metabolic regulation of innate immunity. Cell Metab. https://doi.org/10.1016/j.cmet.2019.02.013
Lee Y, Jung Y, Jeong DE et al (2021) Reduced insulin/IGF1 signaling prevents immune aging via ZIP-10/bZIP-mediated feedforward loop. J Cell Biol. https://doi.org/10.1083/jcb.202006174
López-Lluch G, Navas P (2016) Calorie restriction as an intervention in ageing. J Physiol
Okawa T, Nagai M, Hase K (2021) Dietary intervention impacts immune cell functions and dynamics by inducing metabolic rewiring. Front Immunol
Gorbunova V, Seluanov A, Mita P et al (2021) The role of retrotransposable elements in ageing and age-associated diseases. Nature 596
Funding
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) IG (nos. 19885 to A.S. and 15860 to R.M.); AIRC 5 × 1000 no. 22757; Fondazione Cariplo and Ministero Università Ricerca (project no. 2017BA9LM5_001).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bleve, A., Motta, F., Durante, B. et al. Immunosenescence, Inflammaging, and Frailty: Role of Myeloid Cells in Age-Related Diseases. Clinic Rev Allerg Immunol 64, 123–144 (2023). https://doi.org/10.1007/s12016-021-08909-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-021-08909-7