
Abstract
Identification of very long-chain acyl-CoA dehydrogenase deficiency is possible in the expanded newborn screening (NBS) due to the increase in tetradecenoylcarnitine (C14:1) and in the C14:1/C2, C14:1/C16, C14:1/C12:1 ratios detected in dried blood spots. Nevertheless, different confirmatory tests must be performed to confirm the final diagnosis. We have revised the NBS results and the results of the confirmatory tests (plasma acylcarnitine profiles, molecular findings, and lymphocytes VLCAD activity) for 36 cases detected in three Spanish NBS centers during 4 years, correlating these with the clinical outcome and treatment. Our aim was to distinguish unambiguously true cases from disease carriers in order to obtain useful diagnostic information for clinicians that can be applied in the follow-up of neonates identified by NBS.
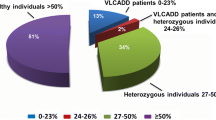
Increases in C14:1 and of the different ratios, the presence of two pathogenic mutations, and deficient enzyme activity in lymphocytes (<12% of the intra-assay control) identified 12 true-positive cases. These cases were given nutritional therapy and all of them are asymptomatic, except one. Seventeen individuals were considered disease carriers based on the mild increase in plasma C14:1, in conjunction with the presence of only one mutation and/or intermediate residual activity (18–57%). In addition, seven cases were classified as false positives, with normal biochemical parameters and no mutations in the exonic region of ACADVL. All these carriers and the false positive cases remained asymptomatic. The combined evaluation of the acylcarnitine profiles, genetic results, and residual enzyme activities have proven useful to definitively classify individuals with suspected VLCAD deficiency into true-positive cases and carriers, and to decide which cases need treatment.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others

References
Andresen BS, Olpin S, Poorthuis BJ, Scholte HR et al (1999) Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet 64:479–494
Arnold GL, Van Hove J, Freedenberg D, Strauss A et al (2009) A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 96:85–90
Boneh A, Andresen BS, Gregersen N, Ibrahim M et al (2006) VLCAD deficiency: pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol Genet Metab 88:166–170
Browning MF, Larson C, Strauss A, Marsden DL (2005) Normal acylcarnitine levels during confirmation of abnormal newborn screening in long-chain fatty acid oxidation defects. J Inherit Metab Dis 28:545–550
Coughlin CR 2nd, Ficicioglu C (2010) Genotype-phenotype correlations: sudden death in an infant with very-long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 33(Suppl 3):S129–S131
Dopazo J, Amadoz A, Bleda M, Garcia-Alonso L et al (2016) 267 Spanish exomes reveal population-specific differences in disease-related genetic variation. Mol Biol Evol 33:1205–1218
Ferrer I, Ruiz-Sala P, Vicente Y, Merinero B et al (2007) Separation and identification of plasma short-chain acylcarnitine isomers by HPLC/MS/MS for the differential diagnosis of fatty acid oxidation defects and organic acidemias. J Chromatogr B 860:121–126
Hall PL, Marquardt G, McHugh DM, Currier RJ et al (2014) Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet Med 16:889–895
Merinero B, Perez-Cerda C, Garcia MJ, Gangoiti J et al (1996) Mitochondrial very long-chain acyl-CoA dehydrogenase deficiency with a mild clinical course. J Inherit Metab Dis 19:173–176
Merinero B, Pascual Pascual SI, Perez-Cerda C, Gangoiti J et al (1999) Adolescent myopathic presentation in two sisters with very long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 22:802–810
Merritt JL 2nd, Vedal S, Abdenur JE, Au SM et al (2014) Infants suspected to have very-long chain acyl-CoA dehydrogenase deficiency from newborn screening. Mol Genet Metab 111:484–492
Osorio JH, Lluch M, Ribes A (2003) Analysis of organic acids after incubation with (16-2H3)palmitic acid in fibroblasts from patients with mitochondrial beta-oxidation defects. J Inherit Metab Dis 26:795–803
Pena LD, van Calcar SC, Hansen J, Edick MJ et al (2016) Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol Genet Metab 118:272–281
Richards S, Aziz N, Bale S, Bick D et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Rocha H, Castineiras D, Delgado C, Egea J et al (2014) Birth prevalence of fatty acid beta-oxidation disorders in Iberia. JIMD Rep 16:89–94
Schiff M, Mohsen AW, Karunanidhi A, McCracken E et al (2013) Molecular and cellular pathology of very-long-chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 109:21–27
Spiekerkoetter U, Lindner M, Santer R, Grotzke M et al (2009) Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis 32:498–505
Spiekerkoetter U, Haussmann U, Mueller M, ter Veld F et al (2010) Tandem mass spectrometry screening for very long-chain acyl-CoA dehydrogenase deficiency: the value of second-tier enzyme testing. J Pediatr 157:668–673
Tajima G, Sakura N, Yofune H, Nishimura Y et al (2005) Enzymatic diagnosis of medium-chain acyl-CoA dehydrogenase deficiency by detecting 2-octenoyl-CoA production using high-performance liquid chromatography: a practical confirmatory test for tandem mass spectrometry newborn screening in Japan. J Chromatogr B 823:122–130
ter Veld F, Mueller M, Kramer S, Haussmann U et al (2009) A novel tandem mass spectrometry method for rapid confirmation of medium- and very long-chain acyl-CoA dehydrogenase deficiency in newborns. PLoS One 4:e6449
Acknowledgments
The authors would like to thank the families involved in this study for giving their consent. This work was funded by a grant from the Fundación Isabel Gemio. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Piero Rinaldo, MD, PhD
Appendices
A Concise One Sentence Take-Home Message
The combined evaluation of the acylcarnitine profiles, genetic results, and residual enzyme activities have proven useful to definitively classify individuals with suspected VLCAD deficiency into true-positive cases and carriers, and to decide which cases need treatment, as well as close clinical and biochemical monitoring.
Compliance with Ethics Guidelines
Conflict of Interests
None of the authors have any conflict of interests to declare.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation and with the Helsinki Declaration of 1975, as revised in 2000 (5). This project was approved by the Universidad Autónoma Madrid Ethics Committee (reference number CEI 74-1349).
Details of the Contributions of Individuals Authors
All authors approved the final manuscript as submitted.
BM: conception and design, drafting, and coordination of the manuscript.
PA: enzyme activity determinations, statistical analysis, and writing the first draft.
EMH: treating metabolic specialist of patients, drafting of the manuscript.
AM: treating metabolic specialist of patients.
MTGS: treating metabolic specialist of patients.
PQF: treating metabolic specialist of patients.
CPG: treating metabolic specialist of patients.
ED: analysis and interpretation of newborn screening data from Madrid.
RY: analysis and interpretation of newborn screening data from Western Andalucia.
JME: analysis and interpretation of newborn screening data from Murcia.
ABQ: treating metabolic specialist of patients.
JBA: treating metabolic specialist of patients.
MLFR: analysis and interpretation of newborn screening data from Madrid.
BB: analysis of newborn screening data from Madrid.
IFL: acylcarnitines and organic acids analysis.
FL: molecular genetic analysis.
MU: critical review of the manuscript for important intellectual content.
PRS: biochemical and enzyme data analysis.
BP: molecular genetic data interpretation, drafting of the manuscript.
CPC: biochemical data interpretation, drafting of the manuscript.
Rights and permissions
Copyright information
© 2017 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Merinero, B. et al. (2017). Four Years’ Experience in the Diagnosis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency in Infants Detected in Three Spanish Newborn Screening Centers. In: Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V. (eds) JIMD Reports, Volume 39. JIMD Reports, vol 39. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2017_40
Download citation
DOI: https://doi.org/10.1007/8904_2017_40
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-57576-5
Online ISBN: 978-3-662-57577-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)