ABSTRACT
INTRODUCTION Hyperphenylalaninemias are inborn errors of phenylalanine metabolism caused by deficiency of L-phenylalanine hydroxylase (the enzyme that converts phenylalanine to tyrosine), resulting in increased serum phenylalanine (>4 mg/dL or 240 µmol/L). Phenylketonuria, or PKU, is the most common form. Untreated PKU is associated with progressive neurodevelopmental delay, evolving towards intellectual impairment.
Cuba introduced a national newborn screening program for PKU in 1986. It has enabled early diagnosis and initiation of dietary treatment, reducing appearance of intellectual impairment in these patients. Originally, confirmatory diagnosis was done only by quantifying serum phenylalanine. In 2010, however, an HPLC method for quantifying serum phenylalanine and tyrosine simultaneously was validated at the National Medical Genetics Center, to perform confirmatory and differential diagnosis of hyperphenylalaninemias, as well as biochemical monitoring of patients diagnosed.
OBJECTIVE Create birth weight percentile distribution tables and curves for neonates by gestational age and sex in Holguín Municipality, capital of the eastern Cuban province of the same name.
METHODS A descriptive retrospective case-series study was conducted from June 2010 through June 2012. The study population comprised 531 infants who tested positive in the National Neonatal Screening Program for Phenylketonuria. Variables used were serum phenylalanine concentration (first criterion of positivity) and tyrosine, phenylalanine/tyrosine ratio (second criterion, both detected by reverse-phase HPLC with direct fluorescence), hyperphenylalaninemia classification, year of diagnosis, sex, and province of origin.
RESULTS Of the samples, 97.7% (519/531) were confirmed as false positives, and 10.4% (55/531) had transient neonatal tyrosinemia. Hyperphenylalaninemia was diagnosed in 12 infants (2.2%): 1.3% (7/531) presented classical PKU, with 34.7 ± 14.7 mg/dL phenylalanine in serum and phenylalanine/tyrosine ratio of 18.9 ± 12.7; and 0.9% (5/531) had persistent hyperphenylalaninemia, with 8.9 ± 3.4 mg/dL of phenylalanine and phenylalanine/tyrosine ratio of 4.5 ± 1.6. Matanzas Province contributed more cases than any of Cuba’s 14 other provinces (3/12, 25%) and there was a slight predominance of male sex (7/12, 58.3%). During biochemical monitoring, 83.3% of patients (10/12) reduced their levels of phenylalanine (≤5 mg/dL or 300 µmol/L): 5 with classical PKU and all 5 with persistent hyperphenylalaninemia. The incidence of neonatal hyperphenylalaninemias was 1/22,503 live births and 1/38,577 for classical PKU.
CONCLUSIONS HPLC for simultaneous quantification of phenylalanine and tyrosine in serum meets the needs of a confirmatory test for patients testing positive in Cuba’s National Neonatal Screening Program for Phenylketonuria (which has high false positive rates). It has enabled introduction in Cuba of a second PKU diagnostic criterion of positivity for both the classification of hyperphenylalaninemias and the biochemical monitoring of diagnosed patients.
KEYWORDS Hyperphenylalaninemias, phenylketonuria, phenylalanine hydroxylase deficiency disease, HPLC, phenylalanine, tyrosine, PKU, PAH deficiency, BH4 deficiency, genetic disease, hereditary disease, screening, Folling disease, Cuba
INTRODUCTION
Hyperphenylalaninemias (HPA) are inborn errors of metabolism of the amino acid phenylalanine (Phe), caused in 98% of cases by mutations in the gene encoding the enzyme L-phenylalanine hydroxylase (PAH) (EC 1.14.16.1), which is responsible for converting Phe into tyrosine (Tyr) by hydroxylation.[1–5] The remaining 1%–2% of cases are attributable to defects in other enzymes involved in synthesis or regeneration of tetrahydrobiopterin (BH4), the natural cofactor for PAH, tyrosine 3-hydroxylase and tryptophan-5-hydroxylase. This group of inherited metabolic diseases exhibits an autosomal recessive inheritance pattern. Without early treatment with a Phe-restricted diet, it leads to progressive neurodevelopmental delay, which can result in intellectual impairment, skin and hair depigmentation, failure to thrive, characteristic urine odor, eczema and epilepsy.[1–11]
HPAs are the second most common cause of preventable intellectual impairment and include several conditions that differ clinically and biochemically, with phenylketonuria (PKU) being the most common and severe. Differential diagnosis of HPA is critical for determining correct treatment.[1,4,5] PKU frequency ranges from 1/143,000 live births in Japan to 1/10,000 in Northern Europe, with 1/4500 in Ireland and 1/2600 in Turkey.[2] Incidence in Cuba has been reported as 1/50,000 live births (1989)[3] and 1/52,590 (2007).[10]
In 1984, neonatal PKU screening began in Havana, using dried blood samples on filter paper to measure Phe concentration with the Guthrie and Susi bacterial inhibition test.[12] This screening program was expanded to the rest of Cuba in 1986,[13] as part of the National Neonatal Screening Program (PNPN). It has facilitated earlier detection of HPA and dietary treatment, and has helped reduce occurrence of intellectual impairment in these patients.[14] In 2000, the PNPN began to use domestic technology for HPA diagnosis, the SUMA ultramicroanalytical system and its UMTEST-PKU reagent kit.[15]
Until 2010, HPA confirmatory diagnosis in Cuba was done with spectrofluorimetry to quantify Phe serum levels only, the first of two diagnostic criteria, since the lack of high-pressure liquid chromatography technology (HPLC) prevented us from introducing the second criterion, the Phe/Tyr ratio, which reduces false positive results and is the most commonly used worldwide for differential diagnosis and biochemical monitoring.[7,16] HPLC is the most widely used quantitative screening method for diagnosing inborn errors of metabolism, chosen for its speed and specificity, and because it permits simultaneous quantification of several biochemical markers using small sample volumes.[1,17–26]
In the first semester of 2010, the National Medical Genetics Center (CNGM) in Havana validated an isocratic reverse-phase HPLC method with direct fluorescent detection for simultaneous quantification of Phe and Tyr in serum, according to modifications of the method described by Kandar and Zakova.[26] It was introduced in the second half of that year for confirmatory and differential HPA diagnosis, as well as for biochemical monitoring of diagnosed patients under dietary treatment.[27,28]
The aim of this study was to describe the first two years’ experience after introducing HPLC for both confirmatory diagnosis of PKU-positive cases detected by PNPN and biochemical monitoring of patients undergoing dietary treatment.
METHODS
Study type and universe A descriptive retrospective study of a series of confirmed HPA cases based on administrative data was carried out at CNGM from June 2010 through June 2012. The universe consisted of 531 infants who tested PKU-positive (Phe >4 mg/dL or 240 µmol/L), from a total of 270,041 newborns screened in the PNPN (70% of total live births n 2010–2012).[29] Infants’ serum samples were received at CNGM for confirmatory diagnosis and subsequent biochemical monitoring of cases diagnosed, both by HPLC.
Variables
Phe and Tyr serum concentration and Phe/Tyr concentration ratio For HPLC confirmatory diagnosis of HPA, laboratory cutoff points were determined for the two diagnostic criteria: first criterion, serum Phe of 4 mg/dL (240 µmol/L); and second criterion Phe/Tyr = 2; higher values were considered pathological.[27,28] For diagnosis of transient neonatal tyrosinemia (TNT), the reference interval determined for serum Tyr levels was 0.9–1.4 mg/dL (50–77 µmol/L) and for the Phe/Tyr ratio, 0.5–1.7; Tyr concentrations >1.4 mg/dL and Phe/Tyr ratios <0.5 were considered pathological.[30]
HPA classification The literature describes different criteria for HPA classification (serum Phe, dietary Phe tolerance and enzyme activity).[1,4–9] In this paper, only Phe levels, assessed in serum during diagnostic confirmation, were considered. HPAs were classified as:
- transient HPA, if Phe >4–10 mg/dL (>240–600 µmol/L) and only in the neonatal period;
- persistent HPA, if Phe >4–16.6 mg/dL (>240–1000 µmol/L); and
- PKU, if Phe >16.6 mg/dL (>1000 µmol/L).
Other variables Sex, province of origin and year of HPA diagnosis.
CNGM laboratory methods
Sample collection and transportation Patients identified as HPA positive by the PNPN have 2 mL of peripheral blood extracted at their corresponding Provincial Medical Genetics Centers for confirmatory diagnosis. Then 2 mL of peripheral blood are extracted monthly from confirmed PKU patients for biochemical monitoring; the serum samples, frozen at -20 oC, are sent to CNGM for HPLC analysis.
Sample preparation for HPLC quantification A deproteinization pretreatment is carried out, using a solution of 10% trichloroacetic acid (1:1, v/v). Samples are centrifuged for 10 minutes at 3000 g and the supernatant is transferred to a clean vial. Subsequently, 100 µL of supernatant are transferred to the glass vial of an autoinjector; 400 µL of distilled water are added, and homogenized in a tube shaker.Phenylalanine and tyrosine quantification by HPLC Isocratic reverse-phase HPLC is used for simultaneous quantification of phenylalanine and tyrosine in serum with direct fluorescence detection, based on the method described by Kandar and Zakova,[26] modified and validated at CNGM. The method meets acceptance criteria for validation parameters established for chromatographic bioanalytical methods: specificity for simultaneous quantification of Phe and Tyr (3.5 min retention time for Tyr and 5 min for Phe), without interference from endogenous serum components; linearity in the range of concentrations studied (0.4–18 mg/dL or 22–990 µmol/L for Tyr and 1–24 mg/mL or 60–1440 µmol/L for Phe); accuracy (100% recovery for both amino acids and variation coefficients lower than 15%); and precision (repeatability and intermediate precision with variation coefficients <15%).[unpublished laboratory data]
A Prominence HPLC system (Shimadzu, Japan) is used, equipped with DGU-20AS degasser, LC-20AT quaternary pump, SIL-20A autoinjector, CTO-20AC oven, RF-10AXL fluorescence detector and CBM-20A communication module. The LC Chromatography Data System Solutions program (Shimadzu, Japan) is used for data acquisition and processing. An isocratic reverse-phase technique with direct fluorescent detection (λEx: 260 nm; λEm: 282 nm) is used for simultaneous quantification of both amino acids in serum.
The mobile phase consists of a mixture of absolute ethanol–distilled water in 5:95 (v/v) ratio. All determinations are performed at 30 oC, in duplicate, using a Shim-pack ODS-GHRS 4 mm x 1 cm pre-column and Shim-pack CLC-ODS 15 cm, 5 µm column (Shimadzu, Japan). Work is carried out with a flow of 1 mL/min and injection volume of 20 µL. Sample quantification relies on Phe and Tyr calibration curves prepared in serum with concentration range of 0.4–20 mg/dL and 1–18 mg/dL, respectively, using Phe and Tyr standards (Merck, Germany).
Biochemical monitoring of diagnosed patients Monitoring is conducted in patients confirmed as positive with the sole aim of quantifying Phe. PKU patients are monitored after initiation of dietary treatment—consisting of a Phe-restricted diet to reduce elevated serum Phe to 5 mg/dL or 300 µmol/L, and maintain these levels over the first 5 years of life,[31] for adequate growth and neurodevelopment, avoiding irreversible intellectual impairment.[1,4,8,31–35] Samples for biochemical monitoring are obtained weekly (in specialized consultations in the patients’ respective Provincial Medical Genetics Centers) until metabolic compensation is reached, and monthly thereafter. Although patients diagnosed with persistent HPA are not treated, they undergo monthly biochemical monitoring until aged 12 months to rule out possible clinical manifestations; if Phe levels >10 mg/dL (600 µmol/L) appear, dietary treatment is started to prevent neurological damage. Patients with HPA are considered biochemically controlled and without risk of complications associated with low Phe (characteristic in malnutrition) when serum Phe levels are approximately 5 mg/dL (300 µmol/L).[8,16,36,37]
Data analysis Percentages were calculated and data grouped by diagnosis, sex, year, and province of origin. The STATISTICA 6.0 program was used to analyze data collected and to create patients’ biochemical monitoring graphs, HPA and PKU incidence (number of patients/number of live births occurred during the study period for denominator).
For biochemical monitoring, only data from the first assessments conducted until metabolic control was achieved were used; these were considered sufficient to demonstrate the usefulness of the method for our objectives.
Ethics Patient anonymity was maintained during data processing. The study and the publication of its results were approved by the CNGM ethics committee.
RESULTS
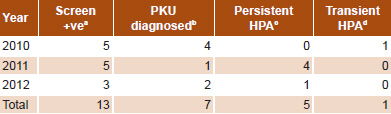
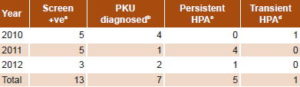
Initially, 13 infants were confirmed with HPA (2.4%); 7 with PKU (1.3%) and 6 with persistent HPA (1.1%) (Table 1). One male infant from Holguín Province had persistent HPA in 2010. However, since analysis of the third sample from this patient (who was already beyond the neonatal period and without dietary treatment) showed phenylalanine concentration < 4 mg/dL and Phe/Tyr ratio < 2, he was diagnosed as transient HPA (Table 1). Therefore, only 12 truly positive patients (2.2%) were finally diagnosed. All remaining cases (519/531) were false positives (97.7%) for HPA; transient neonatal tyrosinemia was detected in 10.3% (55/531). PKU was diagnosed in 58.3% of positive cases (7/12), or 1.3% of the total (7/531) (Table 2).
Table 1: Infants diagnosed with HPA by year in Cuba (n = 13)
a Phe >4 mg/dL b Phe >16.6 mg/dL c Phe >4.0–16.6 mg/dL
dPhe >4.0–10 mg/dL, limited to neonatal period
HPA: hyperphenylalaninemia Phe: phenylalanine
Table 2: PKU screening results and HPLC-confirmed cases in Cuba, 2010–2012 (n = 531)

HPA: hyperphenylalaninemias
TNT: transient neonatal tyrosinemia
a Positive PKU samples from National Neonatal Screening Program
b Serum phenylalanine >16.6 mg/dL
c Serum phenylalanine >4–16.6 mg/dL
d HPLC cutoffs: phenylalanine, 4 mg/dL; phenylalanine/tyrosine ratio = 2
e Serum phenylalanine >4–10) mg/dL (only during neonatal period)
f Phenylalanine/tyrosine ratio <0.5
Overall incidence of HPA at birth over the study period was 1/22,503 live births, 1/38,577 live births for PKU specifically. The provinces where most HPA cases were diagnosed were Matanzas with 3 cases (2 PKU and 1 persistent HPA) and Artemisa with 2 cases (1 PKU and 1 persistent HPA). Male sex was slightly more represented (7/12) than female (5/12), 58.3% vs. 41.7%. The most diagnoses were made in 2011 with 41.7% (5/12), followed by 2010 with 33.3% (4/12) and 2012 with 25% (3/12) (Table 3).
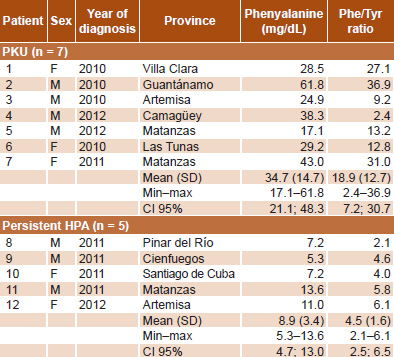
Table 3: Phe levels and Phe/Tyr ratios in patients with HPLC-confirmed HPA in Cuba, 2010–2012 (n = 12)
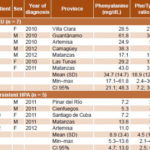
Phe: phenylalanine HPA: hyperphenylalaninemia Tyr: tyrosine HPLC cutoffs for positive diagnosis: phenylalanine >4 mg/dL or 240 µmol/L, phe/tyr ratio >2
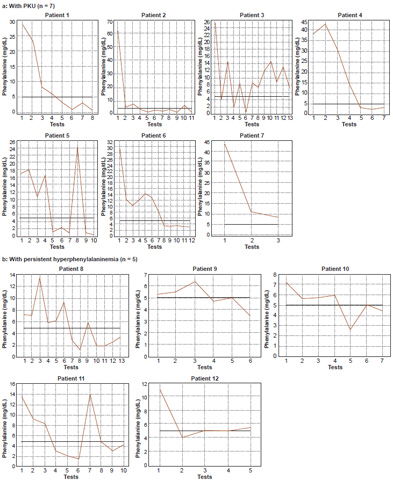
In PKU patients, mean serum Phe was 34.7 mg/dL (SD 14.7) and the mean Phe/Tyr ratio was 18.9 (SD 12.7), much higher than those of patients with persistent hyperphenylalaninemia (Table 3). Biochemical monitoring found a substantial decrease and control of serum Phe levels in 10 of 12 HPA patients (83.3%) (Figure 1).
DISCUSSION
Introduction of HPLC confirmatory of HPA diagnosis revealed high numbers of false-positive patients detected in the PNPN, which has led to performance of confirmatory diagnosis in an excessive number of samples.
PNPN methods for PKU screening may play a role in the high percentage of false positives, both the technique itself and the cutoff level chosen. It is essential to review the cutoff point chosen, since a low threshold may significantly increase the number of false positives.[15,38,39]
The problem could also be related to the low specificity of the technique, whereby an increase in the levels of Phe could be associated with an increase in Tyr.[1,37] For future PNPN screening, more advanced screening technology should be considered, such as tandem mass spectrometry, which provides greater analytic efficiency and speed, high sensitivity, high specificity and low detection limit (1 nmol/mL). The false positive rate using this technique is very low (0.2%), and it allows simultaneous quantification of Phe, Tyr and computation of the Phe/Tyr ratio, permitting screening for large numbers of inborn errors of metabolism in the same specimen.[1,21,36,40]
False positives can also be due to other related factors, such as incorrect blood extraction or failure of patients to fast before collection. To reduce false positive PKU rates, PNPN must continue systematic training of the staff collecting the samples, since incorrect collection of dried blood samples on filter paper may cause both false positive and false negative results.[12,13,21,36,37,41]
Figure 1: Biochemical monitoring profiles of hyperphenylalaninemias patients in Cuba, 2010–2012 (n = 12)

It is important to record the timing and conditions of sampling on the test requisition. Sample collection immediately after breastfeeding, or improper collection of dried blood samples on filter paper could lead to false positive results. Other factors regarding blood sample collection could also be involved, such as application of more than one drop of blood from the heel in the circle of the collection card, card contamination, or samples collected later in the newborn period, when the newborn’s protein intake is higher. Phe concentration in newborns may be normal until the fourth day of life but increase rapidly when protein intake begins or in those infants who receive mixed feeding.[37,42,43] This is the main reason most PKU screening programs use neonatal samples from the fifth to the seventh days of life.[44]
Detecting infants who were false positive for PKU in the PNPN owing to TNT was possible because the HPLC method allows simultaneous quantification of Phe and Tyr. Risk factors for TNT include prematurity, hyper-protein diet (mixed feeding) and deficiency of vitamin C. In studies carried out in newborns, TNT occurs between 0.2% and 10%, predominating in premature infants because of an impairment or immaturity of the liver enzyme p-hydroxyphenylpyruvate oxidase, caused by variations in its formation in the perinatal period. Unlike tyrosinemia types I and II, the transient variant is asymptomatic in most cases.[45–47] For infants who tested positive for TNT, the CNGM clinical laboratory requested that geneticists in each province send a second serum sample to confirm whether it was TNT or another type of tyrosinemia. In the latter case, the patient is followed up clinically to determine whether clinical signs characteristic of each variant (Type I and II) appear; none of the infants assessed proved to have either type of tyrosinemia.
Biochemical monitoring of PKU patients is essential for enabling timely and personalized dietary treatment, thus reducing the risk of complications associated with low Phe, characteristic of malnutrition.[1,8,16,35] The method applied is critical because it shows significant reduction in Phe levels to about 5 mg/dL (300 µmol/L), as seen in this study. Phe decrease to the indicated values promotes the infant’s adequate growth and neurodevelopment, avoiding onset of irreversible intellectual impairment.[1,5,8,16,31,36] In only two PKU patients (3 and 7) was optimal metabolic control not achieved; one (7) had just begun dietary treatment at the time of the study and had had only three Phe serum determinations. Since the laboratory does not conduct clinical care, the causes for failure to control Phe in patient 3 are unknown. The results of our assessment, however, allow us to consider that HPLC provides reliable information on the outcome of early dietary treatment, which proved to be effective in most patients assessed.
PKU incidence at birth was higher than previously reported in the Cuban population.[3,10] In this study, an increase in the incidence of PKU was observed when compared to that reported with the use of the fluorimetric method.[3] This increase may be associated with introduction of a more sensitive and specific method such as HPLC, and also with introduction of the second criterion for confirmatory diagnosis and/or classification of HPA. Reducing false positives has unquestionably improved diagnosis. In Latin America, only four countries have newborn screening programs with >20 years of experience and >98% coverage: Uruguay (99.5%), Cuba (99%), Chile (98.5%), and Costa Rica (98.3%). Prior to the implementation of HPLC, Cuba had the lowest PKU incidence reported in the region, specifically, 1:52,590 (2007), compared to 1:20,891 in Uruguay (2010), 1:19,566 in Chile (2010), and 1:49,176 in Costa Rica (2010).[10] Available data from these neonatal screening programs show the average incidence of PKU to be 1:23,518 and persistent HPA 1:20,759.[48] Incidence in European countries varies between 1:5000 and 1:15,000.[49]
In our study, mean Phe levels and Phe/Tyr ratios of PKU patients were four times higher than those of patients with persistent HPA. This was expected, and consistent with reports from the literature, because of the absence of enzyme phenylalanine hydroxylase activity in patients with PKU, the HPA variant with greatest negative impact on neurodevelopment.[1,4,5,7,8]
One study limitation is related to cases diagnosed with TNT, as it was not possible to establish whether patients were premature newborns, since the confirmatory HPA test requisition does not collect this data. Even with its limitations, however, this study provides insight into the performance of PNPN’s PKU screening program. The results have been applied in PNPN, informing program managers as they analyze the various stages in the process and identify problems that need correcting.
CONCLUSION
HPLC for simultaneous quantification of phenylalanine and tyrosine in serum provides a necessary addition to PKU screening by PNPN for more accurate confirmation of positivity in patients. It has enabled the introduction in Cuba of a second PKU diagnostic criterion of positivity for both classification of hyperphenylalaninemias and biochemical monitoring of diagnosed patients.