 Fiona M. Healy
Fiona M. Healy Lekh N. Dahal
Lekh N. Dahal Jack R.E. Jones1
Jack R.E. Jones1 John F. Woolley
John F. Woolley- 1Department of Pharmacology & Therapeutics, University of Liverpool, Liverpool, United Kingdom
- 2Department of Molecular & Clinical Cancer Medicine, University of Liverpool, Liverpool, United Kingdom
- 3The Clatterbridge Cancer Centre NHS Foundation Trust, Liverpool, United Kingdom
Myeloid malignancies are a heterogeneous group of clonal haematopoietic disorders, caused by abnormalities in haematopoietic stem cells (HSCs) and myeloid progenitor cells that originate in the bone marrow niche. Each of these disorders are unique and present their own challenges with regards to treatment. Acute myeloid leukaemia (AML) is considered the most aggressive myeloid malignancy, only potentially curable with intensive cytotoxic chemotherapy with or without allogeneic haematopoietic stem cell transplantation. In comparison, patients diagnosed with chronic myeloid leukaemia (CML) and treated with tyrosine kinase inhibitors (TKIs) have a high rate of long-term survival. However, drug resistance and relapse are major issues in both these diseases. A growing body of evidence suggests that Interferons (IFNs) may be a useful therapy for myeloid malignancies, particularly in circumstances where patients are resistant to existing front-line therapies and have risk of relapse following haematopoietic stem cell transplant. IFNs are a major class of cytokines which are known to play an integral role in the non-specific immune response. IFN therapy has potential as a combination therapy in AML patients to reduce the impact of minimal residual disease on relapse. Alongside this, IFNs can potentially sensitize leukaemic cells to TKIs in resistant CML patients. There is evidence also that IFNs have a therapeutic role in myeloproliferative neoplasms (MPNs) such as polycythaemia vera (PV) and primary myelofibrosis (PMF), where they can restore polyclonality in patients. Novel formulations have improved the clinical effectiveness of IFNs. Low dose pegylated IFN formulations improve pharmacokinetics and improve patient tolerance to therapies, thereby minimizing the risk of haematological toxicities. Herein, we will discuss recent developments and the current understanding of the molecular and clinical implications of Type I IFNs for the treatment of myeloid malignancies.
Clonal Malignancies in Myeloid Lineages
Myeloid malignancies, consisting of myeloproliferative neoplasms (MPNs), myelodysplastic syndrome (MDS), and acute myeloid leukaemia (AML), are a heterogeneous group of clonal haematopoietic disorders caused by abnormalities in HSCs and myeloid progenitor cells, originating in the bone marrow niche (1, 2). Each myeloid malignancy is unique and therefore presents different challenges with regards to treatment. MPNs have been categorized by the WHO as: chronic myeloid leukaemia (CML), polycythaemia vera (PV), primary myelofibrosis(PMF) and essential thrombocythemia (ET) and are characterized by the presence of the BCR-ABL gene, JAK2, MPL or CALR mutations, excessive erythrocyte production, fibrotic bone marrow degeneration, and excessive megakaryocyte lineage proliferation, respectively (3). MDS is characterized by abnormalities in normal haematopoiesis with resulting progression to bone marrow failure and genetic instability with potential to develop into AML (4) which is often considered the most aggressive myeloid malignancy. Older AML patients, those with poor-risk associated karyotypes, or with an unfavourable mutational burden (e.g. TP53 mutations) have a median survival as low as 4-6 months (5–7). In comparison, patients who are diagnosed with CML and treated with tyrosine kinase inhibitors (TKIs), such as imatinib, have an overall long-term (8-year) survival rate as high as 93% (8–10). This vast difference in survival between different malignancies warrants a novel and innovative approach to the treatment of certain myeloid malignancies.
There are currently a variety of therapies available for myeloid malignancies, dependent on the disease in question. Most CML patients will experience long term responses with tyrosine kinase inhibitors (TKIs) targeting the BCR-ABL oncogene and can in many cases be considered functionally cured of their disease (9, 11). PV is associated with blood hyperviscosity due to the expansion of the erythrocyte mass, and therefore standard therapy involves blood withdrawal to reduce mass as well as treatment with hydroxyurea, or ruxolitinib (Janus activated kinase 1/2 (JAK1/JAK2) inhibitor) as a second-line chemotherapy option (12, 13). PMF can also be treated using JAK2 inhibitors, however this is considered only a symptom relieving measure – PMF is only potentially curable with an allogeneic haematopoietic stem cell transplant (HCT) and is associated with a substantial risk of treatment-related mortality (14). Finally, MDS and AML can also be treated with high dose, aplasia-inducing chemotherapy and consolidated with HCT. However, relapse remains a problem and many factors such as poor cytogenetic risk (15); monosomal karyotype (16); measurable residual disease (MRD) positivity (17, 18); FLT3-ITD and other mutations (19–21) can influence the risk of relapse in AML patients after HCT.
A large portion of AML patients are elderly and are considered unfit to undergo intensive chemotherapy (22). Current treatment for these malignancies is therefore unfavourable for many patients. Alternatively, hypomethylating agents in combination with BCL2 inhibitors (23, 24) are becoming an increasingly better option to reduce toxicity (25), although a new treatment for MDS and AML is still urgently required.
For decades, cytokines belonging to the Interferon (IFN) family have been shown to play an integral role in co-ordinating immune responses against viruses, intracellular pathogens and tumour control (26). IFNs are divided into three types: Type I (including IFN-α and IFN-β), Type II (IFN-γ only) and Type III (IFN-λ). A summary of these subtypes and potential therapeutics is described in Table 1 (27–29). Type I IFNs are produced by most cells, following detection of pathogen-associated molecular patterns (PAMPs), such as foreign or self-nucleic acids (30). Interestingly, Type I IFNs differ in their binding affinity to the same cell surface receptor (IFNAR1/IFNAR2) and consequently trigger different antiviral, antiproliferative, and immunomodulatory outcomes (31). It is these antiproliferative and immunomodulatory outcomes which highlight IFNs as a potential treatment for myeloid malignancies (32). Herein we discuss to what extent IFNs can be used therapeutically to manage myeloid malignancies.
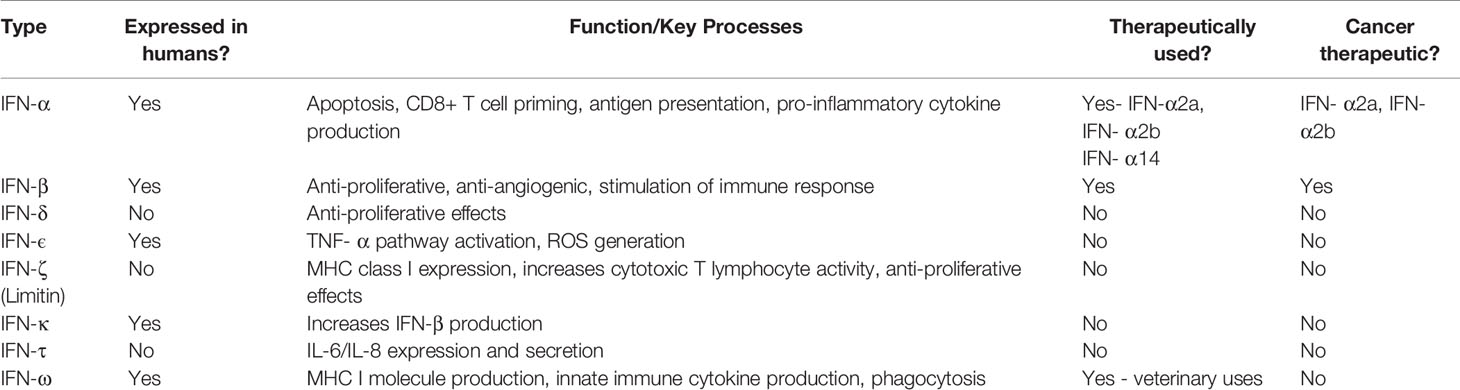
Table 1 Summary of the Canonical Functions of Type I Interferons and Therapeutic Potential.
Type I Interferon Signaling
Following binding of Type I IFNs to the IFNAR1/IFNAR2 receptor complex, the associated JAK1 and tyrosine kinase 2 (TYK2) activation causes the tyrosine phosphorylation of STAT1 (signal transducer and activator of transcription 1) and STAT2 (Figure 1). This ultimately results in the formation of an IFN-stimulated gene factor 3 (ISGF3) complex which is translocated to the nucleus. Here, it binds IFN-stimulated response elements (ISREs), initiating transcription of IFN-stimulated genes (ISGs) (33). These ISGs have a wide range of functions, and can be involved in immune system regulation and cell death, amongst other processes. ISGs are also involved in both positive and negative feedback loops within the IFN signalling system, with the potential to increase or decrease IFN signalling, depending on cellular context such as mutational status (34).
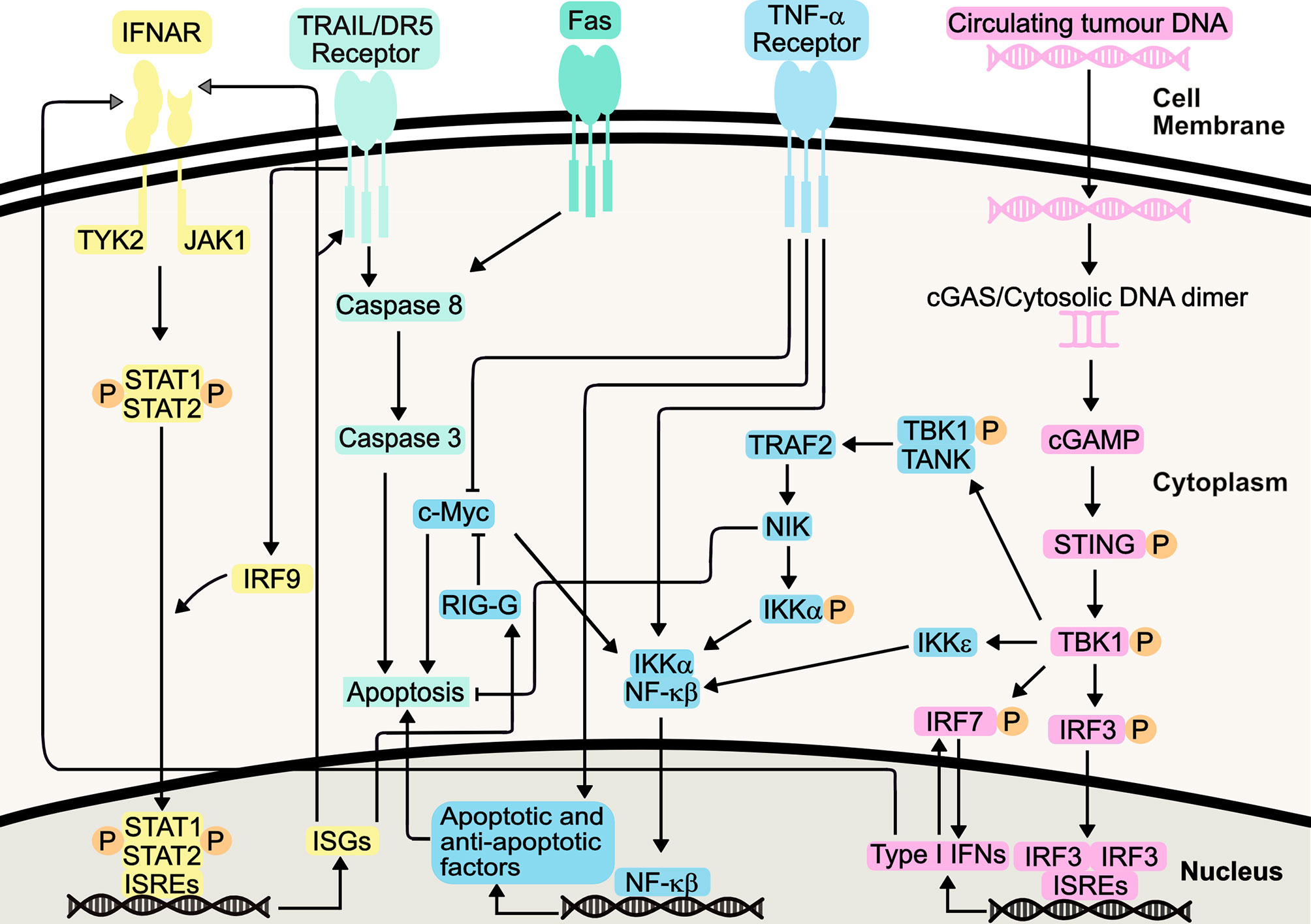
Figure 1 Type I Interferon Signalling Pathways. Type I IFNs bind the IFNAR1/IFNAR2 receptors, causing heterodimerization and TYK2 and JAK1 activation, initiating STAT1/STAT2 heterodimerization and phosphorylation. Following IRF-9-mediated translocation of this heterodimer to the nucleus, it binds interferon-stimulated response elements (ISREs), resulting in transcription of interferon-stimulated genes (ISGs). Resulting proteins include death receptor ligands (including TRAIL and Fas), which bind their respective receptors. Apoptosis is induced, either directly, or indirectly via NF-κβ signalling. These pathways also synergize with TNF-α pathway, increasing apoptosis. The STING pathway, initiated by detection of circulating tumour DNA, can mediate production of Type I IFNs or induce apoptosis, through canonical and non-canonical pathways following TBK1 activation. This includes its role in positive feedback loops involving IRF7, further increasing Type I IFN production.
IFN signaling results in the transcription and translation of cell death-inducing ISGs, and it has been hypothesized that IFN-α works by potentiating Tumour Necrosis Factor (TNF)-induced apoptosis through suppression of nuclear factor-κβ (NF-κβ) (35). Co-treatment of AML cell lines with TNF-α and IFN-α increased apoptosis compared to IFN-α alone, indicating IFN-α enhancement of TNF-α apoptosis via distinct and common pathways (36). Increased IFN production has been shown to inhibit activity of NF-κβ through the NIK/TRAF2 pathway, yet, this can be somewhat of a ‘double-edged sword’, with this pathway also implicated in cellular protection against IFN-mediated apoptosis (37). Furthermore, IFN-mediated NF-κβ inhibition leads to downregulation of cellular Myc (c-Myc) by increasing IFIT3 (RIG-G) gene expression, promoting apoptosis and inducing cell cycle arrest, via enhancement of CD95/Fas expression, triggering increased cellular caspase 3 activity (38–40). Indeed, IFN-α has been known to sensitize cells to TRAIL-induced apoptosis, through activation of DR5 receptor-mediated death signaling and subsequent suppression of TRAIL-mediated NF-κβ activation (41).
NF-κβ is also responsible for the production of cytokines in response to the detection of cytosolic DNA. Through the stimulator of interferon genes (STING) pathway, activation of interferon regulatory factor 3 (IRF-3) and NF-κβ leads to transcription of ISGs and cytokines. This includes IFN-α and IFN-β, and STING activation ultimately promotes varying mechanisms of cell death, including apoptosis (42–44). It can therefore be argued that the STING pathway activation of NF-κβ forms an opposing mechanism, perhaps to maintain homeostasis as well as desensitizing the body to further IFN stimulation (45). This could also explain why some IFN therapies require frequent administration of IFNs to maintain a therapeutic effect. Nevertheless, the STING pathway presents itself as a promising target to promote immunity in cancer, including the treatment of AML (46).
It has been proposed that antiproliferative effects stimulated by Type I IFN on cancerous cells can enhance the immune response in mouse models of haematological malignancies (47), through induction of cell cycle arrest. This is largely through an increase in p21 activity, as well as CDK2 inhibition, though perhaps to a lesser extent (48, 49). Type I IFN-mediated cell cycle arrest in S phase appears to be regulated through IRF9 (48–50). Increased levels of IRF9 have been attributed to increased TRAIL activity, which itself is an ISG (50, 51).
Type I IFNs can also negatively regulate angiogenesis, thereby reducing oxygen supply to malignant cells. This can be through increased transcription of anti-angiogenic factors, including the ISGs IRF7 and PML. Alternatively, Type I IFNs can reduce production of pro-angiogenic factors, including basic fibroblast growth factor (bFGF), a factor increased in myeloid leukaemia (52, 53). Furthermore, it has been suggested that IFN therapy could reduce levels of pro-angiogenic vascular epidermal growth factor (VEGF), which is a biomarker used for CML severity (54, 55). Again, this primarily occurs through IFN-mediated reduction in VEGFA and HIF1α transcription, with lower levels of VEGF mRNA detected in tumour samples following Type I IFN therapy, as compared to diagnostic samples (52, 56).
Immunomodulatory effects of Type I IFNs can be mediated through dendritic cells, with Type I IFNs driving activation and maturation of dendritic cells (57). Following IFN-stimulated dendritic cell maturation, there can be an increase in MHC molecule production and antigen presentation to CD8+ T cells, thereby promoting CD8+ T cell mediated responses, ultimately leading to tumour destruction (58). Furthermore, IFN-stimulated dendritic cell maturation also induces expression of costimulatory ligands CD80 and CD86 (59–61). CD80 and CD86 are needed for full T cell activation, through their binding to CD28. Furthermore, increased CD80 expression induces IRF-7 expression, thereby stimulating further Type I IFN production, in somewhat of a positive feedback mechanism. Such pathway can be mediated by TRADD, TRAF3 and TANK activation, as well as IFN-β and IRF3 (31, 60, 62). Aside from these aforementioned signalling pathways, IFN-α also increases production of pro-inflammatory cytokines, including IL-1β, IFN-γ and TNF-α (31, 63). As previously stated, increased TNF-α contributes to increased apoptosis seen in response to Type I IFN. Thus, it can be deduced that, unsurprisingly, there is synergy between the various IFN-stimulated pathways, converging in key elements responsible for the apoptotic, anti-angiogenic, immunomodulatory and antiproliferative responses.
Do IFNs Reduce the Rates of AML Relapse?
Treatment options for AML patients rely heavily on a combination of anthracyclines (e.g. daunorubicin, idarubicin) and the pyrimidine nucleoside cytarabine (64), which deliver an unsatisfactory 5-year survival rate of <20% in older patients (65–68). A goal of treating AML is obtaining a complete remission and preferably eradicating MRD, which is defined as the presence of residual leukaemic cells in bone marrow (BM) or peripheral blood (PB) in patients who have achieved morphologic complete remission (69). MRD positivity is associated with increased relapse risk and shorter survival in AML (70–73). One recent study found that measuring the transcript levels of RUNX1/RUNX1T1 fusion gene in AML patients with MRD (post-HCT) allowed for a significant discrimination between patients that would continue remission or enter relapse (74). Other studies have supported this finding and argue that monitoring MRD using transcript levels of RUNX1/RUNX1T1 fusion gene is favourable over the measurement of other mutations including c-KIT mutations for the risk stratification of AML patients with MRD (75). IFN therapy could potentially target the leukaemic stem cells (LSCs) at the root of AML development and relapse. It has been demonstrated that IFN-α triggers cell cycle entry in dormant HSCs (76). This raises the possibility that treating AML patients with IFN could force quiescent LSCs to proliferate, and thus amenable to standard chemotherapy. It has been shown that MRD-directed IFN-α treatment was able to significantly decrease the risk of cumulative incidence of relapse and improve survival in MRD-positive AML patients post-HCT, compared to MRD patients with no intervention (77, 78). Indeed, a recent retrospective study examined the use of IFN maintenance therapy following induction and consolidation chemotherapy in favourable-risk AML patients. Of patients who were MRD positive at baseline measurements, 78% of those treated with IFN-α2b became MRD negative, after a median time of 5.5 months. In contrast, only 27% of patients not treated with IFN-α2b (monitored only) showed MRD conversion from positive to negative. This corresponded to an increase in relapse-free survival, with 87% IFN-α2b treated patients relapse free after 4 years, compared to 56% in the control group (79). An ongoing phase I/II trial has demonstrated that pegylated IFN-α, when administered prophylactically after myeloablative conditioning in an AML cohort at high risk for relapse, did not significantly alter toxicity or acute graft versus host disease (GVHD) risk and produced relatively low rates of relapse suggesting a robust graft versus leukaemia (GVL) response (80). Separately a recent trial has explored the use of IFN-α2b therapy following MRD detection post induction and consolidation chemotherapy. A significant decrease in the number of patients with MRD in the IFN-α2b treated group was reported after a median follow-up time of 5 months, compared to a significant increase in patients with MRD detection in the trial arm that did not receive IFN after chemotherapy (patients observed only) (81). Importantly, event-free survival was also considerably higher in the IFN-α2b treated patients. IFN therapies to prevent relapse in AML potentially offer significant benefit to AML patients and this warrants further study.
IFNs for TKI Resistant CML Patients
TKIs including imatinib, nilotinib, dasatinib, bosutinib, and most recently ponatinib, are the current preferred treatment for CML patients, and have dramatically improved the outcome of CML patients (82). Despite the success of TKIs, there is still a possibility that some CML patients become resistant to front-line treatment with imatinib, and to second generation TKIs (e.g. nilotinib, dasatinib) through the BCR-ABL T315I mutation wherein only third generation TKIs (e.g. ponatinib) are efficacious. Even still, resistance to imatinib has been seen since the earliest trials and has recently been shown for third-generation TKIs (83, 84). IFN therapy for the treatment of CML pre-dates TKI therapy, and is characterized by its ability to induce haematologic remission and durable cytogenetic response (85). Combination therapy of IFNs with cytarabine for many CML patients showed promising results, however the advent of TKIs meant that IFN therapy was largely replaced (86). The IRIS trial compared the use of imatinib or IFN-α plus cytarabine. Whilst there was no significant difference in survival after 18 months between either group, patients treated with IFN-α and cytarabine showed a markedly poorer cytogenetic response, coupled with a greater risk of disease progression, compared to those who received imatinib alone. Indeed, many patients who initially received IFN-α and cytarabine crossed over to imatinib therapy (9, 87, 88). Response to imatinib continued over 5 years, with a complete cytogenetic response seen in 69% of all patients who received imatinib (and up to 96% in those still taking imatinib after 5 years) (87). However, approximately 17% patients did lose complete cytogenetic response over the follow-up period (9).
This is in line with current understanding which suggests that TKIs are not entirely curative as monotherapies in CML, as LSC exposure to many TKIs fails to induce apoptosis in the most primitive quiescent CML LSCs (89, 90). Combined therapy of IFN-α with TKIs could address this issue by stimulating cytotoxic T cells to target LSCs refractory to standard TKI therapy. Initial studies comparing patients treated with imatinib and IFNα, to those treated with imatinib alone, showed that the combination therapy resulted in a higher rate of complete cytogenetic remission at 6 months, and similar rates at 4 years (91). Similarly, a trial of imatinib and IFN-α combination therapy showed 75% of patients showed sustained remission following imatinib discontinuation at 2.4 years (92). However, other trials involving the combination of IFNs with imatinib have shown mixed results, with no improvement in cytogenetic response rates or progression-free survival and increased adverse events (93, 94). For example, in extended (10 year) follow-up of the aforementioned IRIS trial, patients treated with imatinib alone had similar overall and progression-free survival than those treated with imatinib and IFN-α (95).
Whilst imatinib is considered a ‘game-changing’ drug for the extremely high survival rate it is associated with, second generation TKIs have shown further improvements in these rates in certain cases (10). IFN-α combination therapy with the second generation TKI dasatinib has also shown promising results (96). Low dose pegylated IFN-α following dasatinib treatment was well tolerated, and demonstrated rapid increased response rates with major molecular response (MMR) rate of 89% after 18 months. Encouragingly pegylated IFN-α was well tolerated in this study, and further investigation is warranted with second generation TKIs. Earlier studies of combination therapy comprising pegylated IFN-α formulations with imatinib, had shown good clinical response (significantly higher MMR of 82% compared to 54% for imatinib monotherapy after 12 months), yet were associated with toxicity also in a majority of patients (54). NiloPeg, a small 2011 phase II trial looking at the use of nilotinib and IFN-α2a showed a good molecular response in patients treated with this regimen (64% after 12 months), although adverse effects were, again, a frequent occurrence (97). The currently ongoing TIGER trial of nilotinib combination therapy with pegylated IFN-α is attempting to address these concerns, and will evaluate the feasibility of discontinuing drug therapy in patients showing stable deep molecular response after nilotinib versus IFN maintenance therapy (98).
Treating MPNs With IFNs
The WHO classification system for MPNs includes seven subcategories (3), but more commonly this refers to the JAK2 mutation-enriched diseases polycythaemia vera (PV), essential thrombocythaemia (ET) and primary myelofibrosis (PMF). PV is characterized by an elevated red blood cell mass (and potentially overproduction of white blood cells and platelets). PV is also associated with an increased risk for thromboembolic events, leukaemic transformation, and/or myelofibrosis.
Type I IFN signalling can contribute to the pathology of MPN, as well as being used as a therapeutic, as mentioned previously. In PMF, levels of IFN-α, and associated proinflammatory cytokines such as IL-1β, can be increased compared to healthy bone marrow or plasma. This contributes to the inflammatory phenotype associated with PMF (99). This coincides with the effects of TET2 mutations, which are common in MPNs and are also associated with inflammation (100). In a similar way, whilst myelodysplastic syndrome (MDS) has a vast range of causes, increased pro-inflammatory signalling is thought to be a key driver, again mediated by Type I IFNs. Type I IFN-associated genes were found to be upregulated in MDS patients’ bone marrow, including IRF-7 and ISG-15, suggesting their involvement in MDS pathology (101).
Standard therapy for PV involves phlebotomy and cytoreductive drugs (hydroxyurea, busulfan) but recently IFN-α and JAK1/2 inhibitors have been examined as potential treatments (102, 103). Although busulfan has been effective over long periods (104), resistance to hydroxyurea is associated with increased mortality and progression to more advanced MPNs or secondary AML (13). IFN therapy is an accepted alternative to hydroxyurea or busulfan in PV, and is particularly useful for younger patients or those who are pregnant (105, 106). In fact, IFN-α2 has been shown to induce haematological responses in about 80% of PV patients while also reducing splenomegaly, and relieving pruritus (107). A study of patients treated with IFN-α compared to a control group showed an MMR of 89%, with a stable circulating JAK2 mutated allele (%V617F) in the other 11% of patients (108).
Recent trial data have demonstrated that 76% of advanced PV patients treated with IFN-α2a showed complete haematologic response, while the complete molecular response rate was 19% at 42 months (109). Similarly, good efficacy for pegylated IFN-α in hydroxyurea resistant patients was seen, with an overall response rate of 60% at 12 months (110). Significantly this trial was based on relatively broad inclusion criteria e.g., advanced age, prolonged disease duration, and a high prevalence of splenomegaly. However, 14% of patients discontinued therapy due to adverse events. However, the PROUD-PV and Continuation-PV trials revealed a need for prolonged IFN-α therapy as a means of reaching both molecular response and complete haematological response. Whilst therapy with ropeginterferon-alfa-2b (pegylated form of IFN-α2b) increased the proportion of patients achieving complete haematological response and improved disease burden after 36 months compared to those patients treated with hydroxyurea (53% versus 38%), this was not the case after shorter time periods (46% versus 51% at 12 months). Thus, there is a need for sustained treatment with IFN-α to achieve best response). Importantly, the toxicity profile associated with prolonged ropeginterferon-alfa-2b therapy was not significantly worse than that of hydroxyurea (111).
Primary Myelofibrosis (PMF) can be classified into two stages: pre-fibrotic (pre-PMF) and fibrotic PMF (overt-PMF) diagnosed by bone marrow morphology, fibrosis grade and clinical features including leukoerythroblastosis, anaemia and splenomegaly (112). Aside from HCT, there are limited treatment options for very high- and high-risk patients and 10-year survival is estimated at <13%. The use of JAK2 inhibitors is considered mostly palliative, and the treatment of clinical features includes the use of androgens, hydroxyurea and ruxolitinib (113). However, IFNs may provide a means of increasing the survival of patients with PMF. Preliminary results of a Phase II study using a combination therapy between pegylated IFN-α and ruxolitinib on 50 MPN patients (32 PV and 18 PMF patients), most of whom were resistant and/or intolerant to IFN-α as a monotherapy, saw complete haematologic responses in 44% and 58% of PV and PMF patients respectively (114). Despite a 20% discontinuation rate for this trial, there is clear indication that the inflammatory mediated toxicity which often limits IFN-α was reduced by ruxolitinib, known for its potent anti-inflammatory properties. It is also reported that levels of the V617F allele declined significantly in both groups. This supports previous findings that monitoring the %V617F is a reliable marker for MPNs and the detection of MRD.
Essential thrombocythaemia (ET) is a clonal MPN characterized by excess production of platelets and mature hyperlobulated megakaryocytes, with the presence of mutations in JAK2, CALR, or MPL and the absence of BCR-ABL (3). Most ET patients have a normal life expectancy, and the goals of treatment are the prevention of thrombotic and bleeding complications while minimizing the risk of progression, the effective control of systemic symptoms, and the appropriate management of complications and risk situations (105). Treatment of ET typically involves combinations of aspirin, ticlopidine, hydroxyurea and anagrelide to reduce platelet number and manage clotting (115, 116). IFN therapy in ET is also effective for reducing platelet numbers while also reducing the risk of thrombotic complications (107, 117) but does not appear to restore polyclonal haematopoiesis (118). Studies that will directly compare pegylated IFN-α2a to hydroxyurea in ET and PV are currently ongoing (110). Initial reports show an overall response rate of 69% for pegylated IFN-α2a in ET patients at 12 months, with acceptable safety profile (major thrombotic events at 1 year was 1%, with no major bleeding events).
A recent study comprising patients with PV, PMF or ET indicated IFN-α has a stronger effect on HSCs than mature blood cells. Within this compartment, those with JAK2 V617F-mutated HSCs responded better than those with other mutations, such as CALR. This is thought to be a result of the JAK2 V617F increasing the sensitivity of HSCs to IFN signalling, through increased ISG activation (119). Whilst it has been widely understood that IFN-α can reduce the HSC compartment through cell cycle activation and subsequent HSC exhaustion, recent findings suggest this is only the case in JAK2 V617F mutated HSCs. In contrast, JAK2 wild-type HSCs generally remain quiescent in the presence of IFN-α. This is consistent with the ability of IFN-α to arrest the cell cycle in bulk tumor cells (48). Current understanding suggests IFN-α-mediated promotion of JAK2 V617F-mutated HSC cell cycle not only exhausts the cells, but uses induces differentiation to the erythroid lineage, making them susceptible to IFN-α-mediated apoptosis (120). Nevertheless, the effects of IFN-α demonstrated here may not be wholly specific to the V617F mutant, since stem cell derived erythrocytes containing JAK2 with exon 12 mutations (also causing constitutive JAK2 activation, as seen in PV) were also sensitive to IFN-α treatment (121).
Conclusions & Future Directions
Similar to symptoms experienced during normal IFN release in the presence of a foreign pathogen, acute toxicity to IFN therapy usually presents as flu-like symptoms including nausea, fatigue, and myalgia (122). Most side effects associated with IFN therapy are dose-dependent, and these can be mitigated by administering low dose pegylated-IFNs in addition to managing inflammation with NSAIDs. Acute toxicity is experienced by nearly all patients, while sub-acute and chronic side effects have been reported in almost all organ systems throughout the body. A 2018 study conducted by Mo et al. using IFN-α for the treatment for MRD in AML for example, reported that all patients reported transient fevers and six patients (14%) showed grade ≥3 toxicity, two of which were haematological (77). A study of CML patients treated with low-dose pegylated IFN-α reported that 20% and 25% of patients experienced grade 3-4 neutropenia and thrombocytopenia in the first 12 months respectively (96). While some toxicity is still observed, pegylated IFNs show increased patient tolerance, and modify the immunological, pharmacokinetic and pharmacodynamic properties of the drug to minimize off-target adverse effects and increase efficacy (123). Pegylated IFN-α lengthens plasma half-life and reduces sensitivity to proteolysis, allowing it to be administered less frequently (122).
It is important that other adverse events in IFN therapy including flu-like symptoms and myalgia are also controlled using anti-inflammatory drugs. A common trend throughout the studies discussed herein is a high percentage of patients who do not complete IFN therapy because it is not well tolerated alongside chemotherapy, which can be as high as 61% of patients discontinuing treatment (54).
Another confounding factor for IFN therapy is the variability of the bone marrow niche between patients. IFN therapy relies on the innate, non-specific immune system to induce an immune response meaning it is not well tailored to one specific individual. This may partially explain why the responsiveness to IFN therapy in a small patient group varies greatly. To account for this, larger studies such as the TIGER study (98) are vital to better understand the causes for intolerance and resistance to therapy.
There is also a considerable economic barrier for uptake of IFN therapy. Analysis has shown that imatinib is a cost-effective first line therapy versus IFN-α plus low-dose cytarabine for newly diagnosed CML (124). If significant cohorts of patients will ultimately discontinue IFN therapy in favour of TKIs, then coupled with cost-based concerns, then IFNs become less favourable drugs to develop.
It is apparent in AML that carefully targeted IFN therapy can provide a significant improvement to patient outcome especially in the management of MRD (27, 77, 78). The sensitization of leukaemic cells in the bone marrow niche by IFNs to classic chemotherapy agents will possibly improve the prospects of many AML patients should the treatment become more widely available (Figure 2), especially in patients post-HCT for the eradication of MRD. This is an extremely important goal, as multiple studies have shown the presence of MRD is associated with increased relapse risk and shorter survival in both paediatric and adult AML (71).
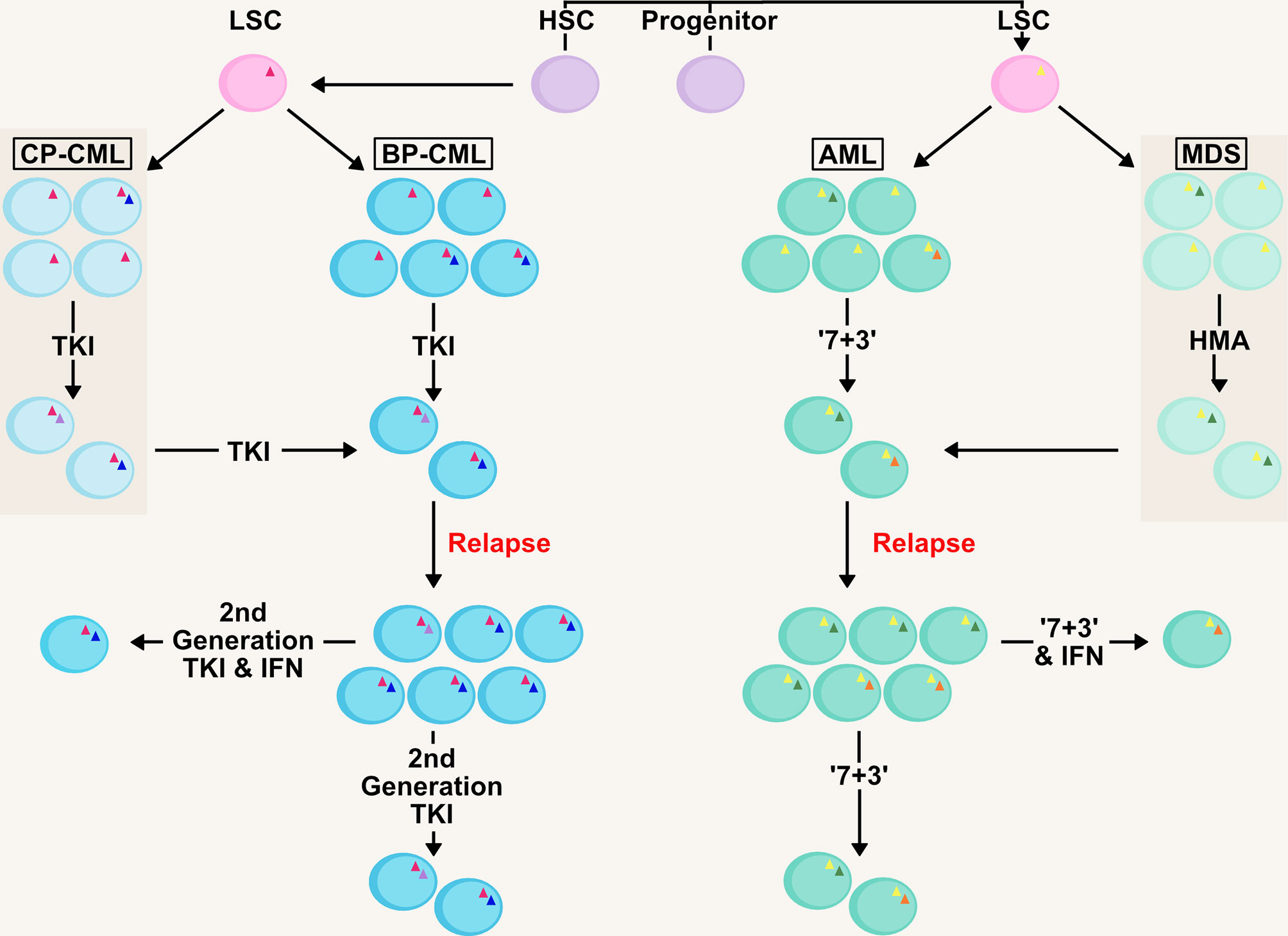
Figure 2 Management of Clonality and MRD in AML/CML with IFN therapy. AML and CML are clonal diseases, presenting at diagnosis with a major immature blast clone and minor clones (denoted by mutations as triangles within cells) which develop due to mutations in the haematopoietic stem and progenitor pool generating the LSC. Initial therapy in both chronic-phase (CP-CML) and blast-phase (BP-CML) patients with a first-generation tyrosine kinase inhibitor (TKI) against the BCR-ABL oncogene (pink triangle), such as imatinib, will reduce the bulk of CML blasts in the bone marrow and peripheral blood. However, it is not uncommon for resistant clones to persist that are refractory to this treatment. Later generation TKIs (e.g. nilotinib, dasatanib, ponatinib) may eradicate these clones but some mutations (e.g. BCR-ABL T315I; green circle) drive resistance to novel TKIs. Similarly, initial ‘7+3’ therapy in AML (7 day treatment with the pyrimidine analogue cytarabine, in conjunction with an anthracycline, such as daunorubicin, for the first 3 days) and HMA-therapy (hypomethylating agent) in MDS will eradicate the bulk of leukaemic blasts (such as those containing initiating mutations; yellow triangle) in the patient. However, clones refractory to these therapies (including those with signalling mutations; blue and orange circles) will drive relapse in MDS/AML patients, as well as transformation from MDS to AML. Management of measurable residual disease (MRD) in myeloid malignancies is critical to preventing relapse. Therapies based on IFNs could target the clones resistant to front-line therapies and deliver longer remissions.
Similarly in CML, further development of IFN therapy provides an opportunity alongside current standards. Continuing focus on development of novel TKIs targeting BCR-ABL has meant that IFN therapy has not advanced greatly over the last decade for CML patients. There is potential for IFN therapy as a combination therapy with TKIs to sensitize cells and aid the action of the primary treatment. Just as in the case of AML, targeting the LSC to overcome resistance to TKIs is a fundamental goal and essential to achieving long-term remissions.
In PV and PMF, where the side effects of IFNs are tolerated, treatment using IFNs in combination with JAK2 inhibitors is certainly a possibility for the prevention of disease progression to other myeloid malignancies. As in AML, the use of IFNs for the treatment of PV and PMF to control MRD post-HCT would be beneficial for patients.
In conclusion, the benefits of using IFN therapy for the treatment of myeloid malignancies warrants further research. Despite intolerance seen in many small studies, IFNs are to be considered as an alternative therapy to traditional chemotherapy regimens or in combination with existing regimens for the sensitization of leukaemic cells to treatment and the reduction of MRD. It is important that other formulations of IFNs are developed in addition to pegylation, as this will determine whether pharmaceutical companies recognize this therapy as a reliable candidate for future treatment – the findings of large, ongoing Phase III trials will become a critical turning point. In short, the re-emergence of IFN therapy does indeed provide new hope for leukaemia patients, more specifically in the treatment of MRD.
Author Contributions
JW conceived the study. FH, JJ, and JW wrote the manuscript. FH and JW produced the figures. YF and LD edited and contributed to the final submitted manuscript, and provided critical insights. All authors contributed to the article and approved the submitted version.
Funding
JW and LD are supported by North West Cancer Research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ABL, Abelson proto-oncogene; AML, Acute Myeloid Leukaemia; ara-C, Cytarabine; bFGF, basic fibroblast growth factor; BCR, Breakpoint cluster region gene; cGAMP, 2’3’-Cyclic GMP-AMP; cGAS, cyclic GMP-AMP synthase; CML, Chronic Myeloid Leukaemia; cMyc, cellular Myc protein; COX, Cyclooxygenase; DNA, Deoxyribonucleic acid; DR5, Death receptor 5; ET, Essential thrombocythaemia; GVHD, Graft versus host disease; GVL, Graft versus leukaemia; HCT, Haematopoietic stem cell transplant; HSCs, Haematopoietic stem cells; IFNAR1/2, Interferon-alpha/beta receptor 1/2; IFNs, Interferons; IFN-α/β/γ, Interferon alpha/beta/gamma; IRF3/9, Interferon regulatory factor 3/9; ISGF3, Interferon-stimulated gene factor 3; ISGs, Interferon-stimulated genes; ISREs, Interferon-stimulated response elements; IκB, Inhibitor of kappa B kinase; JAK1/2, Janus Kinase 1/2; LSCs, Leukaemic stem cells; MDS, Myelodysplastic syndrome; MMR, Molecular response rate; MPNs, Myeloproliferative neoplasms; MRD, Measurable/minimal residual disease; MSCs, Mesenchymal stromal cells; NF-κB, Nuclear factor-kappa B; NSAIDs, Non-steroidal anti-inflammatory drugs; PAMPs, Pathogen-associated molecular patterns; PMF, Primary myelofibrosis; PV, Polycythaemia Vera; Rig-G, Retinoic acid-induced gene G; RUNX1, Runt-related transcription factor 1; STAT1/2, Signal transducer and activator of transcription 1/2; STING, Stimulator of interferon genes; TKIs, Tyrosine Kinase Inhibitors; TNF-α, Tumour necrosis factor alpha; TRAIL, Tumour necrosis factor-related apoptosis inducing ligand; TYK2, Tyrosine kinase 2; VEGF, vascular epidermal growth factor.
References
1. Korn C, Méndez-Ferrer S. Myeloid Malignancies and the Microenvironment. Blood (2017) 129(7):811–22. doi: 10.1182/blood-2016-09-670224
2. Zeidner JR,D, Perl A, Gojo I. The American Cancer Society’s Oncology in Practice: Clinical Management. Newark, United States: John Wiley & Sons, Incorporated (2018).
3. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
4. Haferlach T. The Molecular Pathology of Myelodysplastic Syndrome. Pathobiology (2019) 86(1):24–9. doi: 10.1159/000488712
5. Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 Alterations in Acute Myeloid Leukemia With Complex Karyotype Correlate With Specific Copy Number Alterations, Monosomal Karyotype, and Dismal Outcome. Blood (2012) 119(9):2114–21. doi: 10.1182/blood-2011-08-375758
6. Hou HA, Chou WC, Kuo YY, Liu CY, Lin LI, Tseng MH, et al. TP53 Mutations in De Novo Acute Myeloid Leukemia Patients: Longitudinal Follow-Ups Show the Mutation Is Stable During Disease Evolution. Blood Cancer J (2015) 5:e331 p. doi: 10.1038/bcj.2015.59
7. Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N Engl J Med (2016) 375(21):2023–36. doi: 10.1056/NEJMoa1605949
8. Sacha T. Imatinib in Chronic Myeloid Leukemia: An Overview. Mediterr J Hematol Infect Dis (2014) 6(1):e2014007–e. doi: 10.4084/mjhid.2014.007
9. Hughes TP, Hochhaus A, Branford S, Müller MC, Kaeda JS, Foroni L, et al. Long-Term Prognostic Significance of Early Molecular Response to Imatinib in Newly Diagnosed Chronic Myeloid Leukemia: An Analysis From the International Randomized Study of Interferon and STI571 (IRIS). Blood (2010) 116(19):3758–65. doi: 10.1182/blood-2010-03-273979
10. García-Gutiérrez V, Hernández-Boluda JC. Tyrosine Kinase Inhibitors Available for Chronic Myeloid Leukemia: Efficacy and Safety. Front Oncol (2019) 9(603). doi: 10.3389/fonc.2019.00603
11. Suttorp M, Bornhäuser M, Metzler M, Millot F, Schleyer E. Pharmacology and Pharmacokinetics of Imatinib in Pediatric Patients. Expert Rev Clin Pharmacol (2018) 11(3):219–31. doi: 10.1080/17512433.2018.1398644
12. Dale E, Davis M, Faustman DL. A Role for Transcription Factor NF-kappaB in Autoimmunity: Possible Interactions of Genes, Sex, and the Immune Response. Adv Physiol Educ (2006) 30(4):152–8. doi: 10.1152/advan.00065.2006
13. Nazha A, Gerds AT. Where to Turn for Second-Line Cytoreduction After Hydroxyurea in Polycythemia Vera? Oncologist (2016) 21(4):475–80. doi: 10.1634/theoncologist.2015-0380
14. Tefferi A, Guglielmelli P, Pardanani A, Vannucchi AM. Myelofibrosis Treatment Algorithm 2018. Blood Cancer J (2018) 8(8):72. doi: 10.1038/s41408-018-0109-0
15. Cornelissen JJ, Breems D, van Putten WL, Gratwohl AA, Passweg JR, Pabst T, et al. Comparative Analysis of the Value of Allogeneic Hematopoietic Stem-Cell Transplantation in Acute Myeloid Leukemia With Monosomal Karyotype Versus Other Cytogenetic Risk Categories. J Clin Oncol (2012) 30(17):2140–6. doi: 10.1200/JCO.2011.39.6499
16. Fang M, Storer B, Estey E, Othus M, Zhang L, Sandmaier BM, et al. Outcome of Patients With Acute Myeloid Leukemia With Monosomal Karyotype Who Undergo Hematopoietic Cell Transplantation. Blood (2011) 118(6):1490–4. doi: 10.1182/blood-2011-02-339721
17. Leung W, Pui CH, Coustan-Smith E, Yang J, Pei D, Gan K, et al. Detectable Minimal Residual Disease Before Hematopoietic Cell Transplantation Is Prognostic But Does Not Preclude Cure for Children With Very-High-Risk Leukemia. Blood (2012) 120(2):468–72. doi: 10.1182/blood-2012-02-409813
18. Walter RB, Gyurkocza B, Storer BE, Godwin CD, Pagel JM, Buckley SA, et al. Comparison of Minimal Residual Disease as Outcome Predictor for AML Patients in First Complete Remission Undergoing Myeloablative or Nonmyeloablative Allogeneic Hematopoietic Cell Transplantation. Leukemia (2015) 29(1):137–44. doi: 10.1038/leu.2014.173
19. Brunet S, Labopin M, Esteve J, Cornelissen J, Socié G, Iori AP, et al. Impact of FLT3 Internal Tandem Duplication on the Outcome of Related and Unrelated Hematopoietic Transplantation for Adult Acute Myeloid Leukemia in First Remission: A Retrospective Analysis. J Clin Oncol (2012) 30(7):735–41. doi: 10.1200/JCO.2011.36.9868
20. Schmid C, Labopin M, Socié G, Daguindau E, Volin L, Huynh A, et al. Outcome of Patients With Distinct Molecular Genotypes and Cytogenetically Normal AML After Allogeneic Transplantation. Blood (2015) 126(17):2062–9. doi: 10.1182/blood-2015-06-651562
21. Estey EH. Acute Myeloid Leukemia: 2019 Update on Risk-Stratification and Management. Am J Hematol (2018) 93(10):1267–91. doi: 10.1002/ajh.25214
22. Heiblig M, Elhamri M, Tigaud I, Plesa A, Barraco F, Labussière H, et al. Treatment With Low-Dose Cytarabine in Elderly Patients (Age 70 Years or Older) With Acute Myeloid Leukemia: A Single Institution Experience. Mediterr J Hematol Infect Dis (2016) 8(1):e2016009–e. doi: 10.4084/mjhid.2016.009
23. DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax Combined With Decitabine or Azacitidine in Treatment-Naive, Elderly Patients With Acute Myeloid Leukemia. Blood (2019) 133(1):7–17. doi: 10.1182/blood-2018-08-868752
24. Pollyea DA, Pratz K, Letai A, Jonas BA, Wei AH, Pullarkat V, et al. Venetoclax With Azacitidine or Decitabine in Patients With Newly Diagnosed Acute Myeloid Leukemia: Long Term Follow-Up From a Phase 1b Study. Am J Hematol (2021) 96(2):208–17. doi: 10.1002/ajh.26039
25. Gerds AT, Gooley TA, Estey EH, Appelbaum FR, Deeg HJ, Scott BL. Pretransplantation Therapy With Azacitidine vs Induction Chemotherapy and Posttransplantation Outcome in Patients With MDS. Biol Blood Marrow Transplant (2012) 18(8):1211–8. doi: 10.1016/j.bbmt.2012.01.009
26. Parkin J, Cohen B. An Overview of the Immune System. Lancet (2001) 357(9270):1777–89. doi: 10.1016/S0140-6736(00)04904-7
27. Li S, Gong M, Zhao F, Shao J, Xie Y, Zhang Y, et al. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell Physiol Biochem (2018) 51(5):2377–96. doi: 10.1159/000495897
28. Pestka S, Krause CD, Walter MR. Interferons, Interferon-Like Cytokines, and Their Receptors. Immunol Rev (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
29. Budhwani M, Mazzieri R, Dolcetti R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-Tumor Immunity to Resistance. Front Oncol (2018) 8(322). doi: 10.3389/fonc.2018.00322
30. Musella M, Manic G, De Maria R, Vitale I, Sistigu A. Type-I-Interferons in Infection and Cancer: Unanticipated Dynamics With Therapeutic Implications. Oncoimmunology (2017) 6(5):e1314424. doi: 10.1080/2162402X.2017.1314424
31. Medrano RFV, Hunger A, Mendonça SA, Barbuto JAM, Strauss BE. Immunomodulatory and Antitumor Effects of Type I Interferons and Their Application in Cancer Therapy. Oncotarget (2017) 8(41):71249–84. doi: 10.18632/oncotarget.19531
32. Alsufyani A, Alanazi R, Woolley JF, Dahal LN. Old Dog, New Trick: Type I IFN-Based Treatment for Acute Myeloid Leukemia. Mol Cancer Res (2021) 19(5):753–6. doi: 10.1158/1541-7786.MCR-20-0871
33. Platanias LC. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat Rev Immunol (2005) 5(5):375–86. doi: 10.1038/nri1604
34. Porritt RA, Hertzog PJ. Dynamic Control of Type I IFN Signalling by an Integrated Network of Negative Regulators. Trends Immunol (2015) 36(3):150–60. doi: 10.1016/j.it.2015.02.002
35. Manna SK, Mukhopadhyay A, Aggarwal BB. IFN-α Suppresses Activation of Nuclear Transcription Factors NF-κb and Activator Protein 1 and Potentiates TNF-Induced Apoptosis. J Immunol (2000) 165(9):4927. doi: 10.4049/jimmunol.165.9.4927
36. Nakashima A, Kumakura S, Mishima S, Ishikura H, Kobayashi S. IFN-Alpha Enhances TNF-Alpha-Induced Apoptosis Through Down-Regulation of C-Myc Protein Expression in HL-60 Cells. J Exp Clin Cancer Res (2005) 24(3):447–56.
37. Pfeffer LM. The Role of Nuclear Factor κb in the Interferon Response. J Interferon Cytokine Res (2011) 31(7):553–9. doi: 10.1089/jir.2011.0028
38. Kimura M, Haisa M, Uetsuka H, Takaoka M, Ohkawa T, Kawashima R, et al. TNF Combined With IFN-Alpha Accelerates NF-kappaB-Mediated Apoptosis Through Enhancement of Fas Expression in Colon Cancer Cells. Cell Death Differ (2003) 10(6):718–28. doi: 10.1038/sj.cdd.4401219
39. Sarkar D, Park ES, Fisher PB. Defining the Mechanism by Which IFN-β Dowregulates C-Myc Expression in Human Melanoma Cells: Pivotal Role for Human Polynucleotide Phosphorylase (Hpnpaseold-35). Cell Death Differ (2006) 13(9):1541–53. doi: 10.1038/sj.cdd.4401829
40. Xiao S, Li D, Zhu HQ, Song MG, Pan XR, Jia PM, et al. RIG-G as a Key Mediator of the Antiproliferative Activity of Interferon-Related Pathways Through Enhancing P21 and P27 Proteins. Proc Natl Acad Sci USA (2006) 103(44):16448–53. doi: 10.1073/pnas.0607830103
41. Shigeno M, Nakao K, Ichikawa T, Suzuki K, Kawakami A, Abiru S, et al. Interferon-Alpha Sensitizes Human Hepatoma Cells to TRAIL-Induced Apoptosis Through DR5 Upregulation and NF-Kappa B Inactivation. Oncogene (2003) 22(11):1653–62. doi: 10.1038/sj.onc.1206139
42. Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, et al. Signalling Strength Determines Proapoptotic Functions of STING. Nat Commun (2017) 8(1):427. doi: 10.1038/s41467-017-00573-w
43. Chen D, Tong J, Yang L, Wei L, Stolz DB, Yu J, et al. PUMA Amplifies Necroptosis Signaling by Activating Cytosolic DNA Sensors. Proc Natl Acad Sci USA (2018) 115(15):3930–5. doi: 10.1073/pnas.1717190115
44. Sun F, Liu Z, Yang Z, Liu S, Guan W. The Emerging Role of STING-Dependent Signaling on Cell Death. Immunol Res (2019) 67(2-3):290–6. doi: 10.1007/s12026-019-09073-z
45. Schneider WM, Chevillotte MD, Rice CM. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
46. Curran E, Chen X, Corrales L, Kline DE, Dubensky TW Jr., Duttagupta P, et al. STING Pathway Activation Stimulates Potent Immunity Against Acute Myeloid Leukemia. Cell Rep (2016) 15(11):2357–66. doi: 10.1016/j.celrep.2016.05.023
47. Escobar G, Barbarossa L, Barbiera G, Norelli M, Genua M, Ranghetti A, et al. Interferon Gene Therapy Reprograms the Leukemia Microenvironment Inducing Protective Immunity to Multiple Tumor Antigens. Nat Commun (2018) 9(1):2896. doi: 10.1038/s41467-018-05315-0
48. Murphy D, Detjen KM, Welzel M, Wiedenmann B, Rosewicz S. Interferon-Alpha Delays S-Phase Progression in Human Hepatocellular Carcinoma Cells via Inhibition of Specific Cyclin-Dependent Kinases. Hepatology (2001) 33(2):346–56. doi: 10.1053/jhep.2001.21749
49. Szeps M, Erickson S, Gruber A, Castro J, Einhorn S, Grandér D. Effects of Interferon-α on Cell Cycle Regulatory Proteins in Leukemic Cells. Leukemia Lymphoma (2003) 44(6):1019–25. doi: 10.1080/1042819031000077133
50. Tsuno T, Mejido J, Zhao T, Schmeisser H, Morrow A, Zoon KC. IRF9 Is a Key Factor for Eliciting the Antiproliferative Activity of IFN-Alpha. J Immunother (2009) 32(8):803–16. doi: 10.1097/CJI.0b013e3181ad4092
51. Leaman DW, Chawla-Sarkar M, Jacobs B, Vyas K, Sun Y, Ozdemir A, et al. Novel Growth and Death Related Interferon-Stimulated Genes (ISGs) in Melanoma: Greater Potency of IFN-Beta Compared With IFN-Alpha2. J Interferon Cytokine Res (2003) 23(12):745–56. doi: 10.1089/107999003772084860
52. Borden EC. Interferons α and β in Cancer: Therapeutic Opportunities From New Insights. Nat Rev Drug Discovery (2019) 18(3):219–34. doi: 10.1038/s41573-018-0011-2
53. Song M-Z, Mao Y-M, Wu J, Pan H-F, Ye Q-L. Increased Circulating Basic Fibroblast Growth Factor Levels in Acute Myeloid Leukemia: A Meta-Analysis. Hematology (2020) 25(1):186–93. doi: 10.1080/16078454.2020.1766865
54. Simonsson B, Gedde-Dahl T, Markevärn B, Remes K, Stentoft J, Almqvist A, et al. Combination of Pegylated IFN-α2b With Imatinib Increases Molecular Response Rates in Patients With Low- or Intermediate-Risk Chronic Myeloid Leukemia. Blood (2011) 118(12):3228–35. doi: 10.1182/blood-2011-02-336685
55. Legros L, Guilhot J, Huault S, Mahon FX, Preudhomme C, Guilhot F, et al. Interferon Decreases VEGF Levels in Patients With Chronic Myeloid Leukemia Treated With Imatinib. Leukemia Res (2014) 38(6):662–5. doi: 10.1016/j.leukres.2014.01.010
56. von Marschall Z, Scholz A, Cramer T, Schäfer G, Schirner M, Öberg K, et al. Effects of Interferon Alpha on Vascular Endothelial Growth Factor Gene Transcription and Tumor Angiogenesis. JNCI: J Natl Cancer Institute (2003) 95(6):437–48. doi: 10.1093/jnci/95.6.437
57. Gardner A, Ruffell B. Dendritic Cells and Cancer Immunity. Trends Immunol (2016) 37(12):855–65. doi: 10.1016/j.it.2016.09.006
58. Schiavoni G, Mattei F, Gabriele L. Type I Interferons as Stimulators of DC-Mediated Cross-Priming: Impact on Anti-Tumor Response. Front Immunol (2013) 4:483–. doi: 10.3389/fimmu.2013.00483
59. Simmons DP, Wearsch PA, Canaday DH, Meyerson HJ, Liu YC, Wang Y, et al. Type I IFN Drives a Distinctive Dendritic Cell Maturation Phenotype That Allows Continued Class II MHC Synthesis and Antigen Processing. J Immunol (2012) 188(7):3116–26. doi: 10.4049/jimmunol.1101313
60. Peteranderl C, Herold S. The Impact of the Interferon/TNF-Related Apoptosis-Inducing Ligand Signaling Axis on Disease Progression in Respiratory Viral Infection and Beyond. Front Immunol (2017) 8:313. doi: 10.3389/fimmu.2017.00313
61. Mehrotra A, D’Angelo JA, Romney-Vanterpool A, Chu T, Bertoletti A, Janssen HLA, et al. IFN-α Suppresses Myeloid Cytokine Production, Impairing IL-12 Production and the Ability to Support T-Cell Proliferation. J Infect Dis (2020) 222(1):148–57. doi: 10.1093/infdis/jiaa064
62. Bauvois B, Nguyen J, Tang R, Billard C, Kolb JP. Types I and II Interferons Upregulate the Costimulatory CD80 Molecule in Monocytes via Interferon Regulatory Factor-1. Biochem Pharmacol (2009) 78(5):514–22. doi: 10.1016/j.bcp.2009.05.005
63. Rozera C, Carlei D, Lollini PL, De Giovanni C, Musiani P, Di Carlo E, et al. Interferon (IFN)-Beta Gene Transfer Into TS/A Adenocarcinoma Cells and Comparison With IFN-Alpha: Differential Effects on Tumorigenicity and Host Response. Am J Pathol (1999) 154(4):1211–22. doi: 10.1016/S0002-9440(10)65373-4
64. Büchner T, Schlenk RF, Schaich M, Döhner K, Krahl R, Krauter J, et al. Acute Myeloid Leukemia (AML): Different Treatment Strategies Versus a Common Standard Arm–Combined Prospective Analysis by the German AML Intergroup. J Clin Oncol (2012) 30(29):3604–10. doi: 10.1200/JCO.2012.42.2907
65. Burnett AK, Milligan D, Prentice AG, Goldstone AH, McMullin MF, Hills RK, et al. A Comparison of Low-Dose Cytarabine and Hydroxyurea With or Without All-Trans Retinoic Acid for Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome in Patients Not Considered Fit for Intensive Treatment. Cancer (2007) 109(6):1114–24. doi: 10.1002/cncr.22496
66. Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, et al. Anthracycline Dose Intensification in Acute Myeloid Leukemia. N Engl J Med (2009) 361(13):1249–59. doi: 10.1056/NEJMoa0904544
67. Löwenberg B, Ossenkoppele GJ, van Putten W, Schouten HC, Graux C, Ferrant A, et al. High-Dose Daunorubicin in Older Patients With Acute Myeloid Leukemia. New Engl J Med (2009) 361(13):1235–48. doi: 10.1056/NEJMoa0901409
68. De Angelis R, Sant M, Coleman MP, Francisci S, Baili P, Pierannunzio D, et al. Cancer Survival in Europe 1999-2007 by Country and Age: Results of EUROCARE–5-A Population-Based Study. Lancet Oncol (2014) 15(1):23–34. doi: 10.1016/S1470-2045(13)70546-1
69. Schuurhuis GJ, Heuser M, Freeman S, Béné M-C, Buccisano F, Cloos J, et al. Minimal/Measurable Residual Disease in AML: A Consensus Document From the European LeukemiaNet MRD Working Party. Blood (2018) 131(12):1275–91. doi: 10.1182/blood-2017-09-801498
70. Walter RB, Ofran Y, Wierzbowska A, Ravandi F, Hourigan CS, Ngai LL, et al. Measurable Residual Disease as a Biomarker in Acute Myeloid Leukemia: Theoretical and Practical Considerations. Leukemia (2021) 35(6):1529–38. doi: 10.1038/s41375-021-01230-4
71. Hourigan CS, Gale RP, Gormley NJ, Ossenkoppele GJ, Walter RB. Measurable Residual Disease Testing in Acute Myeloid Leukaemia. Leukemia (2017) 31(7):1482–90. doi: 10.1038/leu.2017.113
72. Grimwade D, Freeman SD. Defining Minimal Residual Disease in Acute Myeloid Leukemia: Which Platforms are Ready for "Prime Time"? Blood (2014) 124(23):3345–55. doi: 10.1182/blood-2014-05-577593
73. Ossenkoppele GJ, van de Loosdrecht AA, Schuurhuis GJ. Review of the Relevance of Aberrant Antigen Expression by Flow Cytometry in Myeloid Neoplasms. Br J Haematol (2011) 153(4):421–36. doi: 10.1111/j.1365-2141.2011.08595.x
74. Qin YZ, Wang Y, Xu LP, Zhang XH, Chen H, Han W, et al. The Dynamics of RUNX1-RUNX1T1 Transcript Levels After Allogeneic Hematopoietic Stem Cell Transplantation Predict Relapse in Patients With T (8,21) Acute Myeloid Leukemia. J Hematol Oncol (2017) 10(1):44. doi: 10.1186/s13045-017-0414-2
75. Wang Y, Wu DP, Liu QF, Qin YZ, Wang JB, Xu LP, et al. In Adults With T(8;21)AML, Posttransplant RUNX1/RUNX1T1-Based MRD Monitoring, Rather Than C-KIT Mutations, Allows Further Risk Stratification. Blood (2014) 124(12):1880–6. doi: 10.1182/blood-2014-03-563403
76. Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha Activates Dormant Haematopoietic Stem Cells In Vivo. Nature (2009) 458(7240):904–8. doi: 10.1038/nature07815
77. Mo X-D, Wang Y, Zhang X-H, Xu L-P, Yan C-H, Chen H, et al. Interferon-α Is Effective for Treatment of Minimal Residual Disease in Patients With T(8;21) Acute Myeloid Leukemia After Allogeneic Hematopoietic Stem Cell Transplantation: Results of a Prospective Registry Study. Oncologist (2018) 23(11):1349–57. doi: 10.1634/theoncologist.2017-0692
78. Mo X-D, Zhang X-H, Xu L-P, Wang Y, Yan C-H, Chen H, et al. IFN-A Is Effective for Treatment of Minimal Residual Disease in Patients With Acute Leukemia After Allogeneic Hematopoietic Stem Cell Transplantation: Results of a Registry Study. Biol Blood Marrow Transplant (2017) 23(8):1303–10. doi: 10.1016/j.bbmt.2017.04.023
79. Jiang H, Liu X-H, Kong J, Wang J, Jia J-S, Lu S-Y, et al. Interferon-α as Maintenance Therapy can Significantly Reduce Relapse in Patients With Favorable-Risk Acute Myeloid Leukemia. Leukemia Lymphoma (2021) 1–8. doi: 10.1080/10428194.2021.1948027
80. Magenau JM, Pawarode A, Riwes MM, Parkin B, Anand S, Ghosh M, et al. A Phase I/II Clinical Trial of Type 1 Interferon for Reduction of Relapse After HCT in High Risk AML. Biol Blood Marrow Transplant (2019) 25(3, Supplement):S12–S3. doi: 10.1016/j.bbmt.2018.12.078
81. Liu X, Duan W, Wang J, Jia J, Gong L, Lu >S-Y, et al. Interferon-A-2b As a Maintenance Therapy Significantly Improves Disease-Free Survival in Patients With Low-Risk Acute Myeloid Leukemia. Blood (2019) 134(Supplement_1):3823–. doi: 10.1182/blood-2019-128505
82. Kannaiyan R, Mahadevan D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev Anticancer Ther (2018) 18(12):1249–70. doi: 10.1080/14737140.2018.1527688
83. Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical Resistance to STI-571 Cancer Therapy Caused by BCR-ABL Gene Mutation or Amplification. Science (2001) 293(5531):876–80. doi: 10.1126/science.1062538
84. Lu L, Kok CH, Saunders VA, Wang J, McLean JA, Hughes TP, et al. Modelling Ponatinib Resistance in Tyrosine Kinase Inhibitor-Naïve and Dasatinib Resistant BCR-ABL1+ Cell Lines. Oncotarget (2018) 9(78):34735–47. doi: 10.18632/oncotarget.26187
85. Talpaz M, Kantarjian H, Kurzrock R, Trujillo JM, Gutterman JU. Interferon-Alpha Produces Sustained Cytogenetic Responses in Chronic Myelogenous Leukemia. Philadelphia Chromosome-Positive Patients. Ann Intern Med (1991) 114(7):532–8. doi: 10.7326/0003-4819-114-7-532
86. Kujawski LA, Talpaz M. The Role of Interferon-Alpha in the Treatment of Chronic Myeloid Leukemia. Cytokine Growth Factor Rev (2007) 18(5-6):459–71. doi: 10.1016/j.cytogfr.2007.06.015
87. Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-Year Follow-Up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. New Engl J Med (2006) 355(23):2408–17. doi: 10.1056/NEJMoa062867
88. O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib Compared With Interferon and Low-Dose Cytarabine for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. New Engl J Med (2003) 348(11):994–1004. doi: 10.1056/NEJMoa022457
89. Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, et al. Dasatinib (BMS-354825) Targets an Earlier Progenitor Population Than Imatinib in Primary CML But Does Not Eliminate the Quiescent Fraction. Blood (2006) 107(11):4532–9. doi: 10.1182/blood-2005-07-2947
90. Graham SM, Jørgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, Quiescent, Philadelphia-Positive Stem Cells From Patients With Chronic Myeloid Leukemia Are Insensitive to STI571 In Vitro. Blood (2002) 99(1):319–25. doi: 10.1182/blood.V99.1.319
91. Palandri F, Castagnetti F, Iacobucci I, Martinelli G, Amabile M, Gugliotta G, et al. The Response to Imatinib and Interferon-Alpha Is More Rapid Than the Response to Imatinib Alone: A Retrospective Analysis of 495 Philadelphia-Positive Chronic Myeloid Leukemia Patients in Early Chronic Phase. Haematologica (2010) 95(8):1415–9. doi: 10.3324/haematol.2009.021246
92. Burchert A, Müller MC, Kostrewa P, Erben P, Bostel T, Liebler S, et al. Sustained Molecular Response With Interferon Alfa Maintenance After Induction Therapy With Imatinib Plus Interferon Alfa in Patients With Chronic Myeloid Leukemia. J Clin Oncol (2010) 28(8):1429–35. doi: 10.1200/JCO.2009.25.5075
93. Preudhomme C, Guilhot J, Nicolini FE, Guerci-Bresler A, Rigal-Huguet F, Maloisel F, et al. Imatinib Plus Peginterferon Alfa-2a in Chronic Myeloid Leukemia. N Engl J Med (2010) 363(26):2511–21. doi: 10.1056/NEJMoa1004095
94. Hanfstein B, Müller MC, Hehlmann R, Erben P, Lauseker M, Fabarius A, et al. Early Molecular and Cytogenetic Response Is Predictive for Long-Term Progression-Free and Overall Survival in Chronic Myeloid Leukemia (CML). Leukemia (2012) 26(9):2096–102. doi: 10.1038/leu.2012.85
95. Hehlmann R, Lauseker M, Saußele S, Pfirrmann M, Krause S, Kolb HJ, et al. Assessment of Imatinib as First-Line Treatment of Chronic Myeloid Leukemia: 10-Year Survival Results of the Randomized CML Study IV and Impact of Non-CML Determinants. Leukemia (2017) 31(11):2398–406. doi: 10.1038/leu.2017.253
96. Hjorth-Hansen H, Stentoft J, Richter J, Koskenvesa P, Höglund M, Dreimane A, et al. Safety and Efficacy of the Combination of Pegylated Interferon-α2b and Dasatinib in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia Patients. Leukemia (2016) 30(9):1853–60. doi: 10.1038/leu.2016.121
97. Nicolini FE, Etienne G, Dubruille V, Roy L, Huguet F, Legros L, et al. Nilotinib and Peginterferon Alfa-2a for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukaemia (NiloPeg): A Multicentre, Non-Randomised, Open-Label Phase 2 Study. Lancet Haematol (2015) 2(1):e37–46. doi: 10.1016/S2352-3026(14)00027-1
98. Hochhaus A. TKI and Interferon Alpha Evaluation Initiated by the German Chronic Myeloid Leukemia Study Group - the TIGER Study (TIGER) Clinicaltrials.Gov2012 (2021). Available at: https://ClinicalTrials.gov/show/NCT01657604.
99. Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating Interleukin (IL)-8, IL-2r, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J Clin Oncol (2011) 29(10):1356–63. doi: 10.1200/JCO.2010.32.9490
100. Hasselbalch HC, Holmström MO. Perspectives on Interferon-Alpha in the Treatment of Polycythemia Vera and Related Myeloproliferative Neoplasms: Minimal Residual Disease and Cure? Semin Immunopathol (2019) 41(1):5–19. doi: 10.1007/s00281-018-0700-2
101. Kim M, Hwang S, Park K, Kim SY, Lee YK, Lee DS. Increased Expression of Interferon Signaling Genes in the Bone Marrow Microenvironment of Myelodysplastic Syndromes. PloS One (2015) 10(3):e0120602. doi: 10.1371/journal.pone.0120602
102. Pardanani A, Vannucchi AM, Passamonti F, Cervantes F, Barbui T, Tefferi A. JAK Inhibitor Therapy for Myelofibrosis: Critical Assessment of Value and Limitations. Leukemia (2011) 25(2):218–25. doi: 10.1038/leu.2010.269
103. Muller EW, de Wolf JT, Egger R, Wijermans PW, Huijgens PC, Halie MR, et al. Long-Term Treatment With Interferon-Alpha 2b for Severe Pruritus in Patients With Polycythaemia Vera. Br J Haematol (1995) 89(2):313–8. doi: 10.1111/j.1365-2141.1995.tb03306.x
104. Brodsky I. Busulfan Versus Hydroxyurea in the Treatment of Polycythemia Vera (PV) and Essential Thrombocythemia (ET). Am J Clin Oncol (1998) 21(1):105–6. doi: 10.1097/00000421-199802000-00024
105. Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, et al. Philadelphia-Negative Classical Myeloproliferative Neoplasms: Critical Concepts and Management Recommendations From European LeukemiaNet. J Clin Oncol (2011) 29(6):761–70. doi: 10.1200/JCO.2010.31.8436
106. Kiladjian JJ, Mesa RA, Hoffman R. The Renaissance of Interferon Therapy for the Treatment of Myeloid Malignancies. Blood (2011) 117(18):4706–15. doi: 10.1182/blood-2010-08-258772
107. Kiladjian JJ, Chomienne C, Fenaux P. Interferon-Alpha Therapy in Bcr-Abl-Negative Myeloproliferative Neoplasms. Leukemia (2008) 22(11):1990–8. doi: 10.1038/leu.2008.280
108. Jones AV, Silver RT, Waghorn K, Curtis C, Kreil S, Zoi K, et al. Minimal Molecular Response in Polycythemia Vera Patients Treated With Imatinib or Interferon Alpha. Blood (2006) 107(8):3339–41. doi: 10.1182/blood-2005-09-3917
109. Quintás-Cardama A, Abdel-Wahab O, Manshouri T, Kilpivaara O, Cortes J, Roupie AL, et al. Molecular Analysis of Patients With Polycythemia Vera or Essential Thrombocythemia Receiving Pegylated Interferon α-2a. Blood (2013) 122(6):893–901. doi: 10.1182/blood-2012-07-442012
110. Yacoub A, Mascarenhas J, Kosiorek H, Prchal JT, Berenzon D, Baer MR, et al. Pegylated Interferon Alfa-2a for Polycythemia Vera or Essential Thrombocythemia Resistant or Intolerant to Hydroxyurea. Blood (2019) 134(18):1498–509. doi: 10.1182/blood.2019000428
111. Gisslinger H, Klade C, Georgiev P, Krochmalczyk D, Gercheva-Kyuchukova L, Egyed M, et al. Ropeginterferon Alfa-2b Versus Standard Therapy for Polycythaemia Vera (PROUD-PV and CONTINUATION-PV): A Randomised, Non-Inferiority, Phase 3 Trial and Its Extension Study. Lancet Haematol (2020) 7(3):e196–208. doi: 10.1016/S2352-3026(19)30236-4
112. Rumi E, Sant’Antonio E, Boveri E, Pietra D, Cavalloni C, Roncoroni E, et al. Diagnosis and Management of Prefibrotic Myelofibrosis. Expert Rev Hematol (2018) 11(7):537–45. doi: 10.1080/17474086.2018.1484280
113. Tefferi A. Primary Myelofibrosis: 2019 Update on Diagnosis, Risk-Stratification and Management. Am J Hematol (2018) 93(12):1551–60. doi: 10.1002/ajh.25230
114. Mikkelsen SU, Kjaer L, Bjørn ME, Knudsen TA, Sørensen AL, Andersen CBL, et al. Safety and Efficacy of Combination Therapy of Interferon-α2 and Ruxolitinib in Polycythemia Vera and Myelofibrosis. Cancer Med (2018) 7(8):3571–81. doi: 10.1002/cam4.1619
115. Harrison CN, Campbell PJ, Buck G, Wheatley K, East CL, Bareford D, et al. Hydroxyurea Compared With Anagrelide in High-Risk Essential Thrombocythemia. N Engl J Med (2005) 353(1):33–45. doi: 10.1056/NEJMoa043800
116. Gisslinger H, Gotic M, Holowiecki J, Penka M, Thiele J, Kvasnicka HM, et al. Anagrelide Compared With Hydroxyurea in WHO-Classified Essential Thrombocythemia: The ANAHYDRET Study, a Randomized Controlled Trial. Blood (2013) 121(10):1720–8. doi: 10.1182/blood-2012-07-443770
117. Radin AI, Kim HT, Grant BW, Bennett JM, Kirkwood JM, Stewart JA, et al. Phase II Study of Alpha2 Interferon in the Treatment of the Chronic Myeloproliferative Disorders (E5487): A Trial of the Eastern Cooperative Oncology Group. Cancer (2003) 98(1):100–9. doi: 10.1002/cncr.11486
118. Sacchi S, Gugliotta L, Papineschi F, Liberati AM, Rupoli S, Delfini C, et al. Alfa-Interferon in the Treatment of Essential Thrombocythemia: Clinical Results and Evaluation of Its Biological Effects on the Hematopoietic Neoplastic Clone. Ital Cooperative Group ET Leukemia (1998) 12(3):289–94. doi: 10.1038/sj.leu.2400931
119. Mosca M, Lamrani L, Marzac C, Tisserand A, Edmond V, Dagher T, et al. Differential Impact of Interferon Alpha on JAK2V617F and Calr Mutated Hematopoietic Stem and Progenitor Cells in Classical MPN. Blood (2018) 132(Supplement 1):4333–. doi: 10.1182/blood-2018-99-114744
120. Mullally A, Bruedigam C, Poveromo L, Heidel FH, Purdon A, Vu T, et al. Depletion of Jak2V617F Myeloproliferative Neoplasm-Propagating Stem Cells by Interferon-α in a Murine Model of Polycythemia Vera. Blood (2013) 121(18):3692–702. doi: 10.1182/blood-2012-05-432989
121. Nilsri N, Jangprasert P, Pawinwongchai J, Israsena N, Rojnuckarin P. Distinct Effects of V617F and Exon12-Mutated JAK2 Expressions on Erythropoiesis in a Human Induced Pluripotent Stem Cell (iPSC)-Based Model. Sci Rep (2021) 11(1):5255. doi: 10.1038/s41598-021-83895-6
122. Sleijfer S, Bannink M, Van Gool AR, Kruit WH, Stoter G. Side Effects of Interferon-Alpha Therapy. Pharm World Sci (2005) 27(6):423–31. doi: 10.1007/s11096-005-1319-7
123. Aghemo A, Rumi MG, Colombo M. Pegylated IFN-Alpha2a and Ribavirin in the Treatment of Hepatitis C. Expert Rev Anti Infect Ther (2009) 7(8):925–35. doi: 10.1586/eri.09.70
Keywords: interferon, myeloid malignancies, interferon alfa, interferon beta, type I interferon (IFN) signaling, myeloid neoplasia
Citation: Healy FM, Dahal LN, Jones JRE, Floisand Y and Woolley JF (2021) Recent Progress in Interferon Therapy for Myeloid Malignancies. Front. Oncol. 11:769628. doi: 10.3389/fonc.2021.769628
Received: 14 September 2021; Accepted: 13 October 2021;
Published: 29 October 2021.
Edited by:
Simona Soverini, University of Bologna, ItalyReviewed by:
Ken Mills, Queen’s University Belfast, United KingdomFabio Stagno, University Hospital Polyclinic Vittorio Emanuele, Italy
Copyright © 2021 Healy, Dahal, Jones, Floisand and Woolley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John F. Woolley, john.woolley@liverpool.ac.uk