 Lanay Tierney1† Jörg Linde2† Sebastian Müller2 Sascha Brunke3,4 Juan Camilo Molina3 Bernhard Hube3,5 Ulrike Schöck6 Reinhard Guthke2 Karl Kuchler1*
Lanay Tierney1† Jörg Linde2† Sebastian Müller2 Sascha Brunke3,4 Juan Camilo Molina3 Bernhard Hube3,5 Ulrike Schöck6 Reinhard Guthke2 Karl Kuchler1*- 1 Christian Doppler Laboratory for Infection Biology, Max F. Perutz Laboratories, Medical University of Vienna, Vienna, Austria
- 2 Research Group Systems Biology and Bioinformatics, Leibniz-Institute for Natural Product Research and Infection Biology – Hans-Knoell-Institute, Jena, Germany
- 3 Department Microbial Pathogenicity Mechanisms, Leibniz-Institute for Natural Product Research and Infection Biology – Hans-Knoell-Institute, Jena, Germany
- 4 Center for Sepsis Control and Care, Jena University Hospital, Jena, Germany
- 5 Friedrich Schiller University, Jena, Germany
- 6 GATC Biotech AG, Konstanz, Germany
The ability to adapt to diverse micro-environmental challenges encountered within a host is of pivotal importance to the opportunistic fungal pathogen Candida albicans. We have quantified C. albicans and M. musculus gene expression dynamics during phagocytosis by dendritic cells in a genome-wide, time-resolved analysis using simultaneous RNA-seq. A robust network inference map was generated from this dataset using NetGenerator, predicting novel interactions between the host and the pathogen. We experimentally verified predicted interdependent sub-networks comprising Hap3 in C. albicans, and Ptx3 and Mta2 in M. musculus. Remarkably, binding of recombinant Ptx3 to the C. albicans cell wall was found to regulate the expression of fungal Hap3 target genes as predicted by the network inference model. Pre-incubation of C. albicans with recombinant Ptx3 significantly altered the expression of Mta2 target cytokines such as IL-2 and IL-4 in a Hap3-dependent manner, further suggesting a role for Mta2 in host–pathogen interplay as predicted in the network inference model. We propose an integrated model for the functionality of these sub-networks during fungal invasion of immune cells, according to which binding of Ptx3 to the C. albicans cell wall induces remodeling via fungal Hap3 target genes, thereby altering the immune response to the pathogen. We show the applicability of network inference to predict interactions between host–pathogen pairs, demonstrating the usefulness of this systems biology approach to decipher mechanisms of microbial pathogenesis.
Introduction
Both host and pathogenic species have evolved a plethora of strategies to rapidly adapt to the changing environmental dynamics within the infection milieu. However, the extent of this complexity has only recently been investigated through the use of system biology approaches (reviewed in Rizzetto and Cavalieri, 2011). On the molecular level, these adaptations are mediated by complex interaction networks, which sense these environmental changes and transmit the information throughout the cell, leading to a cascade of changes in gene and eventually protein expression. Understanding these underlying interaction networks is important to elucidate how organisms and defense mechanisms interact during microbial infection processes. Genome-wide integrative approaches for modeling have become increasingly popular (Rizzetto and Cavalieri, 2011) due to the availability of high-throughput sequencing technologies, including RNA sequencing (RNA-seq). These technologies now allow for the parallel sequencing of millions of nucleotide sequences simultaneously (Wang et al., 2009; Zhang et al., 2011). One major advantage to using sequencing approach rather than microarrays is that it is a species-independent platform, allowing for an in-depth investigation of non-model organism species, as well as multiple organisms from a single experiment.
In many cases, the underlying interaction networks between the organisms of interest are unknown. Network inference uses reverse engineering techniques (Hecker et al., 2009b; Marbach et al., 2010) to predict unknown interaction networks based on high-throughput gene expression data. A number of approaches have been established to predict inference networks including Bayesian network modeling (Friedman et al., 2000), information theoretical approaches (Butte and Kohane, 2000; Faith et al., 2007), regression based models (D’Haeseleer et al., 1999; Hecker et al., 2009a), and differential equation models (Holter et al., 2001; Guthke et al., 2005, 2007). Biological networks are scale free networks composed of nodes and edges, where nodes represent the objects of interest and edges show the relations between those objects (Le Novere et al., 2009). Biological interaction networks often use nodes to represent genes or proteins, and edges to show either a direct or indirection interaction, such as protein binding or transcriptional regulation (Barabasi and Oltvai, 2004). Network inference has been successfully applied to a variety of biological scenarios, including the modeling of immune diseases (Guthke et al., 2005; Hecker et al., 2009a), full-genomic models of Escherichia coli (Faith et al., 2007), and more recently, small scale networks describing fungal infections (Linde et al., 2010). So far these model have only focused on a single species and have not addressed host–pathogen interactions.
In the present work, we have generated the first interspecies computational model of molecular host–pathogen interactions. We used RNA-seq expression data from an infection time course of Candida albicans and bone marrow-derived dendritic cells (BMDCs) from M. musculus. C. albicans is one of the most prevalent opportunistic human fungal pathogens. Although C. albicans normally colonizes the human host, a variety of factors, most notably immune suppression, can lead to dissemination of fungal cells throughout the body. This dissemination can lead to a wide range of diseases, from thrush to multi-organ failure (Gudlaugsson et al., 2003). We focused on dendritic cells as our model host based of their function as antigen-presenting cells, their specialization in pathogen recognition, and their greater role in activating and modulating adaptive immune responses (Netea et al., 2008; Bourgeois et al., 2010). We experimentally verified predicted sub-networks of the interspecies inferred regulatory network, which identifies a role of the transcription factor Hap3 in C. albicans during in vitro infection. We find that fungal Hap3 is regulated by murine Ptx3, a soluble pattern recognition receptor acting as an opsonin for pathogens (Diniz et al., 2004). We show that Ptx3 binding to C. albicans regulates fungal Hap3 target genes, altering the immune response in dendritic cells. Based on the regulation of downstream cytokines and the regulation of MTA2 mRNA in a Hap3-dependent manner, we provide indirect evidence for a role for Mta2, a member of the nucleosome remodeling and histone deacetylase complex NuRD (Manavathi et al., 2007). We propose a mechanism whereby Ptx3 binding to C. albicans leads to cell wall remodeling via fungal Hap3 target genes, thereby changing the ability of the fungi to be recognized by immune cells. The experimental verification of the predicted interspecies interactions is proof-of-principle that network inference can be used to investigate microbial pathogenesis. We suggest that this could be a useful method to identify potential antifungal target genes.
Materials and Methods
Candida Strains and Growth Conditions
All strains were routinely grown on YPD plates (1% yeast extract, 2% peptone, 2% glucose, 2% agar) and in standard rich media YPD (1% yeast extract, 2% peptone, 2% glucose) for liquid culture at 30°C. Fungal cells were collected in the logarithmic growth phase by a brief centrifugation, washed in sterile PBS, and diluted for all interaction studies. The following strains were used in this study: C. albicans clinical isolate SC5314 (Gillum et al., 1984) and homozygous knock-out of Hap3 (hap3Δ/hap3Δ) and revertant strain (hap3Δ/hap3Δ + CIp10 (HAP3, URA3), abbreviated in the text as hap3Δ/hap3Δ + HAP3), were generated from the strain BWP17 (ura3::imm434/ura3::imm434 iro1/iro1::imm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG) by stepwise deletion of both alleles using PCR-amplified HAP3::ARG4 and HAP3::HIS1 cassettes (Gola et al., 2003) and a cIP10 plasmid containing HAP3 and its promoter and terminator sequences integrated at the RP10 locus (Murad et al., 2000).The homozygous knock-out of cda2 and revertant were kindly provided by Neil Gow (Aberdeen, UK).
Cell Culture of Primary Immune Cells from Mouse Bone Marrow
Bone marrow was differentiated to either BMDCs or bone marrow-derived macrophages (BMDMs) from the femurs of 7- to 9-week-old wild type C57BL/6 mice and assessed for homogeneity as previously described using a panel of marker antibodies (Bourgeois et al., 2009).
Fungal-Mammalian Cell Co-Culture
Fungal-mammalian cell co-cultures were performed as previously described (Bourgeois et al., 2009). Briefly, immune cells were plated at a density of 1.0 × 105 cells/cm2 in sterile cell culture dishes and incubated with fungal cells at a multiplicity of infection (MOI) of five fungal cells per immune cell. Samples were incubated at 37°C in 5% CO2, 95% humidity for up to 24 h.
cDNA Preparation for RNA-seq
Total RNA was isolated from immune cells and C. albicans using the SV total RNA isolation system (Promega, Madison, MI, USA) following manufacturers instructions. To obtain RNA mixtures from both C. albicans and BMDCs, cells were first scraped in the provided lysis buffer, followed by homogenization with 200 μl of 0.5 mm acid-washed glass beads (Sigma-Aldrich, St. Louis, MO, USA) in a Fast Prep-24 cooling block at 4°C (MP Biomedicals Europe, Illkirch, France) for 45 s at 5 m/s. Ribosomal RNA was depleted from 10 μg of pooled total RNA samples using the RiboMinus eukaryote kit for RNA-seq (Invitrogen, Carlsbad, CA, USA) and concentrated using the corresponding RiboMinus Concentration Module (Invitrogen) following manufactures instructions for three independent biological repeats. For each sample, 1 μg of ribosomal-depleted RNA was converted into cDNA using the SMARTer PCR cDNA Synthesis kit and the Advantage 2 polymerase mix (Clontech, Mountain View, CA, USA). PCR amplifications were performed on 1/10 of the first strand synthesis reaction for 18 cycles of 90°C for 1 min, 95°C for 15 s, 65°C for 30 s, and 68°C for 6 min on a GeneAmp PCR system 9700 (Applied Biosystems, Carlsbad, CA, USA), and purified on ChromaSpin columns (Clontech, Mountain View, CA, USA). The resulting cDNAs were sequenced on the Genome Analyzer IIx at GATC (Konstanz, Germany) using 36 bp, single run, indexed read mode.
Sequence Read Mapping, Pre-Processing, and Data Normalization
All sequencing reads were mapped using TopHat 1.2.0 (Trapnell et al., 2009) against the SC5314 C. albicans assembly 21 (Skrzypek et al., 2010) and the M. musculus UCSC version mm9 from the ENSEMBL database (Flicek et al., 2011). Mapping was carried out using the default settings in which only unique hits were kept for further analysis. The gene expression and normalization analysis was performed as previously described (Mortazavi et al., 2008). Genes were tested for differential expression using the bioconductor package baySeq (Hardcastle and Kelly, 2010) relative to the 0-min infection time point. The analysis was carried out for C. albicans and M. musculus genes individually.
Clustering and Over-Represented Gene Ontology Terms
Fuzzy c-means clustering (Bezdek, 1992) was applied to the two expression matrices of differentially expressed genes from C. albicans and M. musculus. The optimal number of clusters was estimated as previously described (Guthke et al., 2005; Linde et al., 2010). Functional categorization and significantly over-represented categories were identified using the tool FungiFun (Priebe et al., 2011). All four hierarchical levels of Funcat (Ruepp et al., 2004) and Gene Ontology (Ashburner et al., 2000) categorization were used in this study.
Network Inference Prediction and Measuring Interaction Robustness
Network inference was performed as previously described using the NetGenerator tool (Guthke et al., 2005; Linde et al., 2010). Briefly, NetGenerator is based on a set of linear differential equations and models the temporal change of the expression intensity xi(t) of gene i (i = 1…n) at time t as the weighted sum of the expression intensities of all other genes and an external stimulus u(t) at time t. The external stimulus u(t) is modeled as a stepwise constant function representing the change from no host–pathogen interaction to the onset of the interaction. The tool aims to identify a network structure, which best fits to the measured RNA-seq data, while it minimizes the number of predicted interactions (Guthke et al., 2005). Thus, a sparse network is inferred.
NetGenerator offers the possibility to integrate prior knowledge (i.e., putative regulatory interactions based on additional data besides the initial time series expression data). Based on the confidence of the prior knowledge source, it is possible to score each proposed interaction. The confidence of the prior knowledge is based on the level of experimentation used to verify a specific interaction and the number of independent experiments showing the same interaction. Since different data sources might be contradictory, prior knowledge is softly integrated, i.e., if a proposed interaction contradicts the measured data too much it can be removed by NetGenerator. Furthermore, the tool may add new interactions not covered by the prior knowledge in order to fit to the measured data. In this study, prior knowledge from public databases was softly integrated (Guthke et al., 2005). Each proposed interaction was scored in an additive manner based on the confidence of the prior knowledge source as follows: direct evidence that a gene is involved in a host–pathogen interaction (confidence score = 0.5), co-expression of two genes (confidence score = 0.25), and the occurrence of the respective transcription factor binding motif in the upstream intergenic regions of genes (confidence score = 0.125). Prior knowledge was obtained from GeneMania1, IntAct (Aranda et al., 2010), BioGrid (Stark et al., 2011), the C. albicans database (Skrzypek et al., 2010), the mouse genome database (Blake et al., 2011), and a number of peer-reviewed publications (Lane et al., 2001; Doedt et al., 2004; Martchenko et al., 2004; Zhao et al., 2004; Fradin et al., 2005; Oberholzer et al., 2006; Wang et al., 2006; Spira et al., 2007; Thewes et al., 2007; Zakikhany et al., 2007; Almeida et al., 2008; Baek et al., 2008; Nobile et al., 2008; Frohner et al., 2009; Griffin et al., 2009; Raman et al., 2009; Sellam et al., 2009; Hinze et al., 2010; Hou et al., 2010; Smith et al., 2010; Wachtler et al., 2011) summarized in Figure 1C. Putative regulatory interactions were tested for robustness using two methods. First, Gaussian noise was introduced with a mean of 0 and SD 0.05 to the estimated mRNA concentrations for 1000 iterations. Secondly, predicted interactions were screened for robustness against changes in prior knowledge by iterating the modeling approach 1000 times while randomly skipping 10% of all interactions in the set of prior knowledge for each run. Only edges that were confirmed by more than 50% of the iterations were considered to be robust and used in the resultant model.
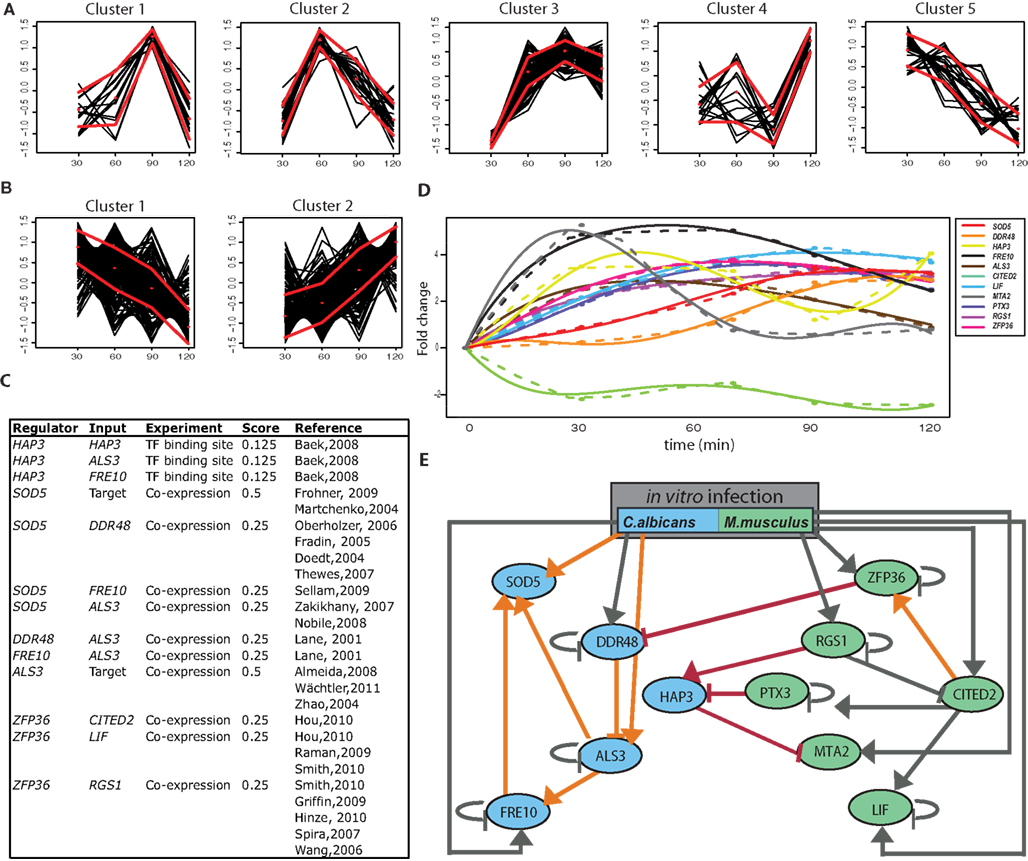
Figure 1. RNA-seq predicts regulatory host-fungus interactions. (A) M. musculus clustering of differentially expressed genes (scaled and centered log2 values) over the time course of the infection. Black lines represent the expression of individual genes. The mean (dotted line, red) and the variance (solid, red) are shown. (B) C. albicans clustering of differentially expressed genes (scaled and centered log2 values) over the time course of the infection. Black lines represent the expression of individual genes. The mean (dotted line, red) and the variance (solid, red) are shown. (C) Prior knowledge incorporated into the inference network for all genes, where the input corresponds to the external perturbation (co-culture) and regulation by target corresponds to induction by the co-culture environment but not associated with a specific gene set. (D) Measured (dots), interpolated (dotted line), simulated (solid line) gene expression for all genes used in the inference model from NetGenerator over the time course of the infection. (E) Inferred network model between C. albicans and M. musculus, where all C. albicans (blue) and M. musculus genes (green) in the study are included. The following interactions are represented on the model: predicted interactions based on the RNA-seq data set from individual species where no prior knowledge exists (gray), predicted interactions between a C. albicans and M. musculus gene where no prior knowledge exists (red), or where prior knowledge exists and corresponds to expression data set (orange). Here, activation is shown as an arrow and a repression with a bar. The “C. albicans/M. musculus” rectangle represents the influence from the external perturbation (co-culture during in vitro infection) on the gene expression level.
Real-Time qPCR Analysis
RNA sample preparation, reverse transcription, and real-time PCR were performed previously described (Bourgeois et al., 2009) using the following primers: mouse β-Actin, forward 5′-GCGTGACATCAAAGAGAAG-3′ reverse 5′-AGGAGCCAGAGCAGTAATC-3′ (RTPrimerDB)2 mouse MTA2, forward 5′-CACTGCTATAGCCTCACGCC-3′, reverse 5′-GCTAGGAGCTGGAACC TCAC-3′, mouse PTX3, forward 5′-CCTGCTTTGTGCTCTCTGGT-3′, reverse 5′-TCTCCAGCATGATGAACAGC-3′ (Diniz et al., 2004), C. albicans TUP1, forward 5′-GACTACGCCTCAAACGAAGC-3′ reverse 5′-TGGTGCCACAATCTGTTGTT-3′, C. albicans FRE6 forward 5′-CCGGTAAACATCCATTCCAC-3′, reverse 5′-TTGATCCAAATGCCATT-CAA-3′, C. albicans SEF1, forward 5′-GTGGAGGACTCGTTCATGGT-3′, reverse 5′-TGAACCAGCACGATTCAGAG-3′, C. albicans RIP1, forward 5′-TGCTGACAGAGTCAAGA-AACC-3′ reverse 5′-GAACCAACCACCGAAATCAC-3′ as determined using the sequence analysis software Vector NTI (Invitrogen, Carlsbad, CA, USA). Results were calculated using the ΔΔct method and are expressed as the fold of the gene expression of interest versus the expression of a housekeeping gene in M. musculus (β-Actin) or C. albicans (RIP1) in treated versus untreated conditions.
Cytokine Quantification from Co-Culture Supernatants
The amount of cytokines released into cell culture supernatants by immune cells during in vitro interaction studies with heat killed C. albicans were assayed after 24 h of co-culture using the mouse IL-2, IL-4, or TNFα Ready-set-go ELISA kit (R&D Systems, Minneapolis, MN, USA) or the Mouse Cytokine Array Panel A kit (R&D Systems) according to the manufacturers instructions.
Binding and Labeling Ptx3 in vitro
Recombinant mouse Ptx3 (rmPtx3) protein (R&D Systems) was reconstituted in sterile PBS and diluted for all experiments. Some 0.5 × 106 fungal cells were incubated for 1 h at 37°C with 5 μg reconstituted rmPtx3. Ptx3 was labeled with the primary antibody against Ptx3 (Abnova, Taiwan) and secondarily labeled with goat-anti-rabbit 649 Dylight (Thermo Scientific, Rockford, Illinois). Fungal cell wall chitin was labeled using 10 μM of Calcofluor White (Sigma-Aldrich). Intracellular labeling of Ptx3 was performed using the BD Cytofix/Cytoperm Fixation/Permeabilization kit with BD GolgiPlug protein transport inhibitor (BD Biosciences, Heidelberg) after 6 h of C. albicans infection following manufactures instructions. Preparations were assessed by flow cytometry or visualized on an Olympus Cell-R live imaging unit (Olympus, Essex, UK) for all experiments.
Statistical Analysis for Inference Model Verification
Statistical analysis of data was performed using the GraphPad Prism graphing and analysis software (GraphPad Software, San Diego, USA) for all in vitro experiments excluding the RNA-seq analysis described above. Statistical significance was assessed using with the Student t-test and a p-value <0.05 was considered significant.
Results
Inferred Regulatory Network Identifies Novel Interspecies Host–Pathogen Interactions
We used massively parallel RNA sequencing of cDNA (RNA-seq) obtained from co-cultures of C. albicans and M. musculus BMDCs over 2 h to model an infection time course from fungal adhesion to early host cell lysis. In total, we obtained approximately 120 million reads, which were mapped to the C. albicans 21 assembly or M. musculus mm9 genome and analyzed each for differential gene expression relative to the pre-infection state. We identified 545 differentially expressed genes for C. albicans and 240 for M. musculus over the complete time course.
The small number of measured data points for each gene over the time course restricts the modeling approach to a limited number of genes. If there was no pre-selection of the genes, or a large number of genes were to be used, it would result in an over-fitting of the measured data that would not produce a robust inference model. For this reason, it is necessary to select a set of relevant genes to be represented by nodes in the network model. To identify candidate genes in C. albicans and M. musculus, all differentially expressed genes were first clustered (Bezdek, 1992) by their kinetics during the time course (Figures 1A,B). From each cluster, one or more representative genes were chosen for use within the model. Several considerations were taken into account for the selection of candidate genes. In C. albicans, we preferentially chose genes that have been either annotated as virulence genes (i.e., adhesion, hyphal formation, or response to host) or strongly respond to infection or infection-like conditions (i.e., temperature stress, nutrient limitation, or iron regulation). For M. musculus, we prioritized genes with phenotypes relating to the immune defense or response, or susceptibility to pathogens in a systemic mouse model of infection.
A number of recent studies have shown the reverse engineering approach is greatly improved by the integration of different data sources (Werhli and Husmeier, 2007; Gustafsson et al., 2008; Hecker et al., 2009a,b). We therefore collected putative regulatory interactions based on additional data obtained from literature, referred to as prior knowledge, for each gene. Based on the confidence of the prior knowledge source, a score is attributed to each interaction (see Materials and Methods). Since different data sources might be contradictory, prior knowledge was softly integrated so that if a proposed interaction contradicts the measured data to a great extent, it can be removed from the resulting network (see Materials and Methods). Genes with no known or predicted function were therefore excluded from the analysis. Based on these criteria, we narrowed our gene lists to five from C. albicans and six from M. musculus. Prior knowledge scores (Figure 1C) and expression kinetics (Figure 1D) for the candidate genes were combined in NetGenerator to generate the final network inference (Figure 1E).
To verify the fit of the model to the actual expression kinetics of the candidate genes, we first used NetGenerator to interpolate and simulate gene expression for the measured data points of each gene (Figure 1D). The closer expression profiles for the individual genes fit to the measured data points, the better the inference prediction is. We found a close relationship between the simulated and measured data points, showing that the NetGenerator model is representative of the measured data. The final interspecies network was based on these predictions (Figure 1E). Only edges that were robust against the addition of Gaussian noise and partial skipping of prior knowledge were used in the construction of the model. The final network predicts 21 putative edges, including 4 interspecies edges.
To specifically test the robustness of the interspecies edges experimentally, we focused on a sub-network composed of a single C. albicans transcription factor Hap3 that was predicted in the inference model to contain two interactions with M. musculus genes. These interactions include a predicted blunt or repressing edge between fungal Hap3 by murine Ptx3, and a predicted blunt edge of murine Mta2 by fungal Hap3 itself.
The Binding of Murine Ptx3 Regulates Hap3 Target Genes
Pentraxin 3 (Ptx3) is a soluble pattern recognition receptor that has been previously shown to function as an opsonin to facilitate pathogen uptake by phagocytic cells in a dectin-1 dependent manner (Diniz et al., 2004). To determine if M. musculus Ptx3 blocked fungal Hap3 function or the expression of Hap3-regulated genes in C. albicans as suggested by the inference model, we first asked whether Ptx3 was induced upon infection with Candida cells. Using intracellular staining for Ptx3, we detected a strong fluorescence signal in BMDMs infected with C. albicans, whereas no signal was detected in BMDMs alone or ptx3−/− macrophages (Figures 2A,B). Furthermore, we found an increase in PTX3 mRNA levels in BMDCs (Figure 2C) and BMDMs (data not shown) after 1 h of C. albicans infection, verifying that Ptx3 is indeed induced in our experimental setup. Interestingly, the amount of PTX3 induced significantly decreased in the absence of Hap3 (Figure 2C). We detected a similarly significant decrease in PTX3 induction in the gene containing the Hap3 binding box, Cda2, a predicted chitin deacetylase in C. albicans.
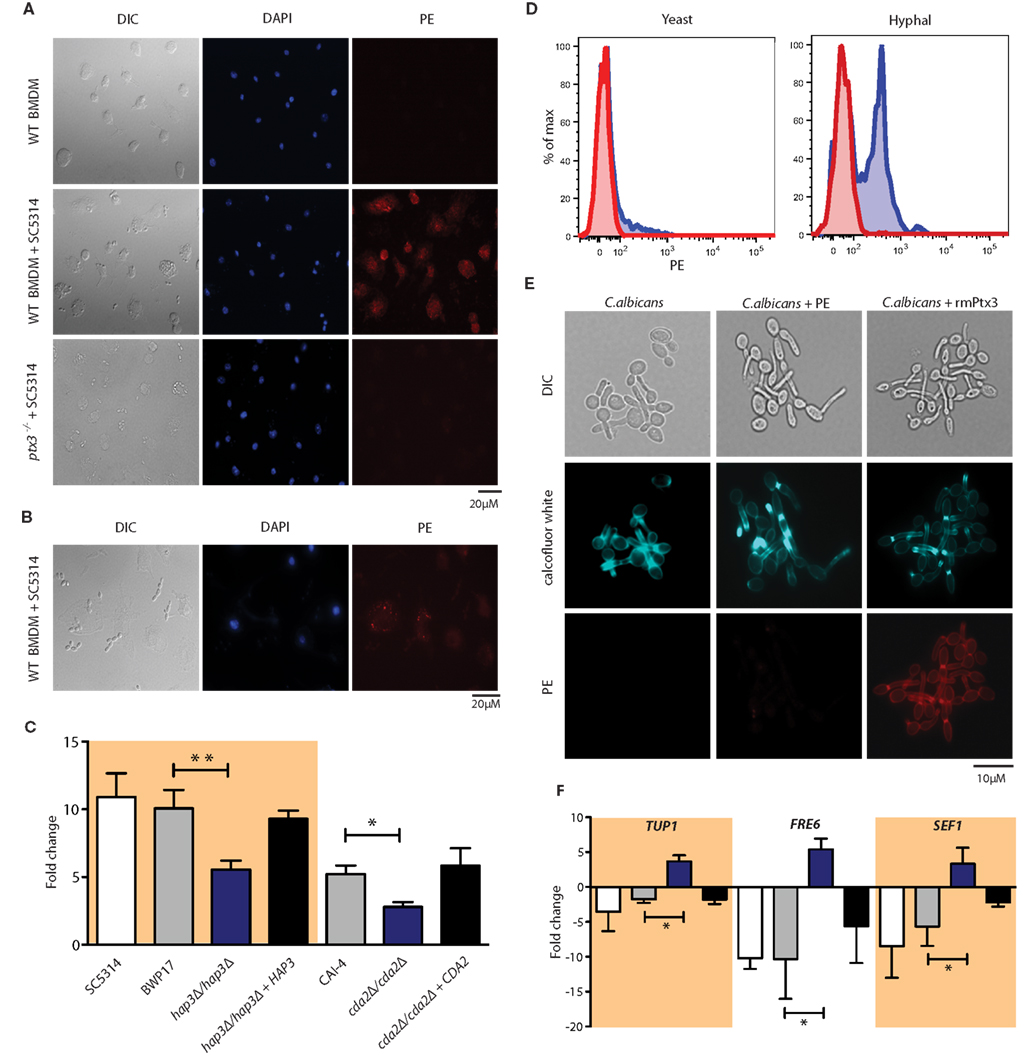
Figure 2. Binding of rmPtx3 to C. albicans mediates the expression of Hap3 target genes. (A) Intracellular labeling of endogenous Ptx3 induction after 6 h of C. albicans stimulation of macrophages derived from wild type or ptx3−/− bone marrow. (B) Intracellular labeling of endogenous Ptx3 after 6 h of C. albicans stimulation of macrophages derived from wild type bone marrow. Macrophages directly associated with fungal cells and show a strong signal for endogenous Ptx3, while those not associated have only background signal levels. (C) qPCR of PTX3 in BMDCs after 1 h of infection with different C. albicans strains. Results represent the mean of 3 pooled experiments ± SD. (D) FACS analysis of wild type strain SC5314 after 1-h treatment with rmPtx3, where untreated cells stained with PE only (red) and rmPtx3 and SC5314 (blue) are shown. Cells were gated by size to differentiate yeast and hyphal morphologies. (E) Fluorescence microscopy of SC5314 after 1 h pre-treatment with 5 μg rmPtx3 (red) or 10 μM Calcofluor White (blue). (F) qPCR of predicted targets genes of Hap3. SC5314 (white), BWP17 (gray), hap3Δ/hap3Δ (blue), and hap3Δ/hap3Δ + HAP3 (black) after 1 h pre-incubation with 5 μg rmPtx3 are shown. Results represent the mean of 3 pooled experiments ± SD.
Ptx3 has been previously shown to bind to numerous fungi, including Aspergillus fumigatus (Jaillon et al., 2007) as well as zymosan-coated particles (Diniz et al., 2004). Therefore, we asked whether recombinant mouse Ptx3 (rmPtx3) could also bind to the C. albicans cell wall. We assessed rmPtx3 binding using fluorescence microscopy and flow cytometry. Fungal cells pre-incubated with rmPtx3 for 1 h at 37°C showed surface localization of Ptx3 to the cell wall. No signal was visible on cells treated with the PE-conjugated secondary alone or the untreated control (Figure 2E). Notably, the labeling pattern of Ptx3 coincided with areas of expected chitin exposure. Hence, we also stained C. albicans cells with Calcofluor White, a fluorescent dye that binds to exposed chitin (Bulawa et al., 1995). Interestingly, the localization of rmPtx3 on the C. albicans overlapped with the signal detected for Calcofluor White alone (Figure 2E), suggesting that Ptx3 might bind to accessible cell surface chitin. We confirmed and quantified the amount of Ptx3 binding to the cell wall using flow cytometry. Based on the binding observed in the fluorescence microscopy, we analyzed our data using both the complete pool of C. albicans cells as well as discriminating yeast and hyphal forms (Figure 2D; Figure A1 in Appendix). We detected a high level of binding in fungal hyphae compared to yeast form cells. This is consistent with our fluorescence microscopy analysis, where we detected much stronger signals on the hyphal cell walls compared to the bud scars on yeast form cells.
Given that rmPtx3 binds to the fungal cell surface, we assessed if rmPtx3 binding influenced the expression of predicted fungal Hap3 target genes as predicted by the inferred network using qPCR (Figure 2F). There are 10 predicted target genes of Hap3 that were recently identified in a network inference study using microarray data from C. albicans during in vitro epithelial infection, where iron is assumed to be limited (Linde et al., 2010). Out of the 10 putative Hap3 target genes, we found three, TUP1, FRE6, and SEF1, whose expressions were significantly decreased in C. albicans after rmPtx3 binding, verifying their functionality as Hap3 target genes. Interestingly, the levels of these genes increased in the Hap3 knock-out. These data strongly suggest that their down-modulation upon binding of rmPtx3 is Hap3-dependent.
Candida albicans Bound by Ptx3 Attenuates the Immune Response in BMDCs
Recently it was shown that the binding of recombinant human Ptx3 increases A. fumigatus conidia phagocytosis and influences cytokine production. Those mice lacking Ptx3 were additionally found to be more susceptible to A. fumigatus infection (Moalli et al., 2011). To determine if the binding of Ptx3 to C. albicans changed the cytokine production of murine immune cells in vitro, we first investigated the gross immune response using a cytokine array after 24 h of co-culture with BMDCs (Figure A2 in Appendix). RmPtx3-bound C. albicans induced multiple cytokines compared to untreated C. albicans including IL-2, a cytokine regulated by Mta2, as well as the inflammatory cytokines KC, JE, and TNFα (Figures A2 in Appendix). Interestingly, when we then compared Hap3 knock-out cells pre-incubated with rmPtx3, we detected a general increase in these cytokines in addition to IL-23, IL-17, IL-16, and IL-10 that were not detected in using wild type C. albicans (Figure A2 in Appendix).
Since the cytokine array is a qualitative assessment of cytokine production with a relatively high detection threshold, we verified the changes in cytokine levels for the cytokines most relevant to the inference model, namely, IL-2 and IL-4, both target cytokines of Mta2, and TNFα, a pleiotropic inflammatory cytokine, by ELISA. Mta2 is a member of the NuRD (nucleosome remodeling and histone deacetylase) complex in M. musculus (Manavathi et al., 2007) and predicted in our network model as repressed by Hap3. The cytokines IL-2 and IL-4 produced during the host immune response were both recently identified as targets of the Mta2/NuRD complex (Lu et al., 2008). We found that in BMDCs, MTA2 increased in the absence of Hap3, suggesting that Hap3 might indirectly regulate expression of Mta2 (Figure 3A). We quantified cellular cytokine release using wild type BMDCs with C. albicans, wild type BMDCs with rmPtx3-bound C. albicans, and ptx3−/− BMDCs. We found that the production of IL-2 and IL-4 significantly increased in the absence of Hap3 (Figures 3B,C). This increase was augmented by C. albicans cells pre-incubated with rmPtx3, confirming our observation from the cytokine array that there is a general increase in the production of these cytokines when there is an increase in Ptx3. Interestingly, compared to wild type BMDCs, the basal level of cytokine production of IL-2 and IL-4 increased in ptx3−/− BMDCs, corresponding to our inferred network prediction that the loss of its predicted negative regulators, Ptx3 and fungal Hap3, would increase the expression of Mta2 and thereby increase the expression of its target cytokines. In Hap3 knock-out cells, we found both on the cytokine arrays and by ELISA a significant decrease in TNFα (Figure 3D). These data show that the binding of Ptx3 to fungal cells alters the cytokine production by immune cells in a Hap3-dependent manner, and the regulation of Mta2 target cytokines indirectly suggests an involvement of Mta2 as predicted by the network inference model.
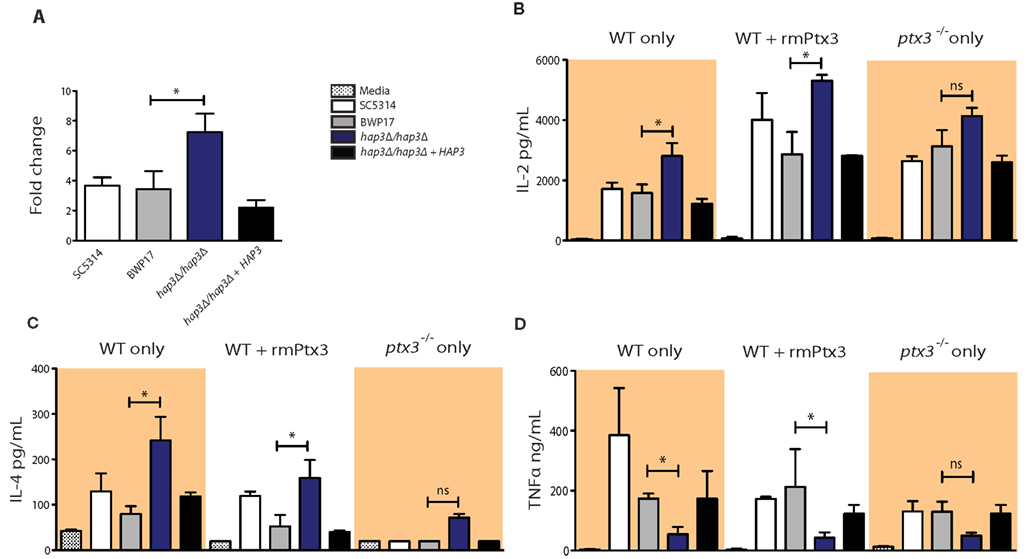
Figure 3. Mouse MTA2, IL-2 and IL-4 increase upon stimulation with C. albicans in a Hap3-dependent manner. (A) qPCR of M. musculus MTA2 expression in BMDCs after 1 h incubation with fungal cells at an MOI of five fungal cells to immune cells are shown. Results represent the mean of 3 pooled experiments ± SD. (B–D) ELISA measurement of IL-2 (B), IL-4 (C), and TNFα (D) from the supernatants of BMDCs after 24 h incubation with fungal cells in WT BMDCs, C. albicans cells pre-incubated with rmPtx3 on wild type BMDCs, or fungal cells on ptx3−/− BMDCs. Incubations were performed at an MOI of five fungal cells to immune cells. Results represent the mean of 2 pooled experiments ± SD.
Identifying Cell Surface Hap3 Target Genes
To identify how immune cells could detect the regulation of the transcription factor Hap3 in C. albicans, we searched for putative Hap3 target genes that could have more direct contact with immune cells, including: cell wall, plasma membrane or secretory proteins. We focused on C. albicans genes of cluster 2, since their expression strongly increased expression over the time course of invasion (Figure 1B). Within this cluster, we scanned for genes harboring the binding site of the Hap-complex in their upstream regulatory regions (Baek et al., 2008). We further narrowed down the candidate list by removing genes that did not have a predicted cellular localization or function in the C. albicans database (Skrzypek et al., 2010). Following these selection criteria, nine candidate genes were left that we used to infer an additional network in combination with Ptx3, Hap3, and Mta2 to determine if an interaction could be inferred with a protein that could come in direct contact with immune cells (Figure A3 in Appendix). To increase the reliability of the putative Hap3 interactions within the new interaction network, we included the validated interactions from our experiments within this study (repression of HAP3 by Ptx3 and MTA2 by Hap3), as additional prior knowledge. Of all of the candidate genes, only the activation of CDA2 (a putative chitin deacetylase in C. albicans) by Hap3, was robust against Gaussian noise and partial skipping of prior knowledge.
Discussion
In this study, we aimed to infer a network that predicts interactions between host and pathogenic species under infection settings. To our knowledge, this is the first network inference approach predicting host–pathogen interactions. This approach allowed for the prediction, identification, and experimental verification of interdependent sub-networks composed of a single C. albicans transcription factor Hap3, and the M. musculus genes Ptx3 and Mta2. The experimental validation suggests a putative mechanism to explain how these interactions could be regulated during infection of immune cells by fungal pathogens.
Our modeling approach was fundamentally based on differential equations, which have been previously used to infer regulatory network models (Toepfer et al., 2007; Linde et al., 2010). This approach is generally suitable for time series data. Nevertheless, this approach is inappropriate for large-scale modeling, because a large number of genes incorporated into a differential equation based model leaves open a number of parameters to be identified. This may result in an over-fitting of the data. The modeling approach uses four attempts to prevent over-fitting. First, we restrict the number of genes within the model such that a smaller number of parameters need to be identified. Second, it aims at inferring a sparse network where many parameters are zero. Thirdly, it makes use of re-sampling techniques where the data are perturbed in a random manner. Finally, we make use of prior knowledge guiding the inferred structure to a knowledge-based solution. Thus skipping incorrect network structures.
Gene expression levels, as well as available prior biological knowledge, were used to aid in the narrowing of genes that we chose to incorporate into the model. For this reason, genes where no biological knowledge was available were excluded from further analysis. However, we cannot exclude the possibility that additional genes of unknown function might also play a role in our inference model. This remains a limitation of the modeling approach, in so far as predictions can only be made for genes where a reasonable amount or prior knowledge is available. The genes incorporated into the model represent only one possible scenario of interactions and we do not exclude the possibility that other genes may play a role under other conditions. We have already started to take first step for a full-genomic network modeling for C. albicans utilizing a compendium of all available expression data (Altwasser et al., 2012). Moreover, we primarily focused on genes acting as putative network “hubs” in their organisms (Bulawa et al., 1995). Hubs are genes such as transcription factors that regulate many other downstream genes within a network either directly or indirectly. Hubs were chosen because they are less likely to have redundant roles. Therefore, we would expect a stronger phenotype than investigating genes that are sparsely connected. This also means that the interactions we are investigating are more likely to be indirect and should be interpreted with caution.
From our original candidate gene list, we inferred HAP3 as a putative network hub targeted by innate immune cells. Interestingly, several putative target genes of Hap3 identified in this study are predicted to localize to the plasma membrane, cell wall, or are involved with cell wall reorganization in C. albicans. The fungal cell wall is a dynamic structure, which undergoes significant structural and molecular composition remodeling throughout its life cycle, as well as in response to a variety of external stimuli (Chaffin, 2008). As Hap3 in C. albicans is a transcription factor up-regulated under iron-limiting conditions (Linde et al., 2010), it is likely that its function during fungal recognition or phagocytosis by immune cells is indirect. Of all of the candidate cell surface Hap3 targets, only Cda2, a putative chitin deacetylase forms a robust interaction with Hap3 within the second network (Figure A3 in Appendix). Chitin deacetylase enzymes exists in both intracellular and secreted forms in different fungi, where they hydrolyzes the acetamido group in the N-acetylglucosamine units of chitin and chitosan, leaving glucosamine units and acetic acid form as byproducts (Zhao et al., 2010). Chitin deacetylases exist in both Saccharomyces cerevisiae (Martinou et al., 2002) and in the opportunistic fungal pathogen Cryptococcus neoformans, where they have been suggested as an antifungal target due to their severe effect on cell wall integrity (Baker et al., 2007). Notably, chitin deacetylases are secreted during different developmental stages of some other fungi (Zhao et al., 2010). For example, in Colletotrichum lindemuthianum, a plant fungal pathogen, chitin deacetylases are exclusively secreted during hyphal penetration into plant tissue (Tokuyasu et al., 1996). We find that Ptx3 induction is decreased in the CDA2 knock-out (Figure 2C), further suggesting a possible connection to the inferred network model. These data are consistent with the overlap of Ptx3 staining and that of Calcofluor White (Figure 2E), which binds to exposed chitin. Therefore, it is tempting to speculate that the recognition of C. albicans by immune cells triggers the production of this enzyme to induce cell wall remodeling as an evasion strategy. However, further work beyond the scope of this study is needed to decipher the specific function of Cda2 in C. albicans and its connection to Hap3.
We observed that upon binding of rmPtx3 to fungal cells, the C. albicans virulence genes TUP1, FRE6, and SEF1 mRNA levels significantly decreased in a Hap3-dependent manner (Figure 2F). Tup1 has a well-characterized role as a key regulator in C. albicans morphogenesis (Braun and Johnson, 1997). We cannot exclude the possibility that Hap3 and Tup1 may have similar or even complementary functions during interaction with immune cells. Interestingly, both Tup1 and Fre6 are either directly or indirectly involved in the C. albicans cell wall homeostasis. Tup1 is a multi-functional transcriptional co-repressor of filamentous growth in C. albicans whose lack leads to constitutive filamentous growth (Braun and Johnson, 1997; Park and Morschhäuser, 2005). Fre6 is an uncharacterized protein, for which in silico predictions suggest it to reside in the plasma membrane with a putative functional similarity to the ferric reductase Fre10, an important protein in iron acquisition (Knight et al., 2005). Therefore, their regulation upon binding or phagocytosis might play an additional role in cell wall remodeling during infection. Since fungal cells experience severe iron-limiting condition within phagosomes of host cells, Hap3 and Fre6 appear as logical candidates involved in this reciprocal interaction. Likewise, Sef1 regulates iron uptake, and has recently been shown to promote virulence in a mouse model of bloodstream infections (Chen et al., 2011). Interestingly, it was shown that knock-out mice lacking Ptx3 are hyper-susceptible to A. fumigatus (Moalli et al., 2010). However, no in vivo work has been performed to date using ptx3−/− mice and C. albicans. A recent study has shown that the activation of the complement system via the lectin pathway can be triggered via a complex of Ptx3 and mannose binding lectin (MBL) on C. albicans mannan in vitro (Ma et al., 2011). They showed the MBL–Ptx3 complex could enhance the deposition of the complement components C3 and C4 and thereby increase phagocytosis of C. albicans by polymorphonuclear leukocytes. It has previously been shown that C3 knock-out mice are additionally more susceptible to C. albicans infections (Han et al., 2001). Therefore, it is possible that the absence of Ptx3 could result in reduced activation of the complement pathway and reduced fungal killing. In vivo studies using ptx3−/− mice would be needed to investigate this hypothesis.
We found that the expression of MTA2 and the regulation of its downstream targets such as cytokines IL-2 and IL-4, are increased during immune cell invasion by C. albicans in a Hap3-dependent manner (Figures 3B,C). Moreover, we found that an altered-immune response is one consequence of rmPtx3 binding. Mta2 knock-out mice display partial embryonic lethality, while the surviving mice develop lupus-like autoimmune symptoms, including severe developmental phenotypes (Lu et al., 2008). Mta2 is uniquely associated with the NuRD chromatin complex, which has both nucleosome remodeling and histone deacetylase activity (Feng and Zhang, 2003). Although there has been no data to date in fungi indicating a role for host chromatin in pathogenesis, recent work in bacteria and viruses (Hamon and Cossart, 2008; Rohde, 2011) shows that chromatin remodeling is induced in host cells during invasion. Consistent with these observations, our data suggests that the regulation of MTA2 may affect chromatin remodeling in immune cells in the response to fungal pathogens. The resultant altered-immune response may be disadvantageous to the pathogen because it would promote fungal clearance.
We propose that Hap3 constitutes a target hub of C. albicans, which actively regulates immune responses through the reorganization of the C. albicans cell wall during invasion of innate immune cells (Figure 4). Specifically, we propose a model in which the binding of Ptx3 released from immune cells to C. albicans cell wall triggering the reorganization of the C. albicans cell wall and plasma membrane via the activation of Hap3 target genes. This reorganization in turn changes the recognition of the fungus by immune cells and attenuates the host immune response. This work demonstrates the possibility to experimentally verify predicted host–pathogen relationships based on an interspecies model of network inference, showing that inference modeling can be used in the investigation of microbial pathogenesis. We propose that this method could be useful for the identification of antifungal target genes.
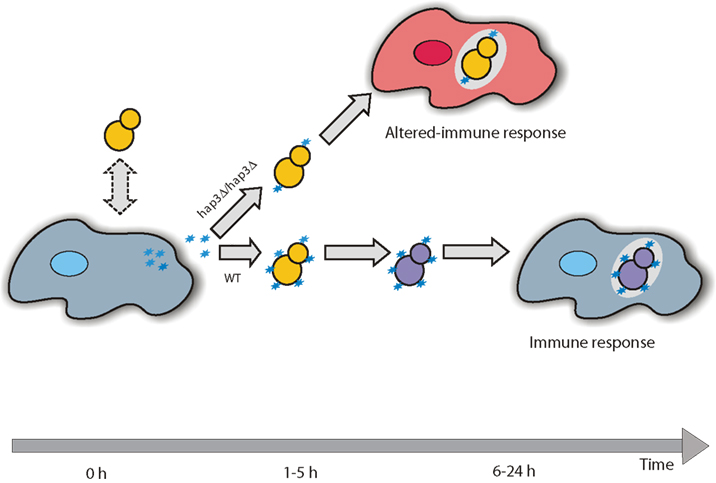
Figure 4. A proposed model for the mode of action of Ptx3 on C. albicans. After mutual recognition between C. albicans (yellow) and host immune cells (blue), Ptx3 (blue, stars) is released into the surrounding milieu where it can bind to the invading fungal cell wall. The binding of Ptx3 induces a change in the C. albicans cell wall (purple) after the activation of Hap3 target genes, influencing its recognition by immune cells and the subsequent immune response. Arrows represent progression of time during an infection.
Contribution
Lanay Tierney designed research, performed experiments, analyzed data and co-wrote the manuscript. Jörg Linde generated network inference maps, analyzed data, and co-wrote the manuscript. Sebastian Müller performed the sequence alignment and normalization. Sascha Brunke, Juan Camilo Molina, and Bernhard Hube provided C. albicans Hap3 deletion strains. Reinhard Guthke and Karl Kuchler designed the research and co-wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Alberto Mantovani and Cecilia Garlanda (Milan, Italy) for kindly providing ptx3−/− bone marrow. We are also indebted to Neil Gow (Aberdeen, UK) for kindly providing the Cda2 knock-out C. albicans strains. This work was supported by a grant from the Christian Doppler Research Society to Karl Kuchler, and in part by the FWF-DACH grant of the Austrian Science Foundation (FWF-Proj.: I-746-B11) to Karl Kuchler and Bernhard Hube. Jörg Linde was supported by the excellence graduate school “Jena School for Microbial Communication (JSMC).” We would like to thank laboratory members for critical reading and helpful comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Microbial_Immunology/10.3389/fmicb.2012.00085/abstract
Table S1. RPKM values for C. albicans genes over the infection time course.
Table S2 RPKM values for M. musculus genes over the infection time course.
Footnotes
References
Almeida, R. S., Brunke, S., Albrecht, A., Thewes, S., Laue, M., Edwards, J. E., Filler, S. G., and Hube, B. (2008). The hyphal-associated adhesin and invasin Als3 of Candida albicans mediates iron acquisition from host ferritin. PLoS Pathog. 4, e1000217. doi:10.1371/journal.ppat.1000217
Altwasser, R., Linde, J., Buyko, E., Hahn, U., and Guthke, R. (2012). Genome-wide scale-free network inference for Candida albicans. Front. Microbiol. 3:51. doi:10.3389/fmicb.2012.00051
Aranda, B., Achuthan, P., Alam-Faruque, Y., Armean, I., Bridge, A., Derow, C., Feuermann, M., Ghanbarian, A. T., Kerrien, S., Khadake, J., Leroy, C., Menden, M., Michaut, M., Montecchi-Palazzi, L., Neuhauser, S. N., Orchard, S., Perreau, V., Roechert, B., van Eijk, K., and Hermjakob, H. (2010). The IntAct molecular interaction database in 2010. Nucleic Acids Res. 38, D525–D531.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M., and Sherlock, G. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29.
Baek, Y. U., Li, M., and Davis, D. A. (2008). Candida albicans ferric reductases are differentially regulated in response to distinct forms of iron limitation by the Rim101 and CBF transcription factors. Eukaryot. Cell 7, 1168–1179.
Baker, L. G., Specht, C. A., Donlin, M. J., and Lodge, J. K. (2007). Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot. Cell 6, 855–867.
Barabasi, A. L., and Oltvai, Z. N. (2004). Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113.
Bezdek, J. (1992). Fuzzy Models for Pattern Recognition: Methods that Search for Structures in Data. New York: Institute of Electrical and Electronics Engineers (IEEE) Press.
Blake, J. A., Bult, C. J., Kadin, J. A., Richardson, J. E., and Eppig, J. T. (2011). The mouse genome database (MGD): premier model organism resource for mammalian genomics and genetics. Nucleic Acids Res. 39, D842–D848.
Bourgeois, C., Majer, O., Frohner, I., and Kuchler, K. (2009). In vitro systems for studying the interaction of fungal pathogens with primary cells from the mammalian innate immune system. Methods Mol. Biol. 470, 125–139.
Bourgeois, C., Majer, O., Frohner, I. E., Tierney, L., and Kuchler, K. (2010). Fungal attacks on mammalian hosts: pathogen elimination requires sensing and tasting. Curr. Opin. Microbiol. 13, 401–408.
Braun, B. R., and Johnson, A. D. (1997). Control of filament formation in Candida albicans by the transcriptional repressor TUP1. Science 277, 105–109.
Bulawa, C. E., Miller, D. W., Henry, L. K., and Becker, J. M. (1995). Attenuated virulence of chitin-deficient mutants of Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 92, 10570–10574.
Butte, A. J., and Kohane, I. S. (2000). Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. Pac. Symp. Biocomput. 5, 418–429.
Chen, C., Pande, K., French, S. D., Tuch, B. B., and Noble, S. M. (2011). An iron homeostasis regulatory circuit with reciprocal roles in Candida albicans commensalism and pathogenesis. Cell Host Microbe 10, 118–135.
D’Haeseleer, P., Wen, X., Fuhrman, S., and Somogyi, R. (1999). Linear modelling of mRNA expression levels during CNS development and injury. Pac. Symp. Biocomput. 4, 41–52.
Diniz, S. N., Nomizo, R., Cisalpino, P. S., Teixeira, M. M., Brown, G. D., Mantovani, A., Gordon, S., Reis, L. F., and Dias, A. A. (2004). PTX3 function as an opsonin for the dectin-1-dependent internalization of zymosan by macrophages. J. Leukoc. Biol. 75, 649–656.
Doedt, T., Krishnamurthy, S., Bockmuhl, D. P., Tebarth, B., Stempel, C., Russell, C. L., Brown, A. J., and Ernst, J. F. (2004). APSES proteins regulate morphogenesis and metabolism in Candida albicans. Mol. Biol. Cell 15, 3167–3180.
Faith, J. J., Hayete, B., Thaden, J. T., Mogno, I., Wierzbowski, J., Cottarel, G., Kasif, S., Collins, J. J., and Gardner, T. S. (2007). Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 5, e8. doi:10.1371/journal.pbio.0050008
Feng, Q., and Zhang, Y. (2003). The NuRD complex: linking histone modification to nucleosome remodeling. Curr. Top. Microbiol. Immunol. 274, 269–290.
Flicek, P., Amode, M. R., Barrell, D., Beal, K., Brent, S., Chen, Y., Clapham, P., Coates, G., Fairley, S., Fitzgerald, S., Gil, L., Gordon, L., Hendrix, M., Hourlier, T., Johnson, N., Kähäri, A. K., Keefe, D., Keenan, S., Kinsella, R., Komorowska, M., Koscielny, G., Kulesha, E., Larsson, P., Longden, I., McLaren, W., Muffato, M., Overduin, B., Pignatelli, M., Pritchard, B., Riat, H. S., Ritchie, G. R., Ruffier, M., Schuster, M., Sobral, D., Tang, Y. A., Taylor, K., Trevanion, S., Vandrovcova, J., White, S., Wilson, M., Wilder, S. P., Aken, B. L., Birney, E., Cunningham, F., Dunham, I., Durbin, R., Fernández-Suarez, X. M., Harrow, J., Herrero, J., Hubbard, T. J., Parker, A., Proctor, G., Spudich, G., Vogel, J., Yates, A., Zadissa, A., and Searle, S. M. (2011). Ensembl 2011. Nucleic Acids Res. 39, D800–D806.
Fradin, C., De Groot, P., MacCallum, D., Schaller, M., Klis, F., Odds, F. C., and Hube, B. (2005). Granulocytes govern the transcriptional response, morphology and proliferation of Candida albicans in human blood. Mol. Microbiol. 56, 397–415.
Friedman, S. R., Kottiri, B. J., Neaigus, A., Curtis, R., Vermund, S. H., and Des Jarlais, D. C. (2000). Network-related mechanisms may help explain long-term HIV-1 seroprevalence levels that remain high but do not approach population-group saturation. Am. J. Epidemiol. 152, 913–922.
Frohner, I. E., Bourgeois, C., Yatsyk, K., Majer, O., and Kuchler, K. (2009). Candida albicans cell surface superoxide dismutases degrade host-derived reactive oxygen species to escape innate immune surveillance. Mol. Microbiol. 71, 240–252.
Gillum, A. M., Tsay, E. Y., and Kirsch, D. R. (1984). Isolation of the Candida albicans gene for orotidine-5’-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol. Gen. Genet. 198, 179–182.
Gola, S., Martin, R., Walther, A., Dunkler, A., and Wendland, J. (2003). New modules for PCR-based gene targeting in Candida albicans: rapid and efficient gene targeting using 100 bp of flanking homology region. Yeast 20, 1339–1347.
Griffin, T. A., Barnes, M. G., Ilowite, N. T., Olson, J. C., Sherry, D. D., Gottlieb, B. S., Aronow, B. J., Pavlidis, P., Hinze, C. H., Thornton, S., Thompson, S. D., Grom, A. A., Colbert, R. A., and Glass, D. N. (2009). Gene expression signatures in polyarticular juvenile idiopathic arthritis demonstrate disease heterogeneity and offer a molecular classification of disease subsets. Arthritis Rheum. 60, 2113–2123.
Gudlaugsson, O., Gillespie, S., Lee, K., Vande Berg, J., Hu, J., Messer, S., Herwaldt, L., Pfaller, M., and Diekema, D. (2003). Attributable mortality of nosocomial candidemia, revisited. Clin. Infect. Dis. 37, 1172–1177.
Gustafsson, M., Björkengren, J., and Tegne, J. (2008). “Soft integration of data for reverse engineering,” in International Conference on Systems Biology, Göteborg, 127–127.
Guthke, R., Albrecht, D., Brackhage, A. A., and Möller, U. (2007). Discovery of gene regulatory networks in Aspergillus fumigatus. Lect. Notes Bioinform. 4366, 22–41.
Guthke, R., Moller, U., Hoffmann, M., Thies, F., and Topfer, S. (2005). Dynamic network reconstruction from gene expression data applied to immune response during bacterial infection. Bioinformatics 21, 1626–1634.
Hamon, M. A., and Cossart, P. (2008). Histone modifications and chromatin remodelling during bacterial infections. Cell Host Microbe 4, 100–109.
Han, Y., Kozel, T. R., Zhang, M. X., MacGill, R. S., Carroll, M. C., and Cutler, J. E. (2001). Complement is essential for protection by an IgM and an IgG3 monoclonal antibody against experimental, hematogenously disseminated candidiasis. J. Immunol. 167, 1550–1557.
Hardcastle, T. J., and Kelly, K. A. (2010). Bayseq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics 11, 422. doi:10.1186/1471-2105-11-422
Hecker, M., Goertsches, R. H., Engelmann, R., Thiesen, H. J., and Guthke, R. (2009a). Integrative modelling of transcriptional regulation in response to antirheumatic therapy. BMC Bioinformatics 10, 262. doi:10.1186/1471-2105-10-262
Hecker, M., Lambeck, S., Toepfer, S., van Someren, E., and Guthke, R. (2009b). Gene regulatory network inference: data integration in dynamic models-a review. BioSystems 96, 86–103.
Hinze, C. H., Fall, N., Thornton, S., Mo, J. Q., Aronow, B. J., Layh-Schmitt, G., Griffin, T. A., Thompson, S. D., Colbert, R. A., Glass, D. N., Barnes, M. G., and Grom, A. A. (2010). Immature cell populations and an erythropoiesis gene-expression signature in systemic juvenile idiopathic arthritis: implications for pathogenesis. Arthritis Res. Ther. 12, R123.
Holter, N. S., Maritan, A., Cieplak, M., Fedoroff, N. V., and Banavar, J. R. (2001). Dynamic modelling of gene expression data. Proc. Natl. Acad. Sci. U.S.A. 98, 1693–1698.
Hou, J., Aerts, J., den Hamer, B., van Ijcken, W., den Bakker, M., Riegman, P., van der Leest, C., van der Spek, P., Foekens, J. A., Hoogsteden, H. C., Grosveld, F., and Philipsen, S. (2010). Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 5, e10312. doi:10.1371/journal.pone.0010312
Jaillon, S., Peri, G., Delneste, Y., Fremaux, I., Doni, A., Moalli, F., Garlanda, C., Romani, L., Gascan, H., Bellocchio, S., Bozza, S., Cassatella, M. A., Jeannin, P., and Mantovani, A. (2007). The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J. Exp. Med. 204, 793–804.
Knight, S. A., Vilaire, G., Lesuisse, E., and Dancis, A. (2005). Iron acquisition from transferrin by Candida albicans depends on the reductive pathway. Infect. Immun. 73, 5482–5492.
Lane, S., Birse, C., Zhou, S., Matson, R., and Liu, H. (2001). DNA array studies demonstrate convergent regulation of virulence factors by Cph1, Cph2, and Efg1 in Candida albicans. J. Biol. Chem. 276, 48988–48996.
Le Novere, N., Hucka, M., Mi, H., Moodie, S., Schreiber, F., Sorokin, A., Demir, E., Wegner, K., Aladjem, M. I., Wimalaratne, S. M., Bergman, F. T., Gauges, R., Ghazal, P., Kawaji, H., Li, L., Matsuoka, Y., Villéger, A., Boyd, S. E., Calzone, L., Courtot, M., Dogrusoz, U., Freeman, T. C., Funahashi, A., Ghosh, S., Jouraku, A., Kim, S., Kolpakov, F., Luna, A., Sahle, S., Schmidt, E., Watterson, S., Wu, G., Goryanin, I., Kell, D. B., Sander, C., Sauro, H., Snoep, J. L., Kohn, K., and Kitano, H. (2009). The systems biology graphical notation. Nat. Biotechnol. 27, 735–741.
Linde, J., Wilson, D., Hube, B., and Guthke, R. (2010). Regulatory network modelling of iron acquisition by a fungal pathogen in contact with epithelial cells. BMC Syst. Biol. 4, 148. doi:10.1186/1752-0509-4-148
Lu, X., Kovalev, G. I., Chang, H., Kallin, E., Knudsen, G., Xia, L., Mishra, N., Ruiz, P., Li, E., Su, L., and Zhang, Y. (2008). Inactivation of NuRD component Mta2 causes abnormal T cell activation and lupus-like autoimmune disease in mice. J. Biol. Chem. 283, 13825–13833.
Ma, Y. J., Doni, A., Skjoedt, M. O., Honore, C., Arendrup, M., Mantovani, A., and Garred, P. (2011). Heterocomplexes of mannose-binding lectin and the pentraxins PTX3 or serum amyloid P component trigger cross-activation of the complement system. J. Biol. Chem. 286, 3405–3417.
Manavathi, B., Singh, K., and Kumar, R. (2007). MTA family of coregulators in nuclear receptor biology and pathology. Nucl. Recept. Signal 5, e010.
Marbach, D., Prill, R. J., Schaffter, T., Mattiussi, C., Floreano, D., and Stolovitzky, G. (2010). Revealing strengths and weaknesses of methods for gene network inference. Proc. Natl. Acad. Sci. U.S.A. 107, 6286–6291.
Martchenko, M., Alarco, A. M., Harcus, D., and Whiteway, M. (2004). Superoxide dismutases in Candida albicans: transcriptional regulation and functional characterization of the hyphal-induced SOD5 gene. Mol. Biol. Cell 15, 456–467.
Martinou, A., Koutsioulis, D., and Bouriotis, V. (2002). Expression, purification, and characterization of a cobalt-activated chitin deacetylase (Cda2p) from Saccharomyces cerevisiae. Protein Expr. Purif. 24, 111–116.
Moalli, F., Doni, A., Deban, L., Zelante, T., Zagarella, S., Bottazzi, B., Romani, L., Mantovani, A., and Garlanda, C. (2010). Role of complement and Fcγ receptors in the protective activity of the long pentraxin PTX3 against Aspergillus fumigatus. Blood 116, 5170–5180.
Moalli, F., Paroni, M., Veliz Rodriguez, T., Riva, F., Polentarutti, N., Bottazzi, B., Valentino, S., Mantero, S., Nebuloni, M., Mantovani, A., Bragonzi, A., and Garlanda, C. (2011). The therapeutic potential of the humoral pattern recognition molecule PTX3 in chronic lung infection caused by Pseudomonas aeruginosa. J. Immunol. 186, 5425–5434.
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628.
Murad, A. M., Lee, P. R., Broadbent, I. D., Barelle, C. J., and Brown, A. J. (2000). CIp10, an efficient and convenient integrating vector for Candida albicans. Yeast 16, 325–327.
Netea, M. G., Brown, G. D., Kullberg, B. J., and Gow, N. A. (2008). An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 6, 67–78.
Nobile, C. J., Solis, N., Myers, C. L., Fay, A. J., Deneault, J. S., Nantel, A., Mitchell, A. P., and Filler, S. G. (2008). Candida albicans transcription factor Rim101 mediates pathogenic interactions through cell wall functions. Cell. Microbiol. 10, 2180–2196.
Oberholzer, U., Nantel, A., Berman, J., and Whiteway, M. (2006). Transcript profiles of Candida albicans cortical actin patch mutants reflect their cellular defects: contribution of the Hog1p and Mkc1p signaling pathways. Eukaryot. Cell 5, 1252–1265.
Park, Y. N., and Morschhäuser, J. (2005). Candida albicans MTLα tup1Δ mutants can reversibly switch to mating-competent, filamentous growth forms. Mol. Microbiol. 58, 1288–1302.
Priebe, S., Linde, J., Albrecht, D., Guthke, R., and Brakhage, A. A. (2011). FungiFun: a web-based application for functional categorization of fungal genes and proteins. Fungal Genet. Biol. 48, 353–358.
Raman, T., O’Connor, T. P., Hackett, N. R., Wang, W., Harvey, B. G., Attiyeh, M. A., Dang, D. T., Teater, M., and Crystal, R. G. (2009). Quality control in microarray assessment of gene expression in human airway epithelium. BMC Genomics 10, 493. doi:10.1186/1471-2164-10-493
Rizzetto, L., and Cavalieri, D. (2011). Friend or foe: using systems biology to elucidate interactions between fungi and their hosts. Trends Microbiol. 19, 509–515.
Ruepp, A., Zollner, A., Maier, D., Albermann, K., Hani, J., Mokrejs, M., Tetko, I., Guldener, U., Mannhaupt, G., Munsterkotter, M., and Mewes, H. W. (2004). The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 32, 5539–5545.
Sellam, A., Al-Niemi, T., McInnerney, K., Brumfield, S., Nantel, A., and Suci, P. A. (2009). A Candida albicans early stage biofilm detachment event in rich medium. BMC Microbiol. 9, 25. doi:10.1186/1471-2180-9-25
Skrzypek, M. S., Arnaud, M. B., Costanzo, M. C., Inglis, D. O., Shah, P., Binkley, G., Miyasato, S. R., and Sherlock, G. (2010). New tools at the Candida genome database: biochemical pathways and full-text literature search. Nucleic Acids Res. 38, D428–D432.
Smith, J. J., Deane, N. G., Wu, F., Merchant, N. B., Zhang, B., Jiang, A., Lu, P., Johnson, J. C., Schmidt, C., Bailey, C. E., Eschrich, S., Kis, C., Levy, S., Washington, M. K., Heslin, M. J., Coffey, R. J., Yeatman, T. J., Shyr, Y., and Beauchamp, R. D. (2010). Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology 138, 958–968.
Spira, A., Beane, J. E., Shah, V., Steiling, K., Liu, G., Schembri, F., Gilman, S., Dumas, Y. M., Calner, P., Sebastiani, P., Sridhar, S., Beamis, J., Lamb, C., Anderson, T., Gerry, N., Keane, J., Lenburg, M. E., and Brody, J. S. (2007). Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat. Med. 13, 361–366.
Stark, C., Breitkreutz, B. J., Chatr-Aryamontri, A., Boucher, L., Oughtred, R., Livstone, M. S., Nixon, J., Van Auken, K., Wang, X., Shi, X., Reguly, T., Rust, J. M., Winter, A., Dolinski, K., and Tyers, M. (2011). The BioGRID interaction database: 2011 update. Nucleic Acids Res. 39, D698–D704.
Thewes, S., Kretschmar, M., Park, H., Schaller, M., Filler, S. G., and Hube, B. (2007). In vivo and ex vivo comparative transcriptional profiling of invasive and non-invasive Candida albicans isolates identifies genes associated with tissue invasion. Mol. Microbiol. 63, 1606–1628.
Toepfer, S., Guthke, R., Driesch, D., Woetzel, D., and Pfaff, M. (2007). The NetGenerator algorithm: reconstruction of gene regulatory networks. Lect. Notes Bioinform. 4366, 119–130.
Tokuyasu, K., Ohnishi-Kameyama, M., and Hayashi, K. (1996). Purification and characterization of extracellular chin deacetylase from Colletotrichum lindemuthianum. Biosci. Biotechnol. Biochem. 60, 1598–1603.
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111.
Wachtler, B., Wilson, D., Haedicke, K., Dalle, F., and Hube, B. (2011). From attachment to damage: defined genes of Candida albicans mediate adhesion, invasion and damage during interaction with oral epithelial cells. PLoS ONE 6, e17046. doi:10.1371/journal.pone.0017046
Wang, Q., Diskin, S., Rappaport, E., Attiyeh, E., Mosse, Y., Shue, D., Seiser, E., Jagannathan, J., Shusterman, S., Bansal, M., Khazi, D., Winter, C., Okawa, E., Grant, G., Cnaan, A., Zhao, H., Cheung, N. K., Gerald, W., London, W., Matthay, K. K., Brodeur, G. M., and Maris, J. M. (2006). Integrative genomics identifies distinct molecular classes of neuroblastoma and shows that multiple genes are targeted by regional alterations in DNA copy number. Cancer Res. 66, 6050–6062.
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63.
Werhli, A. V., and Husmeier, D. (2007). Reconstructing gene regulatory networks with Bayesian networks by combining expression data with multiple sources of prior knowledge. Stat. Appl. Genet. Mol. Biol. 6, 15.
Zakikhany, K., Naglik, J. R., Schmidt-Westhausen, A., Holland, G., Schaller, M., and Hube, B. (2007). In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cell. Microbiol. 9, 2938–2954.
Zhang, J., Chiodini, R., Badr, A., and Zhang, G. (2011). The impact of next-generation sequencing on genomics. J. Genet. Genomics 38, 95–109.
Zhao, X., Oh, S. H., Cheng, G., Green, C. B., Nuessen, J. A., Yeater, K., Leng, R. P., Brown, A. J., and Hoyer, L. L. (2004). ALS3 and ALS8 represent a single locus that encodes a Candida albicans adhesin; functional comparisons between Als3p and Als1p. Microbiology 150, 2415–2428.
Zhao, Y., Park, R. D., and Muzzarelli, R. A. (2010). Chitin deacetylases: properties and applications. Mar. Drugs 8, 24–46.
Appendix
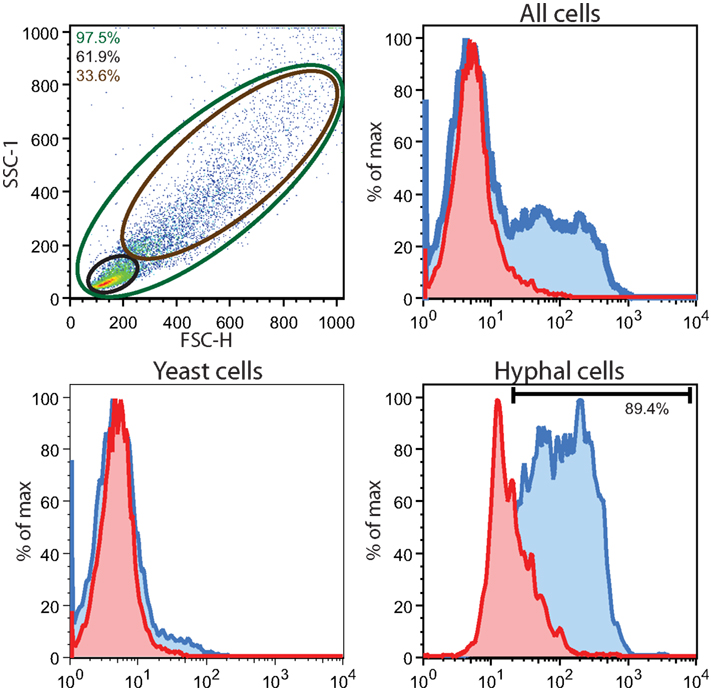
Figure A1. FACS analysis of rmPtx3 binding to C. albicans yeast and hyphal cells. SC5314 after 1-h treatment with rmPtx3. Cells were gated according to morphology based on size, all Candida cells analyzed (green gate), yeast form only (black gate) and hyphal form only (the brown gate). Histograms for untreated cells (red), and treated rmPtx3 and SC5314 (blue) are shown.
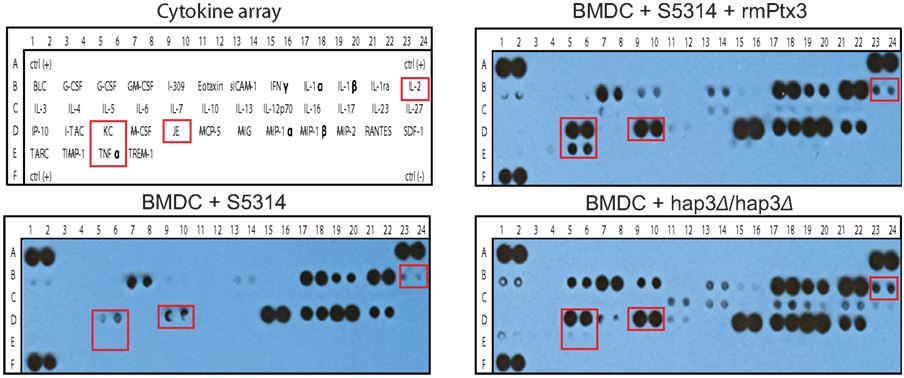
Figure A2. rmPtx3 binds to the C. albicans surface and alters the immune response. The cytokine array panel for M. musculus (R&D systems) where each spot represents an individual cytokine (in duplicate) for supernatants from BMDCs treated fungal cells pre-treated with rmPtx3 after 24 h.
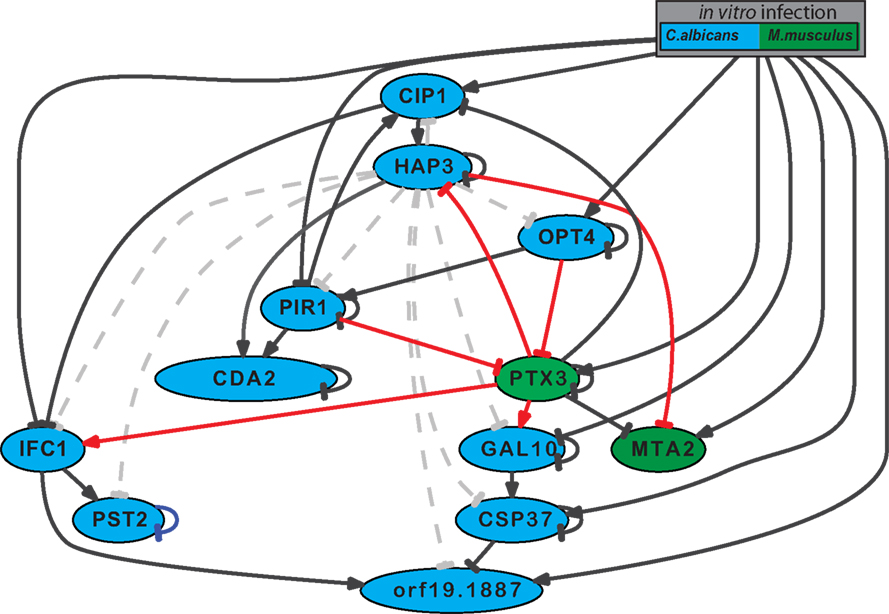
Figure A3. Inferred network model between C. albicans and M. musculus using of candidate Hap3 effector genes. C. albicans (blue) and M. musculus genes (green) included in the model are shown. The following interactions are represented on the model: predicted interactions based on the RNA-seq data set from individual species where no prior knowledge exists (black) or predicted interactions between a C. albicans and M. musculus gene where no prior knowledge exists (red), or where prior knowledge exists but does not corresponds to expression data set (gray, dotted). Here, activation is shown as a pointed arrow and a repression a blunted arrow.
Keywords: host–pathogen, RNA-seq, network inference, modeling, reverse engineering, Candida, dendritic cells
Citation: Tierney L, Linde J, Müller S, Brunke S, Molina JC, Hube B, Schöck U, Guthke R and Kuchler K (2012) An interspecies regulatory network inferred from simultaneous RNA-seq of Candida albicans invading innate immune cells. Front. Microbio. 3:85. doi: 10.3389/fmicb.2012.00085
Received: 08 December 2011; Accepted: 20 February 2012;
Published online: 12 March 2012.
Edited by:
Franziska Mech, Leibniz-Institute for Natural Product Research and Infection Biology – Hans-Knoell-Institute, GermanyReviewed by:
Thomas Dandekar, University of Wuerzburg, GermanyOliver Kurzai, Friedrich Schiller University Jena, Germany
Copyright: © 2012 Tierney, Linde, Müller, Brunke, Molina, Hube, Schöck, Guthke and Kuchler. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Karl Kuchler, Christian Doppler Laboratory for Infection Biology, Max F. Perutz Laboratories, Medical University of Vienna, Campus Vienna Biocenter, Dr. Bohr-Gasse 9/2, A-1030 Vienna, Austria. e-mail: karl.kuchler@meduniwien.ac.at
†Lanay Tierney and Jörg Linde have contributed equally to this work.