 Yanrui Zhang
Yanrui Zhang Taian Jin
Taian Jin Zhangqi Dou
Zhangqi Dou Boxing Wei
Boxing Wei Buyi Zhang
Buyi Zhang Chongran Sun1,3,4*
Chongran Sun1,3,4*- 1Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 2Department of Pathology, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 3Key Laboratory of Precise Treatment and Clinical Translational Research of Neurological Diseases, Hangzhou, Zhejiang, China
- 4Clinical Research Center for Neurological Diseases of Zhejiang Province, Hangzhou, Zhejiang, China
Binding of CD95, a cell surface death receptor, to its homologous ligand CD95L, transduces a cascade of downstream signals leading to apoptosis crucial for immune homeostasis and immune surveillance. Although CD95 and CD95L binding classically induces programmed cell death, most tumor cells show resistance to CD95L-induced apoptosis. In some cancers, such as glioblastoma, CD95-CD95L binding can exhibit paradoxical functions that promote tumor growth by inducing inflammation, regulating immune cell homeostasis, and/or promoting cell survival, proliferation, migration, and maintenance of the stemness of cancer cells. In this review, potential mechanisms such as the expression of apoptotic inhibitor proteins, decreased activity of downstream elements, production of nonapoptotic soluble CD95L, and non-apoptotic signals that replace apoptotic signals in cancer cells are summarized. CD95L is also expressed by other types of cells, such as endothelial cells, polymorphonuclear myeloid-derived suppressor cells, cancer-associated fibroblasts, and tumor-associated microglia, and macrophages, which are educated by the tumor microenvironment and can induce apoptosis of tumor-infiltrating lymphocytes, which recognize and kill cancer cells. The dual role of the CD95-CD95L system makes targeted therapy strategies against CD95 or CD95L in glioblastoma difficult and controversial. In this review, we also discuss the current status and perspective of clinical trials on glioblastoma based on the CD95-CD95L signaling pathway.
Introduction
Glioblastoma (GBM) is the most common and aggressive malignancy in the central nervous system, the efficiency of standard treatment (surgical, radiotherapy, and temozolomide chemotherapy) is limited, and the prognosis is generally poor. Regulating cell death is an attractive target for cancer therapy. CD95 (FAS; APO-1; TNFRSF6) is a member of the tumor necrosis factor (TNF) receptor family of membrane surface proteins ubiquitously expressed in tissues, and its cognate ligand CD95L (FasL/CD178) is expressed primarily by cells of the immune system such as natural killer (NK) cells and activated T lymphocytes. CD95-mediated apoptosis helps to maintain immune system homeostasis and promotes the elimination of malignant cells (1). Although immune cells use CD95L as a mechanism for killing cancer cells, most tumor cells are resistant to CD95L-induced apoptosis. In some cancers, such as GBM, CD95-CD95L can exhibit an atypical function. Previous studies have suggested that CD95 is a specialized death receptor; however, the nonapoptotic functions of CD95 signaling have recently been found to promote tumor cell growth and migration (2). It is essential to explore the dual function of CD95 and the corresponding mechanisms for its application in cancer therapy.
CD95-mediated apoptosis signaling pathway
CD95, a type 1 transmembrane protein, belongs to the TNF receptor superfamily. The extracellular region of CD95 contains three cysteine-rich domains (CRD) that define the ability to recognize and bind CD95 and CD95L. CRD2 and CRD3 of CD95 are involved in CD95L binding, and the preligand assembly domain (PLAD) overlapping CRD1 which is near the N-terminus is required for the assembly of CD95 trimer (3). The death domain (DD) in the intracellular segment of the CD95 molecule is a conserved region of about 80 amino acids, which is crucial for the transmission of apoptotic signals (4). Its ligand, CD95L, is a type II transmembrane protein that can be trimerized by the C-terminal TNF homology domain (THD). The apoptotic ability would be activated when CD95L binds to the death receptor CD95, the binding drives the aggregation of CD95 trimers, which causes DD to cluster together and attracts another protein in the cytosol with the same DD, called Fas-associated protein with death domain (FADD) (5). FADD then ligates the inactive zymogen form of caspase-8 (procaspase-8) through an N-terminal death effector domain (DED) (6), together they constitute the death-inducing signaling complex (DISC) (7), which recruits additional procaspase-8 molecules. Multiple procaspase-8 molecules bind through their tandem DED, thus forming the assembly of DED/caspase-8 filaments (8, 9). The formation of the DED chain drives the dimerization and self-cleavage of procaspase-8 that converts procaspase-8 into activated heterotetramer caspase-8, thus launching executioner caspases (e.g., caspase-3,-6, and-7), ultimately triggering a signaling cascade of apoptosis (Figure 1). However, activation of DR does not produce sufficient amounts of activated caspase-8 to trigger apoptosis in some cases, as has been observed in hepatocytes, pancreatic beta cells, and most cancer cells. In these so-called ‘Type II cells’, mitochondria-dependent intrinsic apoptotic pathways are required to amplify DR-mediated apoptotic signaling. This depends on whether XIAP expression is sufficient to block the enzymatic activities of caspase-3,-9, and -7 (10). The BH3 interacting domain death agonist (Bid) is cleaved by caspase-8 to generate a truncated form (tBid). Translocation of tBid to mitochondria, where it activates pro-apoptotic members of the Bcl-2 family Bax and Bak (11), can mediate mitochondrial outer membrane permeabilization (MOMP), thus releasing cytochrome C, Smac/Diablo and Htra2/OMI into the cytosol (12). This process can also be performed by tBid itself, without relying on Bax/Bak activation (12). The apoptosome is made up of cytochrome C, apoptosis-promoting factor-1 (Apaf1), and procaspase-9 (13), Smac, and Htra2 remove the inhibitory effect of XIAP on caspase-3 (14, 15), leading to caspase-3 activation induction of apoptosis. Thus, unlike type I cells, type II cells can be rescued from CD95-induced apoptosis by inhibiting MOMP (16, 17), for example, GBM overexpress members of the anti-apoptotic Bcl-2 family (18, 19) and downregulate the expression of BAX (20) and pro-apoptotic Bcl-2 proteins. Similarly, disruption of CD95 pro-apoptotic signaling cascades that evade apoptosis has been observed in GBM.
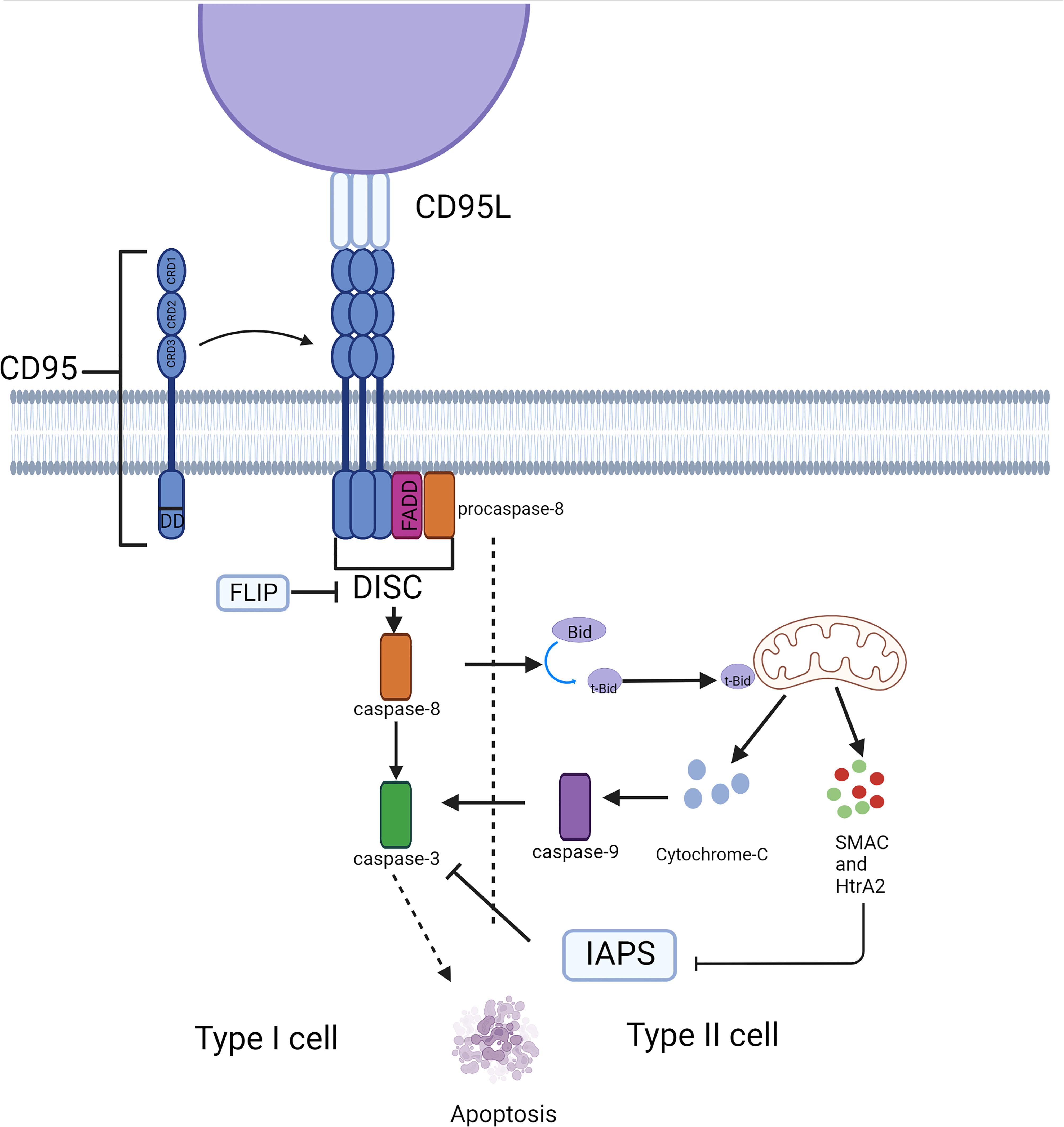
Figure 1 Typical apoptosis signaling pathway mediated by CD95. CD95 induces cell apoptosis through the caspase cascade in different cell types. This process depends on the involvement of mitochondria in type II cells.
CD95-mediated nonapoptotic signaling
The anti-apoptotic function of CD95
The understanding of CD95 activity initially focused on its ability to induce apoptosis, but now it has switched to the nonapoptotic signaling pathway (Figures 2–4). Particularly in cancer, nonapoptotic CD95 signaling has been widely documented and has been associated with cancer cell growth, invasiveness, as well as cancer cell stemness (21).
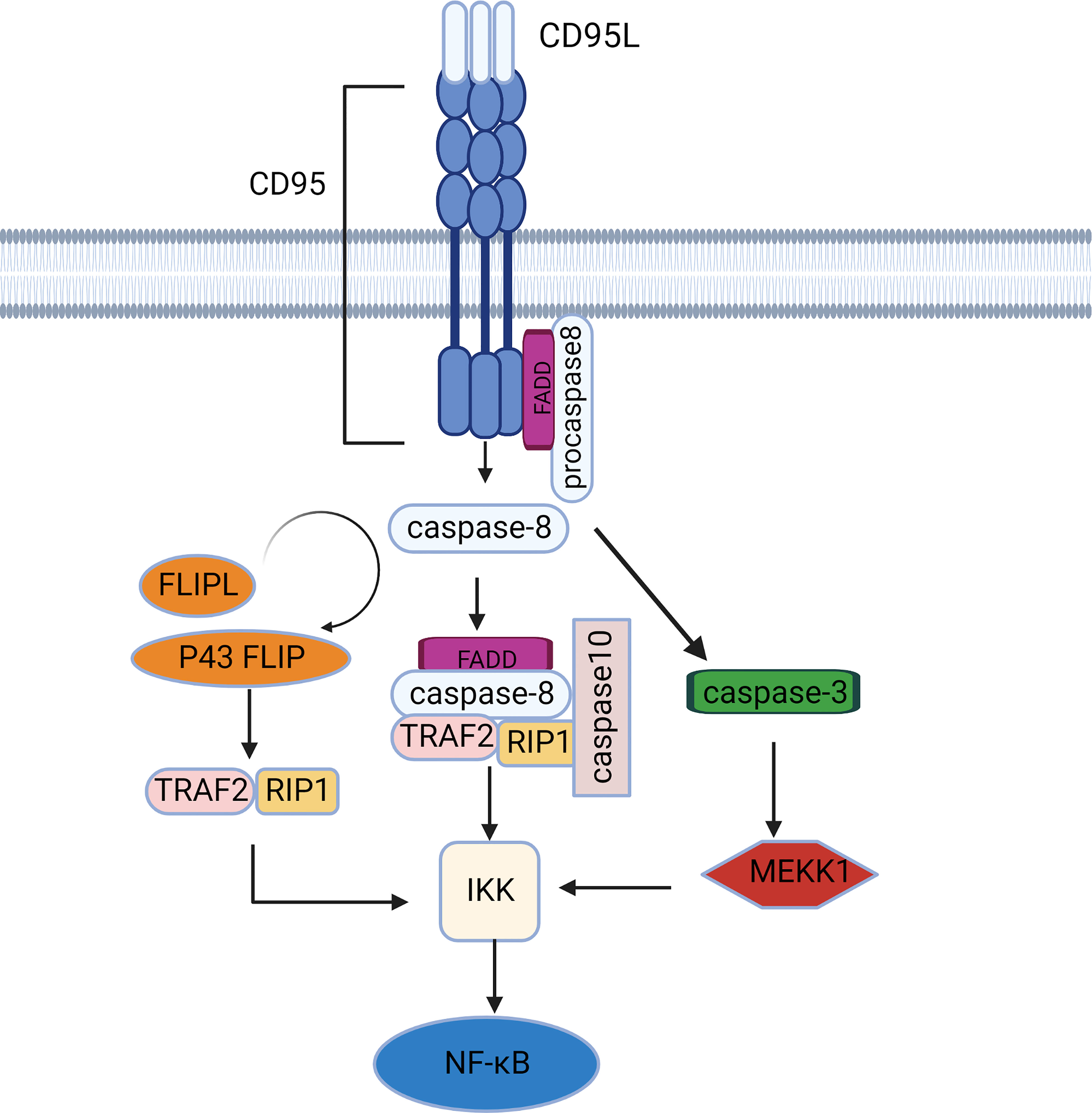
Figure 2 The NF-κB signaling pathway mediated by CD95. CD95 induces caspase-dependent activation of the NF-κB pathway via IKK activation. There are 2 major IKK activation pathways: the RIP1/TRAF2-dependent pathway and the MEKK1 activation pathway.
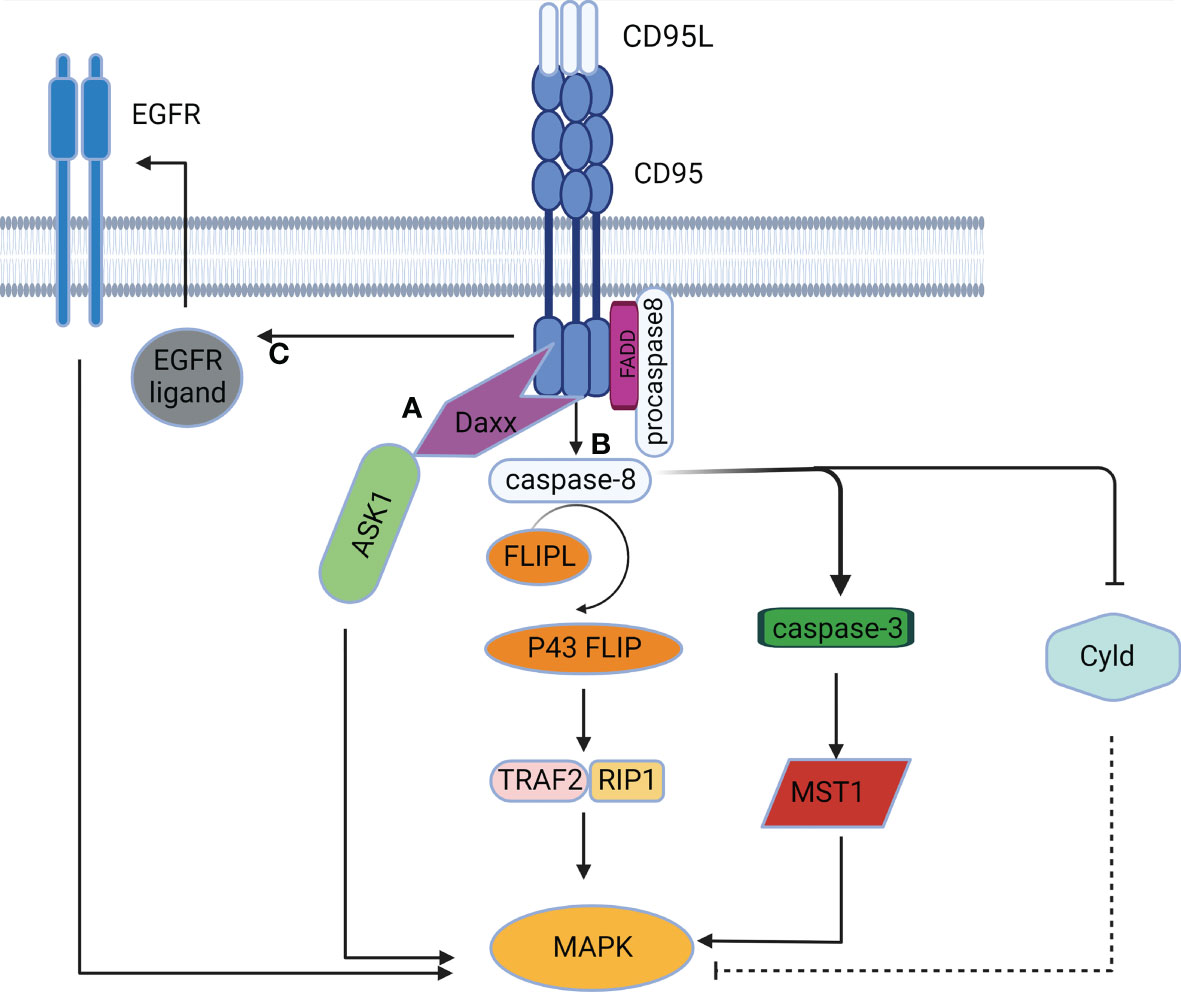
Figure 3 The MAPK signaling pathway mediated by CD95. (A) CD95 induces DD-mediated caspase-independent activation of the MAPK pathway. (B) CD95 induces caspase-dependent activation of the MAPK pathway. (C) CD95 induces activation of the MAPK pathway in a DD-independent manner by stimulating EGFR.
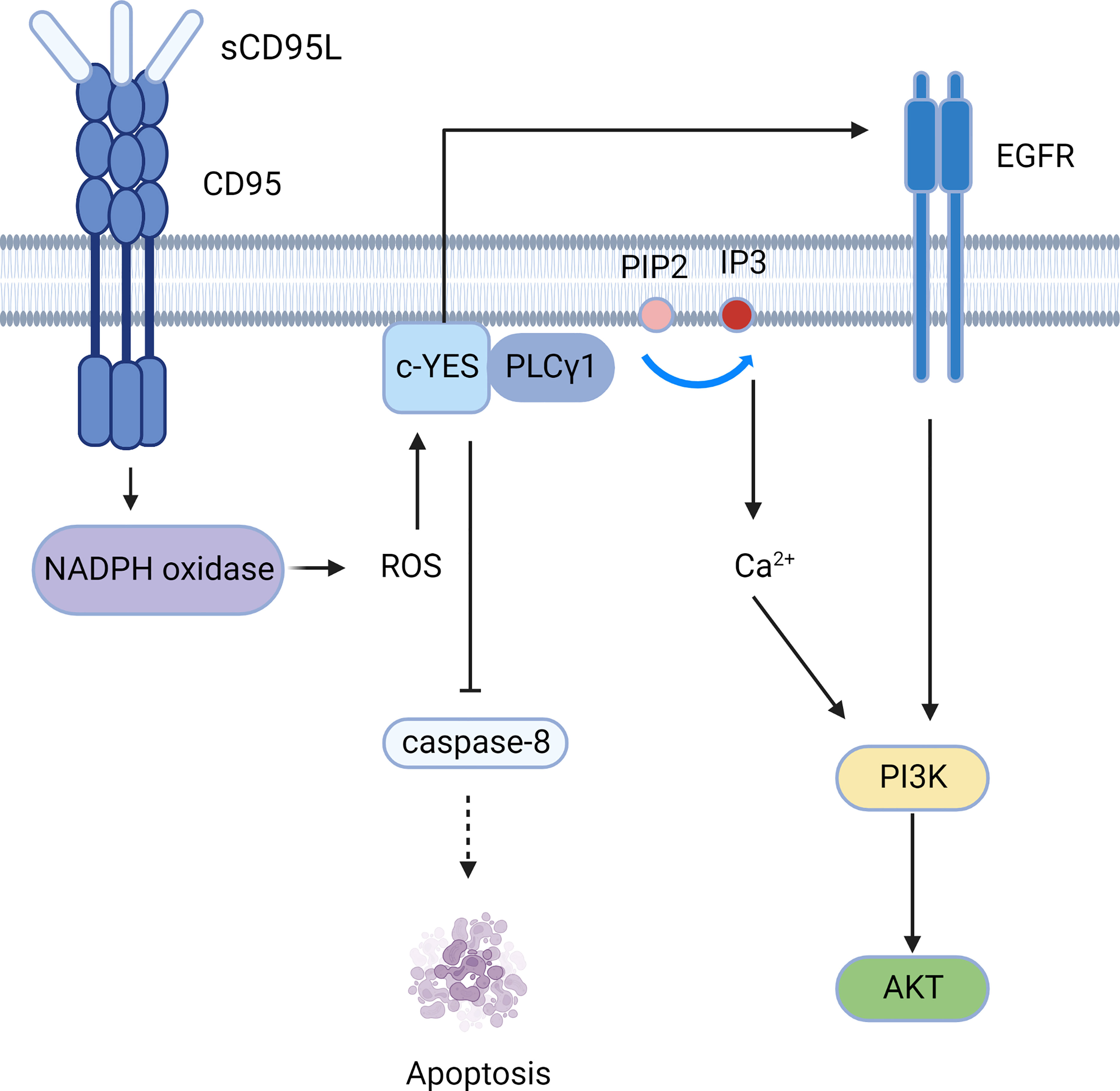
Figure 4 The PI3K-AKT signaling pathway mediated by CD95. CD95 induces the c-YES/calcium/PI3K pathway and c-YES/EGFR/PI3K pathway to mediate cell migration.
Tumor volume and incidence of the mouse with ovarian and liver cancer decreased when CD95 expression was knocked down (22). CD95-mediated Sck/Shc2 activation is essential for pancreatic ductal adenocarcinoma growth and metastasis (23). Balta et al. found that for single tumor cells, CD95 activation promoted their apoptosis. Conversely, the activation of CD95 in tumor cells in tissue promotes their survival, possibly because cell-to-cell contact increases tyrosine kinase activity (24). Recent studies have also provided evidence for the presence of CD95L-independent but CD95-dependent signaling pathways in human glioma initiating cells (Gics) that maintain tumor malignancy (25). The lack of CD95 has been found to enhance immune regulation of tumor cells in triple-negative breast cancer mice, and this suppression of tumor growth is due to enhanced recruitment and activation of NK cells (26). The molecular mechanisms have been elucidated. Independent of CD95L, the C-terminal region of CD95 binds to the Kip1 ubiquitination-promoting complex 2 (KPC2), which in turn recruits the ubiquitin ligase KPC1 and p65, a member of the NF-κB family. NF-κB1 (p105) is ubiquitinated by KPC1 and is degraded to p50 without the transactivation domain, forming a homodimer p50/p50 and thus, inhibits the NF-κB pathway. When CD95 is absent, p50 production is reduced and p65 is released from the plasma membrane to bind to p50 to form a heterodimer that activates the NF-κB pro-inflammatory signaling pathway and regulates cellular immunity (27).
NF-κB signaling pathway
At rest, the inhibitor of NF-κB protein (IκB) combined with NF-κB in an inactive state, activation of IκB kinase (IKK) ubiquitinates, phosphorylates and eventually degrades IκB, transferring NF-κB from the cytoplasm to the nucleus (28). IKK activation is more dependent on caspase-8 activity than CD95 activity, although caspase-8 can be a downstream target of the CD95 signaling pathway (Figure 2). Caspase 8 recruits FADD, TRAF2, RIPK1, and E3 ubiquitin ligases into a complex called FADDosome (29), where effector proteins (e.g., RIP1, TRAF2) bind to the ubiquitin binding domain of IKK in a K63 ubiquitin chain-dependent manner, leading to NF-κB activation and subsequent secretion of pro-inflammatory cytokines. Caspase-10, a homolog of caspase-8, can also be recruited by FADD (30, 31). Early data suggest that caspase-10 is equivalent to caspase-8 as an initiator caspase, leading to cell apoptosis (32). However, recent studies have found that caspase-10 can restrain the activity of caspase-8 in DISC and transform apoptotic signaling into an NF-κB signaling pathway (33). This may be because caspase-10 is a necessary component for the assembly of the FADDosome (34). Furthermore, caspase-8 cleaves its homologue c-FLIPL to produce P43-FLIP that binds NF-κB signaling molecules such as RIP1, TRAF2, and TRAF60 in the absence of CD95 (35). Data also suggest that other cFLIP variants (full-length cFLIPL and cFLIPS) can activate IKK through the interaction of ubiquitinated proteins with IKKγ/NEMO (36). E3 ligases play a key role the degradation of IκB, processing of NF-κB precursors, and activation of the IKK complex. Therefore, the presence of deubiquitinating enzymes such as CYLD cleaves polyubiquitin chains, effectively inhibiting the NF-κB pathway (37, 38).
In addition to promoting inflammation and survival, CD95 controls glioma cell invasion by regulating matrix metalloproteinases (MMP)-2 activation through the NF-κB-TIMP-2 pathway (39). NF-κB is also involved in the expression of decoy receptor (DcR) 3, which inhibits the conduction of the CD95 signal (40). Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is another DR ligand that is highly homologous to CD95L and also activates downstream pathways through FADD and caspase-8. We hypothesize that CD95L can also induce delayed activation of JNK and IKK through caspase-mediated activation of MEKK1 similar to TRAIL, independent of RIP1 and TRAF2 expression. Thus, activation or inhibition of the NF-κB pathway depends on the level of c-FLIPL. When cFLIP was expressed at a low level, fully activated caspase-8 cleaved RIP1 and caspase-3, which was sufficient to inhibit NF-κB signaling and induce apoptosis in type 1 cells. However, in type II cells where caspase-3 is not capable of inducing apoptosis, caspase-3 activates the MEKK1/IKK/NF-κB pathway. In contrast, when cFLIP is overexpressed, caspase-8 with limited activity cannot activate caspase-3, and at the same time, it will cleave RIP1 in a small amount to slow down NF-κB signaling conduction (41).
MAPK signaling pathway
The MAPK chain transfers upstream signals to downstream responding molecules by sequential phosphorylation. The MAPK pathway acts as a pivot in cell proliferation, differentiation, apoptosis, and angiogenesis, and also participates in tumorigenesis (42). CD95 was initially found to activate the MAPK singling pathway in a caspase-independent manner. Death domain-associated protein (Daxx) served as the activator of the Jun NH2-terminal kinase (JNK) pathway. Activation of CD95 induced Daxx to interact with the MAP3K signaling kinase-1 ASK1, thus alleviating the inhibitory intramolecular interaction of ASK1 and activating its kinase activity (43). CD95 can also activate the MAPK singling pathway in a caspase-dependent manner. Caspase-8 and cFLIPL-procaspase-8 heterodimers can trigger MAPK by the formation of p43 FLIP (35), which is associated with the Raf-MAPK cascade reaction (44). Furthermore, caspase-8 can cleave Cyld (45), resulting in the activation of JNK, p38 and ERK signaling (46, 47). Caspase-3 and caspase-7 also participate in CD95-induced MAPK activation by cleaving the mammalian 20-like sterile serine/threonine kinase 1 (MST1) to generate active fragments of MAPK (48). Additionally, CD95 can activate the MAPK pathway independently of DD (Figure 3), since CD95 can induce EGFR activation to mediate subsequent activation of ERK (49). Furthermore, as expressed in the NF-κB signaling pathway above, CD95L can activate the JNK pathway in a TRAIL-like mechanism (41).
PI3K-Akt signaling pathway
The soluble form of CD95L (sCD95L) binds to the receptor and cannot form a DISC, but instead forms a complex called motion-induced signaling complex (MISC); MISC produces ROS through NADPH oxidase (for example, Nox3) (50). Src kinases such as c-YES are subsequently activated, followed by recruitment of PLCγ1 to the plasma membrane, where PLCγ1 hydrolyzes PIP2 to produce IP3 and DAG. Then IP3 activates the intracellular calcium response. Calcium influx can be promoted by CD95-activated Orai1 channels, and elevated calcium concentrations activate DISC inhibitors, such as PKCβ2 (51). Therefore, CD95 can mediate tumor invasion and migration through the c-YES/calcium/PI3K pathway (52).Moreover, c-YES stimulates EGFR in the absence of EGF (53), resulting in activation of the PI3K/Akt signaling pathway (Figure 4). Apart from PLCγ1, Src kinases can phosphorylate other MISC components, including TRIP6, which is involved in NF-κB activation and cell migration (54). Src kinases also phosphorylate caspase-8 at tyrosine 380 (Y380), preventing downstream activation of the caspase apoptotic signaling cascade (55).
Tumor migration and invasion can also be mediated by membrane-bound CD95L (mCD95L). Aggregation of CD95, c-YES, and PI3K has been confirmed in GBM cells, and thus, activation of the PI3K/Akt/GSK3β/MMP pathways can mediate invasion (56).
Disruption of CD95-mediated apoptotic signaling in GBMs
Reduced membrane expression of the CD95 receptor
Simultaneous expression of CD95 and CD95L has been detected in GBMs (57), but GBM has often showed resistance to CD95-mediated apoptosis. Mutations in the CD95 gene have been detected in hematological tumors and some solid tumors, and most are clustered to exons 8 and 9, which encode a major part of the intracellular region of CD95, resulting in the impediment of FADD recruitment (58).
GBM can also modulate CD95 availability on the cell membrane to inhibit CD95-induced apoptosis. The protein tyrosine phosphatase non-receptor 13 (PTPN13), also known as FAP-1, is a widely studied inhibitor of CD95 (59). FAP-1 is highly expressed in GBM (60) and binds to CD95 and to endosome-associated trafficking regulator 1 (ENTR1), thus achieving the allowing the association of CD95 and ENTR1. ENTR1, in turn, promotes CD95 transport to lysosomes (61), sequestering CD95 from the cell membrane and therefore inhibiting cell apoptosis. Additionally, FAP-1 also mediates multiple CD95-independent signaling pathways to inhibit cell proliferation and migration in solid tumors, and thus, plays an important role in cell apoptosis (62).
Lipid rafts are dynamic microdomains of plasma membranes, rich in cholesterol and sphingomyelin (63). Lipid rafts act as a skeleton to isolate and localize cell signaling. After activation, CD95 is recruited into lipid rafts to promote protein-protein interactions and the transmission of apoptotic signals (64). CD95-mediated non-apoptotic PI3K/Akt signaling inhibits CD95 aggregation in lipid rafts (65), thereby promoting tumor survival. In addition, lipid rafts are responsible for the invasion of GBM by collecting CD44 (66) and connexin 43 (67).
The function of a protein can be affected by its post-translational modifications, such as glutathionylation, glycosylation, and nitrosylation, which are conducive to assembly of DISC; instead, phosphorylation inhibits the formation of DISC (68). Palmitoylation positions CD95 in rafts, which is required for effective internalization of the CD95 receptor and subsequent caspase cascade activation (69). The DHHC family is a group of proteins related to protein palmitoylation, and most of these proteins have protein acyltransferase activity. Most DHHC members are expressed at a higher level in GBM, and generally promote tumor survival by regulating CD95-independent signaling pathways (70, 71). In contrast, ZDHHC7, ZDHHC11, and ZDHHC22 are down-regulated in GBM cells (71). ZDHHC7 is the main palmitoyltransferase of residue Cys199 of CD95. Limited CD95 palmitoylation reduces its membrane localization, hindering its distribution in lipid rafts (72).
Interference with CD95-CD95L binding
The phenomenon of gene alternative splicing refers to the formation of different transcripts due to various splicing methods of mRNA precursors (choosing different splicing site combinations). It is a basic and important regulatory mechanism in eukaryotes and controls the expression of cancer-related proteins (73). CD95 premRNA performs alternative splicing by cutting exon 6 which encodes the transmembrane domain to produce soluble isomers sCD95 (74). Soluble CD95 is antiapoptotic by competing with mCD95 for CD95L. Overexpression of sCD95 has been detected in a wide range of cancers (75, 76). Recently, hypoxic microenvironments were found to modulate CD95 splicing and promote the production of anti-apoptotic sCD95 mRNA (77), possibly due to reduced interactions of the splicing factor U2AF-RNA in hypoxic cells (78).
Similarly, CD95L is also available in two forms, but sCD95L is formed by MMP-mediated proteolytic cleavage. sCD95L cannot activate CD95 as mCD95L does (79). It cannot form oligomers with CD95 and, therefore, it cannot induce DISC formation (80). Inhibition of mCD95 cleavage by MMP enhances CD95-mediated apoptosis (81). In addition, it can activate motor signals, which will be described below.
DcR belongs to the TNF receptor family, it is a soluble protein associated with the suppression of death receptors, which overexpression leads to immune escape. DcR3 has been detected in malignant gliomas, and its abnormal expression can inhibit CD95L-induced apoptosis (82, 83). DCR3 relies on four CRDs at the N-terminal to bind CD95L, therefore, three DcR3 and CD95L trimer form the heterohexameric complex. By recognizing the invariant backbone and side chain atom of the ligand, DcR3 specifically binds to CD95L, LIGHT, and TL1A (84). Due to the lack of transmembrane domain, DcR3 disturbs the binding of these receptors to ligands through competitive inhibition, while it also conducts the signaling pathways for tumor cell growth, invasion and epithelial-mesenchymal transition (EMT) (85, 86). Furthermore, through its CD95-independent properties, DcR3 induces local immune suppression, mainly through suppression of Th1 and macrophage propensity to M2 in the tumor microenvironment (87). It was also found to promote effector T cell apoptosis by reverse signaling (88); thus, further enhancing the immune escape of tumor cells.
Blocking caspase-8 activation
The recruitment of procaspase-8 by FADD and subsequent assembly of DISC determine the fate of cell apoptosis. Caspase-8 and FADD mRNA and protein expression levels were lower in GBM tissues than in normal brain tissues (89). Furthermore, caspase-8 was silenced by DNA methylation and gene deletion in pediatric neuroblastoma amplified with MYCN (90). Caspase-8 ability is also highly regulated by post-translational modifications (91), of which phosphorylation is typical (55).
The antiapoptotic protein c-FLIP is a protein containing a DED, which is a homologue of caspase-8 and is overexpressed in a variety of tumors (92–96). The three common isoforms are the short, Raji, and long isoforms of cFLIP (cFLIPS, cFLIPR, and cFLIPL). All of these proteins contain two death effector domains and use them to combine with FADD, recruiting caspase-8/-10 at the DISC. cFLIPL also has a caspase-like domain but lacks a key active site residue (97). CFLIPS has been verified as a dedicated inhibitor of apoptosis, which can form an inactive heterodimer with procaspase-8 (98), and the cleavage of procaspase-8 can be completely blocked (99). There is also evidence that cFLIPS can block caspase-8 activation by breaking the caspase-8 filament to prevent dimerization of the caspase-8 catalytic domain on the DISC (100).
The pro-apoptotic functions of cFLIPL were also verified. Pro-apoptotic activity is induced when expressed at low levels, and the formation of heterodimers with procaspase-8 allows the activation of caspase-8. It is worth noting that the formation of the procaspase-8: cFLIPL heterodimer is preferential to the procaspase-8 homodimer. However, when cFLIPL is highly expressed, c-FLIP without catalytic activity will compete with procaspase-8 for DISC, thus inhibiting apoptosis (101). In addition, a fragment named p43-FLIP is obtained from cleaved c-FLIPL. It interacts with RIP1 and TRAF2 to activate NF-κB (35). Conversely, the expression of c-FLIPL determines the direction of NF-κB as discussed above. It also plays a pseudo-enzyme function independent of caspase-8 (102). Thus, c-FLIPL balances cell survival and death in a complex manner.
For these reasons, the activity of caspase-8 is limited in tumors. Incomplete activation of caspase-8 in tumors contributes to the induction of minority MOMP (103), a limited number of mitochondria undergo MOMP, and the amount of cytochrome c/SMAC released is insufficient to trigger apoptosis, but sufficient to activate downstream caspases at sublethal levels. Subsequent deficient endonuclease activation causes the accumulation of DNA damage without cell death, destabilizing the genome, and promoting tumorigenesis (104, 105).
Upregulation of apoptosis inhibitors
Inhibitors of apoptosis proteins (IAP), characterized by at least one baculoviral IAP repeat (BIR) domain, negatively regulate apoptosis by inhibiting caspase activation. It is frequently observed to be upregulated in GBM samples (106). There are eight proteins in the family, of which XIAP has the most potent antiapoptotic properties and can directly contact and inhibit caspase-3, 7, and 9. XIAP inhibitors have been shown to cause caspase-dependent apoptosis (107). Patients with GBM have a lower survival rate with higher expression of the expression of the XIAP protein (106).
Unlike XIAP, cIAP-1 and cIAP-2 could not directly inhibit caspases, cIAP-1, cIAP-2, as well as XIAP have the same RING domains and possess the Ubiquitin protein ligase (E3) Ubiquitin associated activity (UBA) domain, which mediates caspase-3 and caspase-7 (108). They cause SMAC ubiquitination and subsequent degradation (109). Survivin/BIRC5, the smallest member of the IAP family of proteins, contains only one BIR domain and is the most upregulated IAP in a variety of tumors (110). Survivin binds to Smac to prevent this molecule from inhibiting XIAP, but also exerts antiapoptotic activity based on the ability to protect XIAP from ubiquitination (111). Based on the characteristic inhibition of caspase activity by IAP, an IAP antagonist, also known as a Smac mimetic, has the potential to sensitize tumor cells to apoptosis. However, IAP also influences other cellular processes, such as the regulation of ubiquitin-dependent NF-κB activation for cell survival (112). The interaction with the tumor microenvironment and the crosstalk with various cell signaling pathways lead to the complex role of IAP in cancer and immune regulation (113). The therapeutic results of IAP antagonists need to be further studied.
Powerlessness of endonucleases
Even if the executioner caspases are successfully activated, apoptosis may not be achieved. Apoptotic hydrolysis of DNA requires endonuclease DFF40/CAD, and activation of DFF40/CAD requires its inhibitor ICD to be cleaved and inhibited by caspase-3. This step is limited in the cytosol, and thus the cytosolic level of DFF40/CAD is a determinant for achieving a complete apoptotic reaction (114). Due to abnormal accumulation of DFF40/CAD protein in the nucleus, GBM cells may not be able to undergo apoptosis (115).
Immune privilege and tumor counterattack
CD95 is ubiquitously expressed in the human body and is presented as low tissue specificity. In contrast, CD95L is characteristically expressed in immune cells such as cytotoxic T lymphocytes and NK cells and has long been recognized an essential part of immune homeostasis and immune elimination. CD95L has also been reported to be constitutively expressed in immune-privileged tissues such as the eyes and testes. They mediate apoptosis of immune cells through CD95L, which renders them unable to respond to foreign antigens, including graft antigens (116). Astrocytes in healthy human brains do not express CD95L, but it is expressed by astrocytoma (117). Therefore, it has been proposed that tumors can desensitize themselves to apoptosis and express CD95L to mediate immune escape and realize immune counterattack. This effect is called a ‘tumor counterattack’. However, contradictory phenomena such as rejection and immune induction caused by CD95L expression in grafts or tumor cells have also been reported (118). In recent years, there has been a more profound and comprehensive understanding of tumor counterattack, whereby immune cell killing is not mediated by tumor CD95L, but by CD95L expressed in the tumor microenvironment. CD95L has been reported to be expressed in the human tumor endothelium. These CD95L-expressing endothelial cells selectively kill CD8+ T cells rather than regulatory T cells and establish tumor immune tolerance (119). Some cells educated in tumor microenvironments such as cancer-associated fibroblast (CAF), myeloid-derived suppressor cell (MDSC), microglia, and macrophages also express CD95L (120–122). Therefore, inhibition of the CD95-CD95L pathway in the tumor microenvironment may be a potential therapeutic strategy to improve the efficacy of immunotherapy.
CD95 targeted therapy
Due to the typical pro-apoptotic function of CD95, targeting of the CD95 signaling pathway has been the focus of cancer therapy research. However, CD95 agonists have severe hepatotoxicity (123). To avoid the toxicity caused by its Fc segment, “Mega-Fas-Ligand”, a synthetic CD95 ligand, now known as APO010, was developed. The potential of APO010 to induce human glioma cell apoptosis has been observed in vitro (124, 125), but it is less effective than expected in vivo (126). A phase I trial (ClinicalTrials.gov Identifier: NCT00437736) is underway to determine the recommended dose of APO010.
As the understanding of CD95 tumorigenesis and immune cell killing functions has matured in recent years, inhibition rather than activation of CD95 signaling seems to be a better solution for GBM treatment. APG101 (also known asasunercept) is composed of the extracellular domain of CD95 and the Fc segment of the IG1 antibody, and acts as a CD95 inhibitor. Therefore, APG101 can specifically block CD95L on the tumor cell membrane and the vascular endothelial cell membrane, and the function of CD95L in promoting the invasion and apoptosis of activated T lymphocytes by glioma cells can be neutralized (127). We summarize the phase I/II clinical studies of APG101 in Table 1. Early trials in healthy subjects have demonstrated its safety (128). A phase I study showed that APG101 treatment significantly improved PFS with patients with GBM compared to standard radiotherapy and temozolomide alone, and hypomethylation of the CpG2 site within the CD95L promoter resulted in improved benefits (132).

Table 1 Phase I/II trials of APG101 and their results.
Discussion
CD95-mediated apoptosis and anti-apoptotic signaling within the tumor, contribute to the suppression of apoptotic pathways and the enhancement of survival pathways. Tumor cells also use CD95L expressed in the microenvironment to kill immune cells. Given the above understanding, inhibiting rather than activating the CD95 signals is a better strategy. Because CD95 and its downstream signaling molecules are involved in multiple pathways, direct targeting of effector molecules is also a good option, such as thermal therapy, which promotes the generation of the generation of cytochrome C to induce apoptosis in cancer cells (133). Another factor to consider is the complexity of the interaction between CD95 signaling and other pathways of cell death, such as caspase-8 activation to inhibit necrotizing apoptosis (134). Nonetheless, apoptosis and the cell cycle are closely related and their mutual influence should not be ignored.
Knockdown of CD95-CD95L signaling by si/shRNA induces tumor cell death, this phenomenon called Death-induced by CD95R/L elimination (DICE) has been reported in tumors (135). Putzbach et al. updated the DICE concept to Death-induced by survival gene elimination (DISE), confirming that cell death is not actually due to CD95 or CD95L knockdown. In fact, si/shRNA derived from CD95/CD95L silenced a set of surviving genes through a specific 6mer seed sequence (position 2-7 of the guide strand), resulting in the DISE (136). Furthermore, it has been confirmed that the expression of CD95L mRNA itself was toxic to cells through DISE (137). This could explain the inefficiency of exogenous CD95/CD95L or CD95 agonists, as they fail to trigger DISE. DISE relies on the involvement of RNA induced silencing complex (RISC) where miRNAs target mRNAs. Therefore nonspecific down-regulation of miRNA levels in cancer cells allows the release of RISCs occupied by them and then triggers DISE; whereas in normal tissues, most highly expressed miRNAs carry seed regions with low guanine content, making them less toxic to survival genes (138). Thus, DISE may induce cancer cell death. In vivo induction experiments with DISE have confirmed that normal tissues were not affected by toxicity, thus providing a partial basis for the safety of the strategy (139). As DISE toxicity is independent of CD95L or CD95 receptor expression, the combination of induced overexpression of CD95L and inhibition of CD95 signaling could represent a potential cancer treatment in the future.
Author contributions
YZ was responsible for manuscript writing; TJ contributed to the organization of content; ZD and BW collected relevant data; BZ provided guidance on pathology; CS provided ideas, reviewed, and edited the draft. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Zhejiang Science and Technology Plan Project Key R&D Plan (2021C03067).
Acknowledgments
We wish to thank Qimi Cai from Zhejiang University for support and encouragement of this review. All figures in this review were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
GBM, Glioblastoma; TNF, Tumor necrosis factor; NK, Natural killer; CRD, Cysteine-rich domains; FADD, Fas-associated protein with death domain; DED, Death effector domain; DISC, Death-inducing signaling complex; tBid, Truncated form of the BH3 interacting domain death agonist; MOMP, Mitochondrial outer membrane permeabilization; IκB, Inhibitor of NF-κB protein; IKK, IκB kinase; MMP, Matrix metalloproteinases; DcR, Decoy receptor; TRAIL, Tumor necrosis factor-related apoptosis-inducing ligand; Daxx, Death domain-associated protein; JNK, Jun NH2-terminal kinase; sCD95L, Soluble CD95L; MISC, Motion-induced signaling complex; mCD95L, Membrane-bound CD95L; IAP, Inhibitors of apoptosis proteins; DISE, Death-induced by survival gene elimination.
References
1. Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity (2009) 30:180–92. doi: 10.1016/j.immuni.2009.01.001
2. Yi F, Frazzette N, Cruz AC, Klebanoff CA, Siegel RM. Beyond cell death: New functions for tnf family cytokines in autoimmunity and tumor immunotherapy. Trends Mol Med (2018) 24:642–53. doi: 10.1016/j.molmed.2018.05.004
3. Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science (2000) 288:2351–4. doi: 10.1126/science.288.5475.2351
4. Feinstein E, Kimchi A, Wallach D, Boldin M, Varfolomeev E. The death domain: a module shared by proteins with diverse cellular functions. Trends Biochem Sci (1995) 20:342–4. doi: 10.1016/s0968-0004(00)89070-2
5. Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of fas and initiates apoptosis. Cell (1995) 81:505–12. doi: 10.1016/0092-8674(95)90071-3
6. Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death–inducing signaling complex. Cell (1996) 85:817–27. doi: 10.1016/s0092-8674(00)81266-0
7. Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, et al. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J (1997) 16:2794–804. doi: 10.1093/emboj/16.10.2794
8. Fu TM, Li Y, Lu A, Li Z, Vajjhala PR, Cruz AC, et al. Cryo-EM structure of caspase-8 tandem DED filament reveals assembly and regulation mechanisms of the death-inducing signaling complex. Mol Cell (2016) 64:236–50. doi: 10.1016/j.molcel.2016.09.009
9. Dickens LS, Boyd RS, Jukes-Jones R, Hughes MA, Robinson GL, Fairall L, et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell (2012) 47:291–305. doi: 10.1016/j.molcel.2012.05.004
10. Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature (2009) 460:1035–9. doi: 10.1038/nature08229
11. Sekar G, Ojoawo A, Moldoveanu T. Protein-protein and protein-lipid interactions of pore-forming BCL-2 family proteins in apoptosis initiation. Biochem Soc Transs (2022) 50:1091–103. doi: 10.1042/BST20220323
12. Flores-Romero H, Hohorst L, John M, Albert MC, King LE, Beckmann L, et al. BCL-2-family protein tBID can act as a BAX-like effector of apoptosis. EMBO J (2022) 41:e108690. doi: 10.15252/embj.2021108690
13. Dorstyn L, Akey CW, Kumar S. New insights into apoptosome structure and function. Cell Death Differ (2018) 25:1194–208. doi: 10.1038/s41418-017-0025-z
14. Abbas R, Larisch S. Targeting XIAP for promoting cancer cell death-the story of ARTS and SMAC. Cells (2020) 9(3):663. doi: 10.3390/cells9030663
15. Tait SW, Green DR. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol (2013) 5(9):a008706. doi: 10.1101/cshperspect.a008706
16. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol (2019) 20:175–93. doi: 10.1038/s41580-018-0089-8
17. Zheng JH, Viacava Follis A, Kriwacki RW, Moldoveanu T. Discoveries and controversies in BCL-2 protein-mediated apoptosis. FEBS J (2016) 283:2690–700. doi: 10.1111/febs.13527
18. Tirapelli D, Lustosa IL, Menezes SB, Franco IM, Rodrigues AR, Peria FM, et al. High expression of XIAP and bcl-2 may inhibit programmed cell death in glioblastomas. Arq Neuropsiquiatr (2017) 75:875–80. doi: 10.1590/0004-282X20170156
19. Blahovcova E, Richterova R, Kolarovszki B, Dobrota D, Racay P, Hatok J. Apoptosis-related gene expression in tumor tissue samples obtained from patients diagnosed with glioblastoma multiforme. Int J Mol Med (2015) 36:1677–84. doi: 10.3892/ijmm.2015.2369
20. Wang PG, Li YT, Pan Y, Gao ZZ, Guan XW, Jia L, et al. Lower expression of bax predicts poor clinical outcome in patients with glioma after curative resection and radiotherapy/chemotherapy. J Neurooncol (2019) 141:71–81. doi: 10.1007/s11060-018-03031-9
21. Richards DM, Merz C, Gieffers C, Krendyukov A. CD95L and anti-tumor immune response: current understanding and new evidence. Cancer Manag Res (2021) 13:2477–82. doi: 10.2147/CMAR.S297499
22. Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C, et al. CD95 promotes tumour growth. Nature (2010) 465:492–6. doi: 10.1038/nature09075
23. Teodorczyk M, Kleber S, Wollny D, Sefrin JP, Aykut B, Mateos A, et al. CD95 promotes metastatic spread via sck in pancreatic ductal adenocarcinoma. Cell Death Differ (2015) 22:1192–202. doi: 10.1038/cdd.2014.217
24. Gülcüler Balta GS, Monzel C, Kleber S, Beaudouin J, Balta E, Kaindl T, et al. 3D cellular architecture modulates tyrosine kinase activity, thereby switching CD95-mediated apoptosis to survival. Cell Rep (2019) 29:2295–306.e6. doi: 10.1016/j.celrep.2019.10.054
25. Quijano-Rubio C, Silginer M, Weller M. CD95 gene deletion may reduce clonogenic growth and invasiveness of human glioblastoma cells in a CD95 ligand-independent manner. Cell Death Discovery (2022) 8:341. doi: 10.1038/s41420-022-01133-y
26. Qadir AS, Guégan JP, Ginestier C, Chaibi A, Bessede A, Charafe-Jauffret E, et al. CD95/Fas protects triple negative breast cancer from anti-tumor activity of NK cells. iScience (2021) 24:103348. doi: 10.1016/j.isci.2021.103348
27. Guégan JP, Pollet J, Ginestier C, Charafe-Jauffret E, Peter ME, Legembre P. CD95/Fas suppresses NF-κB activation through recruitment of KPC2 in a CD95L/FasL-independent mechanism. iScience (2021) 24:103538. doi: 10.1016/j.isci.2021.103538
28. Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Ann Rev Immunol (2000) 18:621–63. doi: 10.1146/annurev.immunol.18.1.621
29. Kreuz S, Siegmund D, Rumpf JJ, Samel D, Leverkus M, Janssen O, et al. NFkappaB activation by fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J Cell Biol (2004) 166:369–80. doi: 10.1083/jcb.200401036
30. Kischkel FC, Lawrence DA, Tinel A, LeBlanc H, Virmani A, Schow P, et al. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol Chem (2001) 276:46639–46. doi: 10.1074/jbc.M105102200
31. Sprick MR, Rieser E, Stahl H, Grosse-Wilde A, Weigand MA, Walczak H. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J (2002) 21:4520–30. doi: 10.1093/emboj/cdf441
32. Milhas D, Cuvillier O, Therville N, Clavé P, Thomsen M, Levade T, et al. Caspase-10 triggers bid cleavage and caspase cascade activation in FasL-induced apoptosis. J Biol Chem (2005) 280:19836–42. doi: 10.1074/jbc.M414358200
33. Horn S, Hughes MA, Schilling R, Sticht C, Tenev T, Ploesser M, et al. Caspase-10 negatively regulates caspase-8-mediated cell death, switching the response to CD95L in favor of NF-κB activation and cell survival. Cell Rep (2017) 19:785–97. doi: 10.1016/j.celrep.2017.04.010
34. Mohr A, Deedigan L, Jencz S, Mehrabadi Y, Houlden L, Albarenque SM, et al. Caspase-10: a molecular switch from cell-autonomous apoptosis to communal cell death in response to chemotherapeutic drug treatment. Cell Death Differ (2018) 25:340–52. doi: 10.1038/cdd.2017.164
35. Koenig A, Buskiewicz IA, Fortner KA, Russell JQ, Asaoka T, He YW, et al. The c-FLIPL cleavage product p43FLIP promotes activation of extracellular signal-regulated kinase (ERK), nuclear factor κB (NF-κB), and caspase-8 and T cell survival. J Biol Chem (2014) 289:1183–91. doi: 10.1074/jbc.M113.506428
36. Baratchian M, Davis CA, Shimizu A, Escors D, Bagnéris C, Barrett T, et al. Distinct activation mechanisms of NF-κB regulator inhibitor of NF-κB kinase (IKK) by isoforms of the cell death regulator cellular FLICE-like inhibitory protein (cFLIP). J Biol Chem (2016) 291:7608–20. doi: 10.1074/jbc.M116.718122
37. Harhaj EW, Dixit VM. Regulation of NF-κB by deubiquitinases. Immunol Rev (2012) 246:107–24. doi: 10.1111/j.1600-065X.2012.01100.x
38. Napetschnig J, Wu H. Molecular basis of NF-κB signaling. Annu Rev Biophys (2013) 42:443–68. doi: 10.1146/annurev-biophys-083012-130338
39. Wisniewski P, Ellert-Miklaszewska A, Kwiatkowska A, Kaminska B. Non-apoptotic fas signaling regulates invasiveness of glioma cells and modulates MMP-2 activity via NFkappaB-TIMP-2 pathway. Cell Signal (2010) 22:212–20. doi: 10.1016/j.cellsig.2009.09.016
40. Hou Y, Liang D, Liu Y, Chen H, Lou X. Up-regulation of DcR3 in microbial toxins-stimulated HUVECs involves NF-κB signalling. BMC Biochem (2018) 19:13. doi: 10.1186/s12858-018-0102-z
41. Zhang L, Dittmer MR, Blackwell K, Workman LM, Hostager B, Habelhah H. TRAIL activates JNK and NF-κB through RIP1-dependent and -independent pathways. Cell Signal (2015) 27:306–14. doi: 10.1016/j.cellsig.2014.11.014
42. Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med (2020) 19:1997–2007. doi: 10.3892/etm.2020.8454
43. Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein daxx. Science (1998) 281:1860–3. doi: 10.1126/science.281.5384.1860
44. Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, et al. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and erk signaling pathways. Curr Biol (2000) 10:640–8. doi: 10.1016/s0960-9822(00)00512-1
45. O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol (2011) 13:1437–42. doi: 10.1038/ncb2362
46. Ji YX, Huang Z, Yang X, Wang X, Zhao LP, Wang PX, et al. The deubiquitinating enzyme cylindromatosis mitigates nonalcoholic steatohepatitis. Nat Med (2018) 24(2):213–23. doi: 10.1038/nm.4461
47. Zhu G, Herlyn M, Yang X. TRIM15 and CYLD regulate ERK activation via lysine-63-linked polyubiquitination. Nat Cell Biol (2021) 23:978–91. doi: 10.1038/s41556-021-00732-8
48. Song JJ, Lee YJ. Differential cleavage of Mst1 by caspase-7/-3 is responsible for TRAIL-induced activation of the MAPK superfamily. Cell Signal (2008) 20:892–906. doi: 10.1016/j.cellsig.2008.01.001
49. Farley SM, Purdy DE, Ryabinina OP, Schneider P, Magun BE, Iordanov MS. Fas ligand-induced proinflammatory transcriptional responses in reconstructed human epidermis. recruitment of the epidermal growth factor receptor and activation of MAP kinases. J Biol Chem (2008) 283:919–28. doi: 10.1074/jbc.M705852200
50. Reinehr R, Becker S, Eberle A, Grether-Beck S, Häussinger D. Involvement of NADPH oxidase isoforms and src family kinases in CD95-dependent hepatocyte apoptosis. J Biol Chem (2005) 280:27179–94. doi: 10.1074/jbc.M414361200
51. Khadra N, Bresson-Bepoldin L, Penna A, Chaigne-Delalande B, Ségui B, Levade T, et al. CD95 triggers Orai1-mediated localized Ca2+ entry, regulates recruitment of protein kinase c (PKC) β2, and prevents death-inducing signaling complex formation. Proc Natl Acad Sci U.S.A. (2011) 108:19072–7. doi: 10.1073/pnas.1116946108
52. Tauzin S, Chaigne-Delalande B, Selva E, Khadra N, Daburon S, Contin-Bordes C, et al. The naturally processed CD95L elicits a c-yes/calcium/PI3K-driven cell migration pathway. PloS Biol (2011) 9:e1001090. doi: 10.1371/journal.pbio.1001090
53. Malleter M, Tauzin S, Bessede A, Castellano R, Goubard A, Godey F, et al. CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res (2013) 73:6711–21. doi: 10.1158/0008-5472.CAN-13-1794
54. Lin VT, Lin FT. TRIP6: an adaptor protein that regulates cell motility, antiapoptotic signaling and transcriptional activity. Cell Signal (2011) 23:1691–7. doi: 10.1016/j.cellsig.2011.06.004
55. Cursi S, Rufini A, Stagni V, Condò I, Matafora V, Bachi A, et al. Src kinase phosphorylates caspase-8 on Tyr380: a novel mechanism of apoptosis suppression. EMBO J (2006) 25:1895–905. doi: 10.1038/sj.emboj.7601085
56. Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O, et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell (2008) 13:235–48. doi: 10.1016/j.ccr.2008.02.003
57. Fujita H, Koji T, Kitagawa N, Tsutsumi K, Abe K, Kaminogo M, et al. Possible involvement of fas system in the induction of apoptosis in human astrocytic brain tumors. Cell Mol Neurobiol (2002) 22:393–406. doi: 10.1023/a:1021007503779
58. Tauzin S, Debure L, Moreau JF, Legembre P. CD95-mediated cell signaling in cancer: mutations and post-translational modulations. Cell Mol Life Sci (2012) 69:1261–77. doi: 10.1007/s00018-011-0866-4
59. Ivanov VN, Lopez Bergami P, Maulit G, Sato TA, Sassoon D, Ronai Z. FAP-1 association with fas (Apo-1) inhibits fas expression on the cell surface. Mol Sci Biol (2003) 23:3623–35. doi: 10.1128/MCB.23.10.3623-3635.2003
60. Foehr ED, Lorente G, Vincent V, Nikolich K, Urfer R. FAS associated phosphatase (FAP-1) blocks apoptosis of astrocytomas through dephosphorylation of FAS. J Neurooncol (2005) 74:241–8. doi: 10.1007/s11060-004-7202-x
61. Sharma S, Carmona A, Skowronek A, Yu F, Collins MO, Naik S, et al. Apoptotic signalling targets the post-endocytic sorting machinery of the death receptor Fas/CD95. Nat Commun (2019) 10:3105. doi: 10.1038/s41467-019-11025-y
62. McHeik S, Aptecar L, Coopman P, D'Hondt V, Freiss G. Dual role of the PTPN13 tyrosine phosphatase in cancer. Biomolecules (2020) 10(12):1659. doi: 10.3390/biom10121659
63. Pike LJ. Rafts defined: a report on the keystone symposium on lipid rafts and cell function. J Lipid Res (2006) 47:1597–8. doi: 10.1194/jlr.E600002-JLR200
64. Mollinedo F, Gajate C. Clusters of apoptotic signaling molecule-enriched rafts, CASMERs: membrane platforms for protein assembly in Fas/CD95 signaling and targets in cancer therapy. Biochem Soc Transs (2022) 50:1105–18. doi: 10.1042/BST20211115
65. Bénéteau M, Pizon M, Chaigne-Delalande B, Daburon S, Moreau P, De Giorgi F, et al. Localization of Fas/CD95 into the lipid rafts on down-modulation of the phosphatidylinositol 3-kinase signaling pathway. Mol Cancer Res (2008) 6:604–13. doi: 10.1158/1541-7786.MCR-07-0331
66. Murai T, Maruyama Y, Mio K, Nishiyama H, Suga M, Sato C. Low cholesterol triggers membrane microdomain-dependent CD44 shedding and suppresses tumor cell migration. J Biol Chem (2011) 286:1999–2007. doi: 10.1074/jbc.M110.184010
67. Strale PO, Clarhaut J, Lamiche C, Cronier L, Mesnil M, Defamie N. Down-regulation of Connexin43 expression reveals the involvement of caveolin-1 containing lipid rafts in human U251 glioblastoma cell invasion. Mol Carcinog (2012) 51:845–60. doi: 10.1002/mc.20853
68. Seyrek K, Lavrik IN. Modulation of CD95-mediated signaling by post-translational modifications: towards understanding CD95 signaling networks. Apoptosis Apoptosis (2019) 24:385–94. doi: 10.1007/s10495-019-01540-0
69. Chakrabandhu K, Hérincs Z, Huault S, Dost B, Peng L, Conchonaud F, et al. Palmitoylation is required for efficient fas cell death signaling. EMBO J (2007) 26:209–20. doi: 10.1038/sj.emboj.7601456
70. Liu Z, Xiao M, Mo Y, Wang H, Han Y, Zhao X, et al. Emerging roles of protein palmitoylation and its modifying enzymes in cancer cell signal transduction and cancer therapy. Int J Biol Sci (2022) 18:3447–57. doi: 10.7150/ijbs.72244
71. Chen X, Li H, Fan X, Zhao C, Ye K, Zhao Z, et al. Protein palmitoylation regulates cell survival by modulating XBP1 activity in glioblastoma multiforme. Mol Ther Oncolytics (2020) 17:518–30. doi: 10.1016/j.omto.2020.05.007
72. Rossin A, Durivault J, Chakhtoura-Feghali T, Lounnas N, Gagnoux-Palacios L, Hueber AO. Fas palmitoylation by the palmitoyl acyltransferase DHHC7 regulates fas stability. Cell Death Differ (2015) 22:643–53. doi: 10.1038/cdd.2014.153
73. Ouyang J, Zhang Y, Xiong F, Zhang S, Gong Z, Yan Q, et al. The role of alternative splicing in human cancer progression. Am J Cancer Res (2021) 11:4642–67.
74. Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, et al. Protection from fas-mediated apoptosis by a soluble form of the fas molecule. Science (1994) 263:1759–62. doi: 10.1126/science.7510905
75. Jodo S, Kobayashi S, Nakajima Y, Matsunaga T, Nakayama N, Ogura N, et al. Elevated serum levels of soluble Fas/APO-1 (CD95) in patients with hepatocellular carcinoma. Clin Exp Immunol (1998) 112:166–71. doi: 10.1046/j.1365-2249.1998.00569.x
76. Lee SH, Shin MS, Lee JY, Park WS, Kim SY, Jang JJ, et al. In vivo expression of soluble fas and FAP-1: possible mechanisms of fas resistance in human hepatoblastomas. J Pathol (1999) 188:207–12. doi: 10.1002/(SICI)1096-9896(199906)188:2207::AID-PATH337>3.0.CO;2-8
77. Peciuliene I, Vilys L, Jakubauskiene E, Zaliauskiene L, Kanopka A. Hypoxia alters splicing of the cancer associated fas gene. Exp Cell Res (2019) 380:29–35. doi: 10.1016/j.yexcr.2019.04.015
78. Vilys L, Peciuliene I, Jakubauskiene E, Zinkeviciute R, Makino Y, Kanopka A. U2AF - hypoxia-induced fas alternative splicing regulator. Exp Cell Res (2021) 399:112444. doi: 10.1016/j.yexcr.2020.112444
79. O' Reilly LA, Tai L, Lee L, Kruse EA, Grabow S, Fairlie WD, et al. Membrane-bound fas ligand only is essential for fas-induced apoptosis. Nature (2009) 461:659–63. doi: 10.1038/nature08402
80. Kurma K, Boizard-Moracchini A, Galli G, Jean M, Vacher P, Blanco P, et al. Soluble CD95L in cancers and chronic inflammatory disorders, a new therapeutic target? Biochim Biophys Acta Rev Cancer (2021) 1876:188596. doi: 10.1016/j.bbcan.2021.188596
81. Knox PG, Milner AE, Green NK, Eliopoulos AG, Young LS. Inhibition of metalloproteinase cleavage enhances the cytotoxicity of fas ligand. J Immunol (2003) 170:677–85. doi: 10.4049/jimmunol.170.2.677
82. Roth W, Isenmann S, Nakamura M, Platten M, Wick W, Kleihues P, et al. Soluble decoy receptor 3 is expressed by malignant gliomas and suppresses CD95 ligand-induced apoptosis and chemotaxis. Cancer Res (2001) 61:2759–65.
83. Arakawa Y, Tachibana O, Hasegawa M, Miyamori T, Yamashita J, Hayashi Y. Frequent gene amplification and overexpression of decoy receptor 3 in glioblastoma. Acta Neuropathol (2005) 109:294–8. doi: 10.1007/s00401-004-0956-6
84. Liu W, Ramagopal U, Cheng H, Bonanno JB, Toro R, Bhosle R, et al. Crystal structure of the complex of human FasL and its decoy receptor DcR3. Structure (2016) 24:2016–23. doi: 10.1016/j.str.2016.09.009
85. Toda M, Kawamoto T, Ueha T, Kishimoto K, Hara H, Fukase N, et al. 'Decoy' and 'non-decoy' functions of DcR3 promote malignant potential in human malignant fibrous histiocytoma cells. Int J Oncol (2013) 43:703–12. doi: 10.3892/ijo.2013.1999
86. Liu YP, Zhu HF, Liu DL, Hu ZY, Li SN, Kan HP, et al. DcR3 induces epithelial-mesenchymal transition through activation of the TGF-β3/SMAD signaling pathway in CRC. Oncotarget (2016) 7:77306–18. doi: 10.18632/oncotarget.12639
87. Lin WW, Hsieh SL. Decoy receptor 3: a pleiotropic immunomodulator and biomarker for inflammatory diseases, autoimmune diseases and cancer. Biochem Pharmacol (2011) 81:838–47. doi: 10.1016/j.bcp.2011.01.011
88. Weng SC, Wen MC, Hsieh SL, Chen NJ, Tarng DC. Decoy receptor 3 suppresses T-cell priming and promotes apoptosis of effector T-cells in acute cell-mediated rejection: The role of reverse signaling. Front Immunol (2022) 13:879648. doi: 10.3389/fimmu.2022.879648
89. Wang HB, Li T, Ma DZ, Ji YX, Zhi H. Overexpression of FADD and caspase-8 inhibits proliferation and promotes apoptosis of human glioblastoma cells. BioMed Pharmacother (2017) 93:1–7. doi: 10.1016/j.biopha.2017.05.105
90. Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, et al. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med (2000) 6:529–35. doi: 10.1038/75007
91. Seyrek K, Ivanisenko NV, Richter M, Hillert LK, König C, Lavrik IN. Controlling cell death through post-translational modifications of DED proteins. Trends Cell Biol (2020) 30:354–69. doi: 10.1016/j.tcb.2020.02.006
92. Lee SH, Kim HS, Kim SY, Lee YS, Park WS, Kim SH, et al. Increased expression of FLIP, an inhibitor of fas-mediated apoptosis, in stomach cancer. APMIS (2003) 111:309–14. doi: 10.1034/j.1600-0463.2003.1110203.x
93. Haag C, Stadel D, Zhou S, Bachem MG, Möller P, Debatin KM, et al. Identification of c-FLIP(L) and c-FLIP(S) as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut (2011) 60:225–37. doi: 10.1136/gut.2009.202325
94. Ili CG, Brebi P, Tapia O, Sandoval A, Lopez J, Garcia P, et al. Cellular FLICE-like inhibitory protein long form (c-FLIPL) overexpression is related to cervical cancer progression. Int J Gynecol Pathol (2013) 32:316–22. doi: 10.1097/PGP.0b013e31825d8064
95. Tirapelli DP, Menezes SB, Franco IM, Lustosa IL, Rodrigues AR, Novais PC, et al. High expression of anti-apoptotic genes in grade I and II meningiomas. Arq Neuropsiquiatr (2017) 75:209–15. doi: 10.1590/0004-282X20170027
96. Luebke T, Schwarz L, Beer YY, Schumann S, Misterek M, Sander FE, et al. C-FLIP and CD95 signaling are essential for survival of renal cell carcinoma. Cell Death Dis (2019) 10:384. doi: 10.1038/s41419-019-1609-y
98. Schleich K, Buchbinder JH, Pietkiewicz S, Kähne T, Warnken U, Öztürk S, et al. Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ (2016) 23:681–94. doi: 10.1038/cdd.2015.137
99. Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem (2001) 276:20633–40. doi: 10.1074/jbc.M101780200
100. Fox JL, Hughes MA, Meng X, Sarnowska NA, Powley IR, Jukes-Jones R, et al. Cryo-EM structural analysis of FADD:Caspase-8 complexes defines the catalytic dimer architecture for co-ordinated control of cell fate. Nat Commun (2021) 12:819. doi: 10.1038/s41467-020-20806-9
101. Fricker N, Beaudouin J, Richter P, Eils R, Krammer PH, Lavrik IN. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J Cell Biol (2010) 190:377–89. doi: 10.1083/jcb.201002060
102. Smyth P, Sessler T, Scott CJ, Longley DB. FLIP(L): the pseudo-caspase. FEBS J (2020) 287:4246–60. doi: 10.1111/febs.15260
103. Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell (2015) 57:860–72. doi: 10.1016/j.molcel.2015.01.018
104. Lopez KE, Bouchier-Hayes L. Lethal and non-lethal functions of caspases in the DNA damage response. Cells (2022) 11:1887. doi: 10.3390/cells11121887
105. Xu Y, Surman DR, Ripley RT. Minority MOMP: a toxic, slow demise. Oncotarget (2020) 11:3559–61. doi: 10.18632/oncotarget.27753
106. Emery IF, Gopalan A, Wood S, Chow KH, Battelli C, George J, et al. Expression and function of ABCG2 and XIAP in glioblastomas. J Neurooncol (2017) 133:47–57. doi: 10.1007/s11060-017-2422-z
107. Vellanki SH, Grabrucker A, Liebau S, Proepper C, Eramo A, Braun V, et al. Small-molecule XIAP inhibitors enhance gamma-irradiation-induced apoptosis in glioblastoma. Neoplasia (2009) 11:743–52. doi: 10.1593/neo.09436
108. Choi YE, Butterworth M, Malladi S, Duckett CS, Cohen GM, Bratton SB. The E3 ubiquitin ligase cIAP1 binds and ubiquitinates caspase-3 and -7 via unique mechanisms at distinct steps in their processing. J Biol Chem (2009) 284:12772–82. doi: 10.1074/jbc.M807550200
109. Hu S, Yang X. Cellular inhibitor of apoptosis 1 and 2 are ubiquitin ligases for the apoptosis inducer Smac/DIABLO. J Biol Chem (2003) 278:10055–60. doi: 10.1074/jbc.M207197200
110. Liang J, Zhao W, Tong P, Li P, Zhao Y, Li H, et al. Comprehensive molecular characterization of inhibitors of apoptosis proteins (IAPs) for therapeutic targeting in cancer. BMC Med Genomics (2020) 13:7. doi: 10.1186/s12920-020-0661-x
111. Cheung CHA, Chang YC, Lin TY, Cheng SM, Leung E. Anti-apoptotic proteins in the autophagic world: an update on functions of XIAP, survivin, and BRUCE. J BioMed Sci (2020) 27:31. doi: 10.1186/s12929-020-0627-5
112. Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer (2010) 10:561–74. doi: 10.1038/nrc2889
113. Hanifeh M, Ataei F. XIAP as a multifaceted molecule in cellular signaling. Apoptosis Apoptosis (2022) 27:441–53. doi: 10.1007/s10495-022-01734-z
114. Iglesias-Guimarais V, Gil-Guiñon E, Gabernet G, García-Belinchón M, Sánchez-Osuna M, Casanelles E, et al. Apoptotic DNA degradation into oligonucleosomal fragments, but not apoptotic nuclear morphology, relies on a cytosolic pool of DFF40/CAD endonuclease. J Biol Chem (2012) 287:7766–79. doi: 10.1074/jbc.M111.290718
115. Sánchez-Osuna M, Martínez-Escardó L, Granados-Colomina C, Martínez-Soler F, Pascual-Guiral S, Iglesias-Guimarais V, et al. An intrinsic DFF40/CAD endonuclease deficiency impairs oligonucleosomal DNA hydrolysis during caspase-dependent cell death: a common trait in human glioblastoma cells. Neuro Oncol (2016) 18:950–61. doi: 10.1093/neuonc/nov315
116. Ferguson TA, Griffith TS. A vision of cell death: insights into immune privilege. Immunol Rev (1997) 156:167–84. doi: 10.1111/j.1600-065x.1997.tb00967.x
117. Saas P, Walker PR, Hahne M, Quiquerez AL, Schnuriger V, Perrin G, et al. Fas ligand expression by astrocytoma in vivo: maintaining immune privilege in the brain? J Clin Invest (1997) 99:1173–8. doi: 10.1172/JCI119273
118. Igney FH, Krammer PH. Tumor counterattack: fact or fiction? Cancer Immunol Immunother (2005) 54:1127–36. doi: 10.1007/s00262-005-0680-7
119. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med (2014) 20:607–15. doi: 10.1038/nm.3541
120. Badie B, Schartner J, Prabakaran S, Paul J, Vorpahl J. Expression of fas ligand by microglia: possible role in glioma immune evasion. J Neuroimmunol (2001) 120:19–24. doi: 10.1016/s0165-5728(01)00361-7
121. Zhu J, Powis de Tenbossche CG, Cané S, Colau D, van Baren N, Lurquin C, et al. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat Commun (2017) 8:1404. doi: 10.1038/s41467-017-00784-1
122. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T cells to protect tumour cells. Nat Commun (2018) 9:948. doi: 10.1038/s41467-018-03347-0
123. Kondo T, Suda T, Fukuyama H, Adachi M, Nagata S. Essential roles of the fas ligand in the development of hepatitis. Nat Med (1997) 3:409–13. doi: 10.1038/nm0497-409
124. Eisele G, Roth P, Hasenbach K, Aulwurm S, Wolpert F, Tabatabai G, et al. APO010, a synthetic hexameric CD95 ligand, induces human glioma cell death in vitro and in vivo. Neuro Oncol (2011) 13:155–64. doi: 10.1093/neuonc/noq176
125. Eisele G, Wolpert F, Decrey G, Weller M. APO010, a synthetic hexameric CD95 ligand, induces death of human glioblastoma stem-like cells. Anticancer Res (2013) 33:3563–71.
126. Verbrugge I, Wissink EH, Rooswinkel RW, Jongsma J, Beltraminelli N, Dupuis M, et al. Combining radiotherapy with APO010 in cancer treatment. Clin Cancer Res (2009) 15:2031–8. doi: 10.1158/1078-0432.CCR-08-2125
127. Merz C, Strecker A, Sykora J, Hill O, Fricke H, Angel P, et al. Neutralization of the CD95 ligand by APG101 inhibits invasion of glioma cells in vitro. Anticancer Drugs (2015) 26:716–27. doi: 10.1097/CAD.0000000000000237
128. Tuettenberg J, Seiz M, Debatin KM, Hollburg W, von Staden M, Thiemann M, et al. Pharmacokinetics, pharmacodynamics, safety and tolerability of APG101, a CD95-fc fusion protein, in healthy volunteers and two glioma patients. Int immunopharmacol (2012) 13:93–100. doi: 10.1016/j.intimp.2012.03.004
129. Boch T, Luft T, Metzgeroth G, Mossner M, Jann JC, Nowak D, et al. Safety and efficacy of the CD95-ligand inhibitor asunercept in transfusion-dependent patients with low and intermediate risk MDS. Leuk Res (2018) 68:62–9. doi: 10.1016/j.leukres.2018.03.007
130. Wick W, Fricke H, Junge K, Kobyakov G, Martens T, Heese O, et al. Randomized, study of weekly APG101+reirradiation versus reirradiation in progressive glioblastoma. Clin Cancer Res (2014) 20:6304–13. doi: 10.1158/1078-0432.CCR-14-0951-T
131. Wick W, Krendyukov A, Junge K, Höger T, Fricke H. Longitudinal analysis of quality of life following treatment with asunercept plus reirradiation versus reirradiation in progressive glioblastoma patients. J Neurooncol (2019) 145:531–40. doi: 10.1007/s11060-019-03320-x
132. Wei KC, Hsu PW, Tsai HC, Lin YJ, Chen KT, Toh CH, et al. Safety and tolerability of asunercept plus standard radiotherapy/temozolomide in Asian patients with newly-diagnosed glioblastoma: a phase I study. Sci Rep (2021) 11:24067. doi: 10.1038/s41598-021-02527-1
133. Hermisson M, Wagenknecht B, Wolburg H, Glaser T, Dichgans J, Weller M. Sensitization to CD95 ligand-induced apoptosis in human glioma cells by hyperthermia involves enhanced cytochrome c release. Oncogene (2000) 19:2338–45. doi: 10.1038/sj.onc.1203554
134. Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose-Girma M, Dohse M, et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature (2019) 574:428–31. doi: 10.1038/s41586-019-1548-x
135. Hadji A, Ceppi P, Murmann AE, Brockway S, Pattanayak A, Bhinder B, et al. Death induced by CD95 or CD95 ligand elimination. Cell Rep (2014) 7:208–22. doi: 10.1016/j.celrep.2014.02.035
136. Putzbach W, Gao QQ, Patel M, van Dongen S, Haluck-Kangas A, Sarshad AA, et al. Many si/shRNAs can kill cancer cells by targeting multiple survival genes through an off-target mechanism. Elife (2017) 6:e29702. doi: 10.7554/eLife.29702
137. Putzbach W, Haluck-Kangas A, Gao QQ, Sarshad AA, Bartom ET, Stults A, et al. CD95/Fas ligand mRNA is toxic to cells. Elife (2018) 7:e38621. doi: 10.7554/eLife.38621
138. Haluck-Kangas A, Patel M, Paudel B, Vaidyanathan A, Murmann AE, Peter ME. DISE/6mer seed toxicity-a powerful anti-cancer mechanism with implications for other diseases. J Exp Clin Cancer Res (2021) 40:389. doi: 10.1186/s13046-021-02177-1
Keywords: CD95, apoptosis, glioblastoma, TIL death, tumor immunotherapy
Citation: Zhang Y, Jin T, Dou Z, Wei B, Zhang B and Sun C (2022) The dual role of the CD95 and CD95L signaling pathway in glioblastoma. Front. Immunol. 13:1029737. doi: 10.3389/fimmu.2022.1029737
Received: 27 August 2022; Accepted: 09 November 2022;
Published: 24 November 2022.
Edited by:
Jianguo Xu, Sichuan University, ChinaCopyright © 2022 Zhang, Jin, Dou, Wei, Zhang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Buyi Zhang, 2510174@zju.edu.cn; Chongran Sun, sun.chongran@zju.edu.cn
†These authors have contributed equally to this work and share first authorship