 Fatima Dhalla
Fatima Dhalla Dylan J. Mac Lochlainn
Dylan J. Mac Lochlainn Helen Chapel
Helen Chapel Smita Y. Patel
Smita Y. Patel- 1Department of Clinical Immunology, John Radcliffe Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford, United Kingdom
- 2Developmental Immunology, MRC Weatherall Institute of Molecular Medicine, University of Oxford, Oxford, United Kingdom
- 3Primary Immunodeficiency Unit, Nuffield Department of Medicine and National Institute for Health Research Oxford Biomedical Research Centre, University of Oxford, Oxford, United Kingdom
Interstitial lung disease (ILD) is an important non-infectious complication in several primary immune deficiencies. In common variable immune deficiency (CVID) it is associated with complex clinical phenotypes and adverse outcomes. The histology of ILD in CVID is heterogeneous and mixed patterns are frequently observed within a single biopsy, including non-necrotising granulomatous inflammation, lymphoid interstitial pneumonitis, lymphoid hyperplasia, follicular bronchiolitis, organizing pneumonia, and interstitial fibrosis; ILD has to be differentiated from lymphoma. The term granulomatous-lymphocytic interstitial lung disease (GLILD), coined to describe the histopathological findings within the lungs of patients with CVID with or without multisystem granulomata, is somewhat controversial as pulmonary granulomata are not always present on histology and the nature of infiltrating lymphocytes is variable. In this mini review we summarize the literature on the histology of CVID-related ILD and discuss some of the factors that may contribute to the inter- and intra- patient variability in the histological patterns reported. Finally, we highlight areas for future development. In particular, there is a need for standardization of histological assessments and reporting, together with a better understanding of the immunopathogenesis of CVID-related ILD to resolve the apparent heterogeneity of ILD in this setting and guide the selection of rational targeted therapies in different patients.
Introduction
Common variable immune deficiency (CVID) is the most common of the primary immunodeficiency (PID) syndromes with a prevalence of 1 in 25,000 and 50,000, depending on the population (1, 2). It is characterized by low serum levels of IgG, IgA, and/or IgM, and poor specific antibody production (3). There is no definitive diagnostic test, so diagnosis requires the exclusion of secondary hypogammaglobulinaemia, combined immune defects, and, where appropriate, Mendelian disorders (4, 5). Up to 70% of patients suffer with variable non-infectious complications reflecting broader immune dysregulation, including autoimmunity, most commonly autoimmune cytopaenias; lymphocytic infiltration and/or granulomatous inflammation which can affect the lungs, gastrointestinal tract, spleen, skin or liver; or malignancy, in particular lymphoma (6, 7). Importantly, while bacterial infections are significantly reduced by adequate replacement therapeutic IgG, disease-related complications are not, but are associated with substantially increased mortality (7–9).
Respiratory tract pathology is a major contributor to impaired quality-of-life (10). Bacterial sinopulmonary infections are often the presenting feature, most frequently caused by Haemophilus influenzae or Streptococcus pneumoniae (11, 12). Recurrent and/or severe lower respiratory tract infections, particularly pneumonia, lead to bronchiectasis with an overall estimated prevalence of 30-35% among CVID patients, which, when present in isolation, does not contribute to increased mortality (8, 11–14). Interstitial lung disease (ILD), on the other hand, probably occurs due to immune dysregulation and/or viral infection rather than as a consequence of bacterial infection (7, 15, 16), and occurs alongside other disease-related complications, and shortens survival (7–9, 16). More rarely, the lungs can be the location for extranodal lymphomas, particularly B-cell non-Hodgkin’s lymphomas or MALToma (7, 17–20).
Interstitial Lung Disease in Common Variable Immune Deficiency
Clinical Significance of CVID-Related ILD
ILD is among the more frequent non-infectious complications of CVID, reported in 15%–60% of patients (7, 9, 14, 21–23). Clinical symptoms and high-resolution computed tomography (HRCT) findings of ILD can appear before or after CVID diagnosis (24, 25). The pathogenesis of CVID-related ILD is presumed to be unrelated to bacterial infections because it can be seen in the absence of bronchiectasis and is not significantly associated with a history of pneumonia (21). Patients with ILD have distinct clinical and immunological phenotypes in keeping with immune dysregulation, in contrast to those without ILD or those with bronchiectasis alone (6, 9, 14, 16, 21, 26, 27). Furthermore, there is no current histological or molecular evidence for chronic bacterial, EBV or CMV viral infections as triggers for inflammation (16, 28–30), though granulomas in other PIDs, such as those with DNA repair defects, show evidence of vaccine derived rubella virus (31). Other related complications, including splenomegaly, autoimmune cytopaenias, persistent lymphadenopathy and lymphoproliferation, but not necessarily granulomata, occur more frequently in patients with CVID-related ILD, supporting at least a role for intrinsic immune dysregulation driving these varied features (6, 9, 16, 21, 27, 32, 33).
Since CVID-related ILD causes significant morbidity, can be progressive and contributes to mortality, there is urgent need for effective treatments (8, 9, 34, 35). Because the mechanism(s) underlying CVID ILD have not been elucidated, immunosuppressive treatments have been tried with varying success, including corticosteroids, ciclosporin, methotrexate, sirolimus, cyclophosphamide, hydroxychloroquine, anti-TNF agents, mycophenolate mofetil, abatacept, rituximab and azathioprine (16, 34, 36–38). Corticosteroids are often used first-line, however, response may be short-lived or incomplete, there are significant side effects associated with protracted use and a proportion of patients are refractory (16, 34, 36, 39). Success with Rituximab, both in combination with azathioprine or mycophenolate mofetil, and as monotherapy, has been reported although controlled trials and long-term outcome data are lacking (40–43). Elevated levels of B-cell activating factor (BAFF), a cytokine that promotes the maturation and survival of B-cells, within the serum and lungs of patients with CVID-related ILD levels drives B-cell hyperplasia and may account for disease progression in a small proportion of patients (15) with invasive B cells in inappropriate germinal centers (28, 44).
Nomenclature
Various terminologies are used for CVID-related ILD, reflecting a lack of consensus regarding the naming of this complication and its heterogeneous nature (45). Lymphoid interstitial pneumonitis was first reported in patients with antibody deficiency in 1973 (46). Since then, various histopathological entities have been reported within lung biopsies of CVID ILD patients, from those caused by polyclonal lymphocytic inflammation to well-formed granulomata, organizing pneumonia, or pulmonary fibrosis, often with mixed pathology within individual patient biopsies (7, 9, 16, 27, 33, 35, 44). “Granulomatous-lymphocytic interstitial lung disease” (GLILD), first proposed in 2004, is often used as an overarching term to describe CVID ILD with lymphocytic infiltrates and/or granulomata (9, 45). However, the accuracy of this term has been called into question. Since not all patients have pulmonary granulomata, it does not fully capture the heterogeneity of the histopathology and similar histological patterns fulfilling a GLILD diagnosis are found in non-CVID PIDs (33, 47).
Investigations for CVID-Related ILD
Non-invasive investigations for CVID-related ILD include elevated serum IgM, decreased class-switched memory B-cells and absolute/relative numerical abnormalities of T-cell populations (15, 16, 34, 35, 48). Alongside rising IgM levels, BAFF, soluble IL-2 receptor and β2microgloblin have also been proposed as serum biomarkers for disease activity (15, 34, 49). Lung function tests, particularly the diffusion capacity for carbon monoxide (DLCO), are useful in monitoring for disease progression and response to treatment, but can lack the sensitivity required for diagnosis, particularly early in the disease course (14, 28, 34, 35, 37). HRCT is highly sensitive for the detection of CVID ILD, including at an early stage before symptoms or abnormal pulmonary function have developed (14, 33, 34). Radiographic findings are mixed and include lymphadenopathy, ground glass opacification, nodularity, septal thickening and consolidation (21, 33, 50). The use of CT combined with positron emission technology (PET) has also been reported as useful to identify sites of active disease, guide biopsy sampling, and monitor response to treatment (41). In selected cases, particularly, but not restricted to, pediatric presentations, genetic testing may be warranted. For example, patients with mutations in CTLA4, LRBA, TACI, KMT2D, XIAP, RAG1, and NFKB1 have been found within so called “CVID” cohorts, and ILD is a common feature of other monogenic PIDs (34, 39, 51–57). A molecular diagnosis enables other therapeutic approaches such as CTLA-4 fusion proteins abatacept and belatacept for the inflammatory associations of CTLA-4 and LRBA deficiency (58, 59). Invasive investigations include assessment of bronchoalveolar lavage fluid for infection and lymphocyte phenotyping, often used to avoid possible complications of biopsy (60), or biopsy of lung tissue under imaging for histopathological assessment.
Importance of Histopathological Assessment of Lung Tissue
Histological assessment of affected lung tissue is essential if features of ILD are present on HRCT. Imaging alone is not sufficient because radiographic patterns of parenchymal lung disease do not correlate with pathological features (33). It has been suggested that tissue from more accessible organs could be used in lieu of lung biopsy (34); however, patients with granulomata at other sites do not necessarily display granulomata within areas of ILD, indicating that other organs do not necessary serve as a proxy for the lung (33). Importantly, histological assessment contributes to the exclusion of differential diagnoses including infection and lymphoma and can provide prognostic information, since interstitial fibrosis has been associated with poorer outcomes (7, 17–20, 33). Currently, it is common practice to subject lung biopsy specimens to hematoxylin and eosin (H&E) staining, immunohistochemical staining for CD3, CD4, CD20/19 and EBV and CMV viral infections (37, 44). Understanding the pathological processes at play and the phenotype of infiltrating immune cells can help rationalize the selection of therapeutics used for CVID ILD (40–43).
We have reviewed the published literature of large series (>10 cases) for detailed histological findings of CVID ILD, the most recent being Larsen et al. (46). It is not always possible to know which patients were included in previous reports so only the most recent from each center is used unless marked (Table 1). Variations including the methods used for both biopsy and reporting are discussed in Section 4.
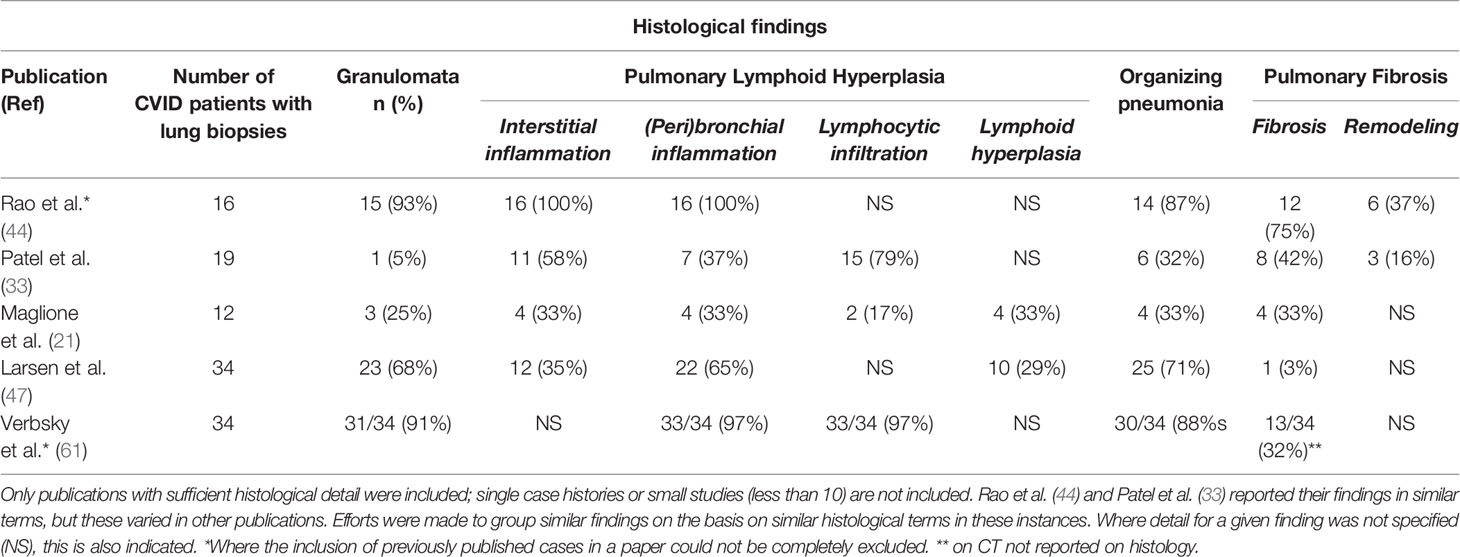
Table 1 Histological lung biopsy findings from common variable immune deficiency (CVID) patients reported in the literature.
Histological Patterns of ILD in CVID
The histological abnormalities reported in CVID ILD vary and overlap extensively. Similar patterns can also be found in numerous other lung diseases, making diagnosis challenging (44). Using a similar structure as Rao et al. (44), we summarize the commonly reported lung biopsy findings, each of which we discuss in turn (Table 1).
Granulomata
The granulomata reported in CVID ILD can vary from poorly- to well-circumscribed, with an apparent predilection for the former (28, 33, 44). Non-infectious CVID granulomatous lung disease shares some similar histological features with sarcoidosis and hypersensitivity pneumonitis; thus, clinical and radiological correlation is important in distinguishing these conditions (44, 62). “Poorly-formed granulomata” have been found within areas of pulmonary lymphoid hyperplasia and are difficult to define, as these are very subjective; additionally, granulomata can be found throughout the lung parenchyma (28, 44). It is worth re-emphasizing that granulomata are not reported in all cases of CVID-related ILD, with frequencies ranging from 0-94% depending on the individual study (Table 1) (7, 33, 44, 47). This suggests that there may be more than one pathological process in CVID-ILD (33, 47) and that the generalized use of overarching term “GLILD” to refer to all CVID-related ILD can be misleading.
Pulmonary Lymphoid Hyperplasia
Lymphoid proliferation has been designated as the “cardinal” feature of CVID ILD, and different patterns of pulmonary lymphoid hyperplasia (PLH) have been described, including follicular bronchiolitis, lymphocytic interstitial pneumonitis (LIP), lymphocytic infiltrates, and nodular lymphoid hyperplasia (28, 38, 40, 44, 47). In one case series where severity was assessed, PLH tended to be toward the moderate to severe end of the spectrum, with peribronchiolar and interstitial lymphocytic inflammation (44). These patterns often occur together and are rarely found in isolation (33, 44). Follicular bronchiolitis and/or LIP are found in around half of the cases reviewed (Table 1), and this is also in keeping with a recent review where 20/46 patients had some form of lymphoid infiltration, though not always specified (7).
Organizing Pneumonia
Organizing pneumonia (OP), intra-alveolar buds of granulation tissue with myofibroblasts and connective tissue, is reported in a substantial number of histological specimens, although to varying degrees between studies (Table 1). Cryptogenic organizing pneumonia (COP) is also found in CVID patients and is an important differential diagnosis when OP is the predominant finding on biopsy (40, 44). However, Rao et al. demonstrated the potential for misdiagnosis of CVID ILD when isolated COP was found on limited biopsy samples obtained by bronchoscopy.
OP can have many aetiologies. Larsen et al. reported that in their cohort OP was accompanied by a “dense lymphoid infiltrate”, which was not seen in biopsies from other causes of OP (47). Therefore, in their cohort of 34 patients with CVID and 4 with IgAD, these authors suggest that the combination of these two findings should suggest CVID or IgA deficiency rather than another etiology.
The lack of overlap between OP and pulmonary fibrosis (1/19 cases) in our cases might indicate separate pathological entities; however, significant overlap was described by Rao et al. (11/16 cases) (33, 44), who suggested evolving pathology.
Pulmonary Fibrosis
Pulmonary fibrosis is described in a quarter of CVID ILD cases (Table 1); however, similar to OP, one case series accounts for most of these cases (44), where the majority of patients had some degree of fibrosis. In contrast, Ho et al. found 6.3% of cases where “extensive pulmonary fibrosis” was the “predominant” finding at the time of biopsy; however, it was not reported whether it was a feature in other biopsies to a lesser degree (7).
Interstitial fibrosis in CVID ILD together with lymphoproliferation may resemble some of the patterns of idiopathic interstitial pneumonia, particularly if significant fibrosis (44). Only two studies looked specifically for architectural remodeling, and one of these found this to be associated with significant interstitial fibrosis (33, 44). The presence of fibrosis is a poor prognostic factor; prospective clinical studies are needed to justify earlier treatment (33).
Immunohistochemistry
Immunohistochemical staining of the lymphocytic infiltrate has produced discordant findings in the cases where it has been performed. CD20+ B-cells were found in a small proportion of cases, in follicles with T-cells circumscribing them, although T-cells are also reported more diffusely and in areas without B-cells (28, 33, 44). Rao et al. found a predominance of CD4+ T-cells within lymphoid infiltrates and also observed the presence of B-cell follicles surrounded by CD4+ T-cells (44). We recently reported a predominance of T-cells in most cases (Figure 1A), either CD4+ or CD8+; only 1 of six had germinal centers within B-cell follicles (Figure 1B) (33). Maglione et al. reported actively proliferating germinal centers in some of their patients with B-cell follicles (28). It is important to differentiate these from pulmonary MALToma, as found in two patients in the Oxford series (33).
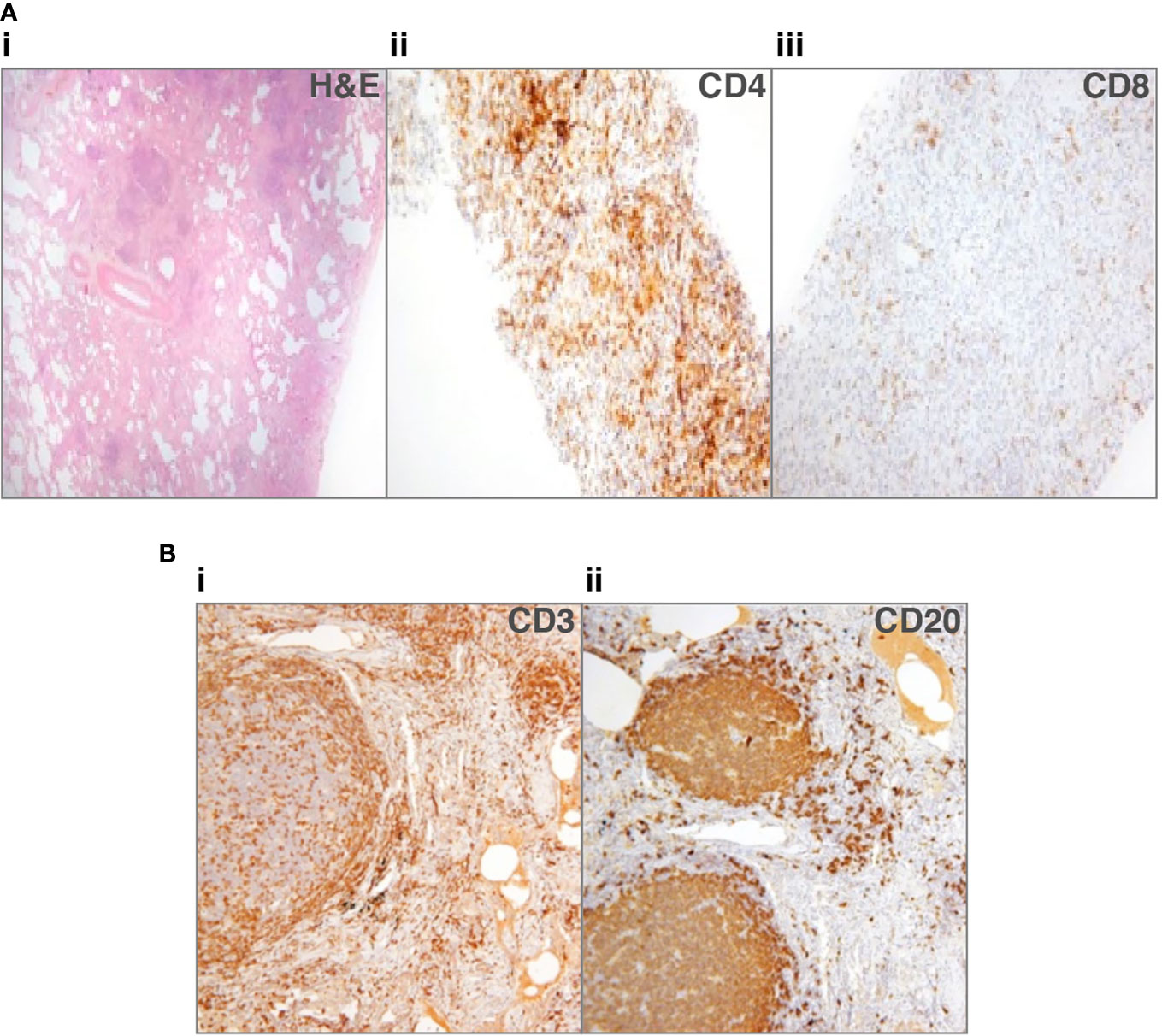
Figure 1 Lung biopsies from patients with common variable immune deficiency (CVID)-related interstitial lung disease (ILD). (A) Patient 1: (i) lung biopsy section stained with hematoxylin and eosin (H&E), to show lack of alveolar spaces, and many lymphocytes infiltrating the interstitium (ii) shows staining for CD4+ cells that predominate, sometimes in nodules, (iii) shows scanty CD8+ cells (33). No granulomata or organizing pneumonia. (B) Patient 2: (i) lung biopsy section stained for CD3+ cells, showing that T-cells surround follicles and are additionally found in discreet nodules, (ii) shows the follicles to consist of CD20+ cells, with only scattered CD20+ B-cells in other areas. No granulomata or organizing pneumonia.
We suggested that since the predominant T-cells were either CD4+ or CD8+, this pointed to different pathological entities (33). Chase et al. hypothesized that the inflammatory infiltrate, including B- and T-cells, might contribute to progressive ILD and pulmonary fibrosis, something that therapy directed against B- and T-cells might possibly prevent (40). Similarly, Maglione et al. suggested B-cells may be responsible for leukocyte accumulation in their role as antigen presenting cells and producers of chemokines and/or cytokines, making them a therapeutic target (28).
Addressing the Heterogeneity of Histopathological Findings CVID-Related ILD
There is a large amount of histopathological heterogeneity in biopsies from CVID-related ILD cases, both from one patient to the next, as well as between different case reports (Table 1). We discuss possible reasons for this in respect to the underlying pathophysiology, the patient populations reported, and factors relating to obtaining and interpreting lung biopsies.
Pathophysiology: A Spectrum of Disease, Separate Diseases, or a Shared Endpoint for Several Diseases?
Since the pathophysiology of CVID ILD is unknown, it is not surprising that there is no explanation for the degree of heterogeneity in the histology (33, 44). CVID-related ILD (or GLILD) was originally defined as a “conglomeration of pulmonary histopathologic abnormalities seen in a subset of patients with CVID (44). The divergent findings may represent a “spectrum” of a single disease (44) or several different pathologies, in addition to the primary antibody deficiency. Another hypothesis is that CVID ILD represents a common “pulmonary reaction pattern” (or “morphological common endpoint”) not only for CVID but also for other PIDs in which similar clinical, radiographical, and histological features have been described (44, 47). None of these hypotheses are mutually exclusive; it may be that the small numbers and the absence of international standardization frustrate the recognition of distinct pathological patterns.
Patient Populations
Geography may influence the variability observed, with different genetic influences in particular populations. It is interesting that three of the large CVID-related ILD case series, one from the UK and two from the USA, show the most divergence, despite a conscious effort on the part of the former to adhere to similar definitions used previously.
Differences in clinical practice, including diagnosis, cannot be totally discounted. Some series are restricted to patients with spontaneous (non-familial) CVID in adults and others include patients diagnosed in childhood. Since no diagnostic details are given, the exclusion of combined immune deficiencies involving T-cell immunity as well as B-cell failure (5), or known mutations in monogenic disease (e.g. CTLA4, LRBA, KMT2D, XIAP, RAG1, NFKB1) (34, 39, 51–57, 63) is unclear.
Biopsy-Related Factors: Technique, Timing, Treatment, and Interpretation
The method by which a biopsy has been obtained may have a significant impact on the clinical conclusions reached (61). Given that several different biopsy techniques have been used across the cases reported, this may be a contributing factor to some of the variation between cases, though in almost all series so far, imaging was used to obtain the biopsy.
A further consideration is the timing of the biopsy with respect to disease progression but most patients do not undergo repeat biopsies. It is likely that once pulmonary fibrosis and possibly organizing pneumonia are present that these may progress (33).
Another potential contributing factor is whether the biopsy was performed prior to or following corticosteroid or immunosuppressive treatment. These drugs could plausibly alter the patterns observed or mask them entirely, particularly those related to inflammation. While some authors have clearly documented when such drugs were used before biopsies were performed (33), this is not always the case, so firm conclusions cannot be drawn.
In the absence of standardized reporting, reading of the biopsy adds a great deal of potential for variation to be introduced. Although some authors have tried to mirror the approach pioneered by others and/or have a second, independent pathologist review the histology, some degree of both intra- and inter-operator variability is inevitable when faced with an uncommonly encountered pathological entity (33, 40).
Conclusions and Future Directions
In summary, there is considerable heterogeneity in the histopathological findings both within individual patients, between patients and between study centers, which include lymphoid hyperplasia, granulomata, organizing pneumonia and pulmonary fibrosis. The term “GLILD” is best avoided as not all patients have pulmonary granulomata (32, 46), and its use may mask the histopathological complexity and/or multiple pathological processes (33, 47).
Possible explanations include differences in the timing of sampling with respect to the disease process or treatments, genetic, geographical and environmental factors (7, 33, 44, 47). Finally, inconsistencies in obtaining histological specimens, treated, immuno-stained and described between studies have contributed (33), highlighting an urgent need for standardization of histopathological findings, to allow fairer comparisons to be made between distinct studies. The ability to compare separate studies is of paramount importance when dealing with a rare disease entity.
We need to expand our understanding of the etiology and immunopathogenesis of ILD in CVID, to provide more accurate prognostication and select appropriate treatments. Future studies will incorporate detailed cellular phenotypic, proteomic, transcriptomic and genomic dissection of CVID-ILD, to shed further light on pathogenesis, identify disease-relevant biomarkers and better guide treatment selection.
Author Contributions
FD and DM prepared the first draft of the manuscript. All authors contributed to editing of subsequent versions and reviewed and authorized the final version. HC and SP played a supervisory role. All authors contributed to the article and approved the submitted version.
Funding
FD is supported by an Academic Clinical Lectureship from the National Institute for Health Research (NIHR). DM is supported by an Academic Clinical Fellowship from the National Institute for Health Research (NIHR). SP is supported by the NIHR Oxford Biomedical Research Centre and Oxford University Hospitals NHS Foundation Trust.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past collaboration with one of the authors, SP.
Acknowledgments
The histological images in Figure 1B were used with kind permission of Dr Natalja Kurjane.
References
1. Shillitoe B, Bangs C, Guzman D, Gennery AR, Longhurst HJ, Slatter M, et al. The United Kingdom Primary Immune Deficiency (UKPID) registry 2012 to 2017. Clin Exp Immunol (2018) 192(3):284–91. doi: 10.1111/cei.13125
2. Westh L, Mogensen TH, Dalgaard LS, Bernth Jensen JM, Katzenstein T, Hansen AE, et al. Identification and Characterization of a Nationwide Danish Adult Common Variable Immunodeficiency Cohort. Scand J Immunol (2017) 85(6):450–61. doi: 10.1111/sji.12551
3. Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol Pract (2019) 7(6):1763–70. doi: 10.1016/j.jaip.2019.02.004
4. Bertinchamp R, Gerard L, Boutboul D, Malphettes M, Fieschi C, Oksenhendler E, et al. Exclusion of Patients with a Severe T-Cell Defect Improves the Definition of Common Variable Immunodeficiency. J Allergy Clin Immunol Pract (2016) 4(6):1147–57. doi: 10.1016/j.jaip.2016.07.002
5. Chapel H. Common Variable Immunodeficiency Disorders (CVID) - Diagnoses of Exclusion, Especially Combined Immune Defects. J Allergy Clin Immunol Pract (2016) 4(6):1158–9. doi: 10.1016/j.jaip.2016.09.006
6. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood (2008) 112(2):277–86. doi: 10.1182/blood-2007-11-124545
7. Ho HE, Cunningham-Rundles C. Non-infectious Complications of Common Variable Immunodeficiency: Updated Clinical Spectrum, Sequelae, and Insights to Pathogenesis. Front Immunol (2020) 11:149. doi: 10.3389/fimmu.2020.00149
8. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood (2012) 119(7):1650–7. doi: 10.1182/blood-2011-09-377945
9. Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol (2004) 114(2):415–21. doi: 10.1016/j.jaci.2004.05.057
10. Hurst JR, Workman S, Garcha DS, Seneviratne SL, Haddock JA, Grimbacher B. Activity, severity and impact of respiratory disease in primary antibody deficiency syndromes. J Clin Immunol (2014) 34(1):68–75. doi: 10.1007/s10875-013-9942-x
11. Oksenhendler E, Gerard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis (2008) 46(10):1547–54. doi: 10.1086/587669
12. Sperlich JM, Grimbacher B, Workman S, Haque T, Seneviratne SL, Burns SO, et al. Respiratory Infections and Antibiotic Usage in Common Variable Immunodeficiency. J Allergy Clin Immunol Pract (2018) 6(1):159–68. doi: 10.1016/j.jaip.2017.05.024
13. Ramzi N, Jamee M, Bakhtiyari M, Rafiemanesh H, Zainaldain H, Tavakol M, et al. Bronchiectasis in common variable immunodeficiency: A systematic review and meta-analysis. Pediatr Pulmonol (2020) 55(2):292–9. doi: 10.1002/ppul.24599
14. Maarschalk-Ellerbroek LJ, de Jong PA, van Montfrans JM, Lammers JW, Bloem AC, Hoepelman AI, et al. CT screening for pulmonary pathology in common variable immunodeficiency disorders and the correlation with clinical and immunological parameters. J Clin Immunol (2014) 34(6):642–54. doi: 10.1007/s10875-014-0068-6
15. Maglione PJ, Gyimesi G, Cols M, Radigan L, Ko HM, Weinberger T, et al. BAFF-driven B cell hyperplasia underlies lung disease in common variable immunodeficiency. JCI Insight (2019) 4(5). doi: 10.1172/jci.insight.122728
16. Schussler E, Beasley MB, Maglione PJ. Lung Disease in Primary Antibody Deficiencies. J Allergy Clin Immunol Pract (2016) 4(6):1039–52. doi: 10.1016/j.jaip.2016.08.005
17. Reichenberger F, Wyser C, Gonon M, Cathomas G, Tamm M. Pulmonary mucosa-associated lymphoid tissue lymphoma in a patient with common variable immunodeficiency syndrome. Respiration (2001) 68(1):109–12. doi: 10.1159/000050475
18. Aghamohammadi A, Parvaneh N, Tirgari F, Mahjoob F, Movahedi M, Gharagozlou M, et al. Lymphoma of mucosa-associated lymphoid tissue in common variable immunodeficiency. Leuk Lymphoma (2006) 47(2):343–6. doi: 10.1080/10428190500285285
19. Cunningham-Rundles C, Cooper DL, Duffy TP, Strauchen J. Lymphomas of mucosal-associated lymphoid tissue in common variable immunodeficiency. Am J Hematol (2002) 69(3):171–8. doi: 10.1002/ajh.10050
20. Cunningham-Rundles C. The many faces of common variable immunodeficiency. Hematol Am Soc Hematol Educ Program (2012) 2012:301–5. doi: 10.1182/asheducation.V2012.1.301.3798316
21. Maglione PJ, Overbey JR, Radigan L, Bagiella E, Cunningham-Rundles C. Pulmonary radiologic findings in common variable immunodeficiency: clinical and immunological correlations. Ann Allergy Asthma Immunol (2014) 113(4):452–9. doi: 10.1016/j.anai.2014.04.024
22. Verma N, Grimbacher B, Hurst JR. Lung disease in primary antibody deficiency. Lancet Respir Med (2015) 3(8):651–60. doi: 10.1016/S2213-2600(15)00202-7
23. López AL, Paolini MV, Fernández Romero DS. Lung disease in patients with common variable immunodeficiency. Allergologia Immunopathol (2020) 20:S0301-0546(20)30063-X. doi: doi: 10.1016/j.aller.2020.04.001
24. Chapel H, Lucas M, Patel S, Lee M, Cunningham-Rundles C, Resnick E, et al. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol (2012) 130(5):1197–8.e9. doi: 10.1016/j.jaci.2012.05.046
25. Hanitsch LG, Wittke K, Stittrich AB, Volk HD, Scheibenbogen C. Interstitial Lung Disease Frequently Precedes CVID Diagnosis. J Clin Immunol (2019) 39(8):849–51. doi: 10.1007/s10875-019-00708-2
26. Hartono S, Motosue MS, Khan S, Rodriguez V, Iyer VN, Divekar R, et al. Predictors of granulomatous lymphocytic interstitial lung disease in common variable immunodeficiency. Ann Allergy Asthma Immunol (2017) 118(5):614–20. doi: 10.1016/j.anai.2017.01.004
27. Weinberger T, Fuleihan R, Cunningham-Rundles C, Maglione PJ. Factors Beyond Lack of Antibody Govern Pulmonary Complications in Primary Antibody Deficiency. J Clin Immunol (2019) 39(4):440–7. doi: 10.1007/s10875-019-00640-5
28. Maglione PJ, Ko HM, Beasley MB, Strauchen JA, Cunningham-Rundles C. Tertiary lymphoid neogenesis is a component of pulmonary lymphoid hyperplasia in patients with common variable immunodeficiency. J Allergy Clin Immunol (2014) 133(2):535–42. doi: 10.1016/j.jaci.2013.08.022
29. Wheat WH, Cool CD, Morimoto Y, Rai PR, Kirkpatrick CH, Lindenbaum BA, et al. Possible role of human herpesvirus 8 in the lymphoproliferative disorders in common variable immunodeficiency. J Exp Med (2005) 202(4):479–84. doi: 10.1084/jem.20050381
30. Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol (2009) 145(6):709–27. doi: 10.1111/j.1365-2141.2009.07669.x
31. Buchbinder D, Hauck F, Albert MH, Rack A, Bakhtiar S, Shcherbina A, et al. Rubella Virus-Associated Cutaneous Granulomatous Disease: a Unique Complication in Immune-Deficient Patients, Not Limited to DNA Repair Disorders. J Clin Immunol (2019) 39(1):81–9. doi: 10.1007/s10875-018-0581-0
32. Bondioni MP, Soresina A, Lougaris V, Gatta D, Plebani A, Maroldi R. Common variable immunodeficiency: computed tomography evaluation of bronchopulmonary changes including nodular lesions in 40 patients. Correlation with clinical and immunological data. J Comput Assist Tomogr (2010) 34(3):395–401. doi: 10.1097/RCT.0b013e3181cad9da
33. Patel S, Anzilotti C, Lucas M, Moore N, Chapel H. Interstitial lung disease in patients with common variable immunodeficiency disorders: several different pathologies? Clin Exp Immunol (2019) 198(2):212–23. doi: 10.1111/cei.13343
34. Baumann U, Routes JM, Soler-Palacin P, Jolles S. The Lung in Primary Immunodeficiencies: New Concepts in Infection and Inflammation. Front Immunol (2018) 9:1837. doi: 10.3389/fimmu.2018.01837
35. Maglione PJ, Overbey JR, Cunningham-Rundles C. Progression of Common Variable Immunodeficiency Interstitial Lung Disease Accompanies Distinct Pulmonary and Laboratory Findings. J Allergy Clin Immunol Pract (2015) 3(6):941–50. doi: 10.1016/j.jaip.2015.07.004
36. Boursiquot JN, Gerard L, Malphettes M, Fieschi C, Galicier L, Boutboul D, et al. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol (2013) 33(1):84–95. doi: 10.1007/s10875-012-9778-9
37. Hurst JR, Verma N, Lowe D, Baxendale HE, Jolles S, Kelleher P, et al. British Lung Foundation/United Kingdom Primary Immunodeficiency Network Consensus Statement on the Definition, Diagnosis, and Management of Granulomatous-Lymphocytic Interstitial Lung Disease in Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract (2017) 5(4):938–45. doi: 10.1016/j.jaip.2017.01.021
38. Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol (2010) 134(2):97–103. doi: 10.1016/j.clim.2009.10.002
39. Pac M, Bielecka T, Grzela K, Komarnicka J, Langfort R, Koltan S, et al. Interstitial Lung Disease in Children With Selected Primary Immunodeficiency Disorders—A Multicenter Observational Study. Front Immunol (2020) 11:1950. doi: 10.3389/fimmu.2020.01950
40. Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol (2013) 33(1):30–9. doi: 10.1007/s10875-012-9755-3
41. Jolles S, Carne E, Brouns M, El-Shanawany T, Williams P, Marshall C, et al. FDG PET-CT imaging of therapeutic response in granulomatous lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Exp Immunol (2017) 187(1):138–45. doi: 10.1111/cei.12856
42. Pecoraro A, Crescenzi L, Galdiero MR, Marone G, Rivellese F, Rossi FW, et al. Immunosuppressive therapy with rituximab in common variable immunodeficiency. Clin Mol Allergy (2019) 17:9. doi: 10.1186/s12948-019-0113-3
43. Cereser L, De Carli R, Girometti R, De Pellegrin A, Reccardini F, Frossi B, et al. Efficacy of rituximab as a single-agent therapy for the treatment of granulomatous and lymphocytic interstitial lung disease in patients with common variable immunodeficiency. J Allergy Clin Immunol Pract (2019) 7(3):1055–7 e2. doi: 10.1016/j.jaip.2018.10.041
44. Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency–histologic and immunohistochemical analyses of 16 cases. Hum Pathol (2015) 46(9):1306–14. doi: 10.1016/j.humpath.2015.05.011
45. Hurst JR, Warnatz K. Collaboration ERSeCR. Interstitial lung disease in primary immunodeficiency: towards a brighter future. Eur Respir J (2020) 55(4):2000089. doi: 10.1183/13993003.00089-2020
46. Liebow AA, Carrington CB. Diffuse Pulmonary Lymphoreticular Infiltrations Associated with Dysproteinemia. Med Clinics North America (1973) 57(3):809–43. doi: 10.1016/S0025-7125(16)32278-7
47. Larsen BT, Smith ML, Tazelaar HD, Yi ES, Ryu JH, Churg A. GLILD Revisited: Pulmonary Pathology of Common Variable and Selective IgA Immunodeficiency. Am J Surg Pathol (2020) 44(8):1073–81. doi: 10.1097/PAS.0000000000001479
48. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood (2008) 111(1):77–85. doi: 10.1182/blood-2007-06-091744
49. Vitale J, Convers KD, Goretzke S, Guzman M, Noyes B, Parkar N, et al. Serum IL-12 and soluble IL-2 receptor levels as possible biomarkers of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency: a case report. J Allergy Clin Immunol Pract (2015) 3(2):273–6. doi: 10.1016/j.jaip.2014.09.019
50. Torigian DA, LaRosa DF, Levinson AI, Litzky LA, Miller WT Jr. Granulomatous-lymphocytic interstitial lung disease associated with common variable immunodeficiency: CT findings. J Thorac Imaging (2008) 23(3):162–9. doi: 10.1097/RTI.0b013e318166d32f
51. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic Diagnosis Using Whole Exome Sequencing in Common Variable Immunodeficiency. Front Immunol (2016) 7:220. doi: 10.3389/fimmu.2016.00220
52. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345(6204):1623–7. doi: 10.1126/science.1255904
53. van de Ven AAJM, de Jong PA, van Konijnenburg DPH, Kessels OAM, Boes M, Sanders EAM, et al. Airway and interstitial lung disease are distinct entities in paediatric common variable immunodeficiency. Clin Exp Immunol (2011) 165(2):235–42. doi: 10.1111/j.1365-2249.2011.04425.x
54. Adams JA, Gallagher JL, Hintermeyer M, Verbsky JW, Routes JM. Granulomatous and Lymphocytic Interstitial Lung Disease (GLILD) Associated with KMT2D Gene Mutation in Kabuki Syndrome. J Allergy Clin Immunol (2016) 137(2):AB118. doi: 10.1016/j.jaci.2015.12.512
55. Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious Mutations in LRBA Are Associated with a Syndrome of Immune Deficiency and Autoimmunity. Am J Hum Genet (2012) 90(6):986–1001. doi: 10.1016/j.ajhg.2012.04.015
56. Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol (2015) 35(2):119–24. doi: 10.1007/s10875-014-0121-5
57. Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol (2018) 142(4):1285–96. doi: 10.1016/j.jaci.2018.01.039
58. Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol (2018) 142(6):1932–46. doi: 10.1016/j.jaci.2018.02.055
59. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (2015) 349(6246):436–40. doi: 10.1126/science.aaa1663
60. Gregersen S, Holm AM, Fevang B, Ueland T, Sikkeland LI, Aalokken TM, et al. Lung disease, T-cells and inflammation in common variable immunodeficiency disorders. Scand J Clin Lab Invest (2013) 73(6):514–22. doi: 10.3109/00365513.2013.819523
61. Verbsky JW, Hintermeyer MK, Simpson PM, Feng M, Barbeau J, Rao N, et al. Rituximab and antimetabolite treatment of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency. J Allergy Clin Immunol (2020) S0091-6749(20)31069-1. doi: 10.1016/j.jaci.2020.07.021
62. Bouvry D, Mouthon L, Brillet PY, Kambouchner M, Ducroix JP, Cottin V, et al. Granulomatosis-associated common variable immunodeficiency disorder: a case-control study versus sarcoidosis. Eur Respir J (2013) 41(1):115–22. doi: 10.1183/09031936.00189011
Keywords: common variable immune deficiency, interstitial lung disease, histology review, literature analysis, primary immune deficiencies
Citation: Dhalla F, Lochlainn DJM, Chapel H and Patel SY (2020) Histology of Interstitial Lung Disease in Common Variable Immune Deficiency. Front. Immunol. 11:605187. doi: 10.3389/fimmu.2020.605187
Received: 11 September 2020; Accepted: 26 October 2020;
Published: 20 November 2020.
Edited by:
Klaus Warnatz, University of Freiburg, GermanyReviewed by:
Asghar Aghamohammadi, Tehran University of Medical Sciences, IranStephen Jolles, University Hospital of Wales, United Kingdom
Copyright © 2020 Dhalla, Lochlainn, Chapel and Patel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helen Chapel, helen.chapel@ndm.ox.ac.uk
†These authors have contributed equally to this work