 Mario Mellado1*
Mario Mellado1* Laura Martínez-Muñoz1
Laura Martínez-Muñoz1 Graciela Cascio1
Graciela Cascio1 Pilar Lucas1
Pilar Lucas1 José L. Pablos2
José L. Pablos2 José Miguel Rodríguez-Frade1
José Miguel Rodríguez-Frade1
- 1Department of Immunology and Oncology, Centro Nacional de Biotecnología, Consejo Superior de Investigaciones, Madrid, Spain
- 2Grupo de Enfermedades Inflamatorias y Autoinmunes, Instituto de Investigación Sanitaria Hospital, Madrid, Spain
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic inflammation in joints, associated with synovial hyperplasia and with bone and cartilage destruction. Although the primacy of T cell-related events early in the disease continues to be debated, there is strong evidence that autoantigen recognition by specific T cells is crucial to the pathophysiology of rheumatoid synovitis. In addition, T cells are key components of the immune cell infiltrate detected in the joints of RA patients. Initial analysis of the cytokines released into the synovial membrane showed an imbalance, with a predominance of proinflammatory mediators, indicating a deleterious effect of Th1 T cells. There is nonetheless evidence that Th17 cells also play an important role in RA. T cells migrate from the bloodstream to the synovial tissue via their interactions with the endothelial cells that line synovial postcapillary venules. At this stage, selectins, integrins, and chemokines have a central role in blood cell invasion of synovial tissue, and therefore in the intensity of the inflammatory response. In this review, we will focus on the mechanisms involved in T cell attraction to the joint, the proteins involved in their extravasation from blood vessels, and the signaling pathways activated. Knowledge of these processes will lead to a better understanding of the mechanism by which the systemic immune response causes local joint disorders and will help to provide a molecular basis for therapeutic strategies.
Rheumatoid Arthritis
Incorrect resolution of inflammation underlies pathologies of clinical importance, including cancer, atherosclerosis, and rheumatic diseases, and precise understanding of inflammatory responses is a major challenge to medical science. Of these conditions, rheumatoid arthritis (RA) is an enormous economic and social problem, highly prevalent in industrialized countries (0.5–1%, with two- to threefold greater incidence in women), resulting in disability, loss in quality of life, and reduced life expectancy.
Rheumatoid arthritis is a systemic autoimmune disease, characterized by non-organ-specific autoantibody production and chronic inflammation of synovial tissues, leading to cartilage and bone destruction. During disease development, other organs can also become inflamed, and as a consequence, systemic cardiovascular, pulmonary, and skeletal complications frequently appear (1). Little is known of the initiating events or factors that perpetuate RA, but advances in understanding the pathogenesis of the disease have contributed notably to development of new therapies. RA is a polygenic disease that involves complex interactions between genetic and environmental factors. The long-established association of RA patients with the human leukocyte antigen (HLA)-DRB1 locus suggests the influence of T cell selection and antigen presentation in the induction of autoreactive immune responses (2–4). Other genetic risk alleles/factors in RA include immune regulation (CD28), NF-κB-dependent signaling (TRAF1), control of T cell activation (PTPN22), and functional differentiation (CTLA-4) (5–9) (Table 1). Many cytokines, chemokines, growth factors, intracellular signaling molecules, and transcription factors have also been implicated in RA pathogenesis (10, 11).
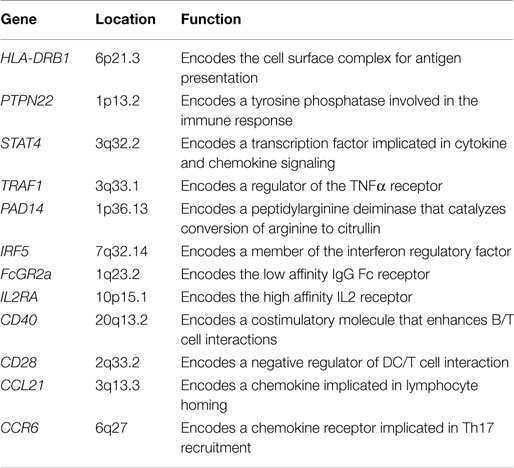
Table 1. Most relevant genes with single-nucleotide polymorphisms associated with susceptibility to rheumatoid arthritis and their functional role.
Synovial inflammation, or synovitis, results from leukocyte infiltration of the synovial compartment, enabled by increased expression of adhesion molecules and chemokines in the endothelium (Figure 1) (12–14). The cellular infiltrate includes granulocytes, monocytes/macrophages, NK cells, B cells, and especially CD4+ and CD8+ T cells, all leading to the production of large amounts of chemokines and proinflammatory cytokines (15–20).
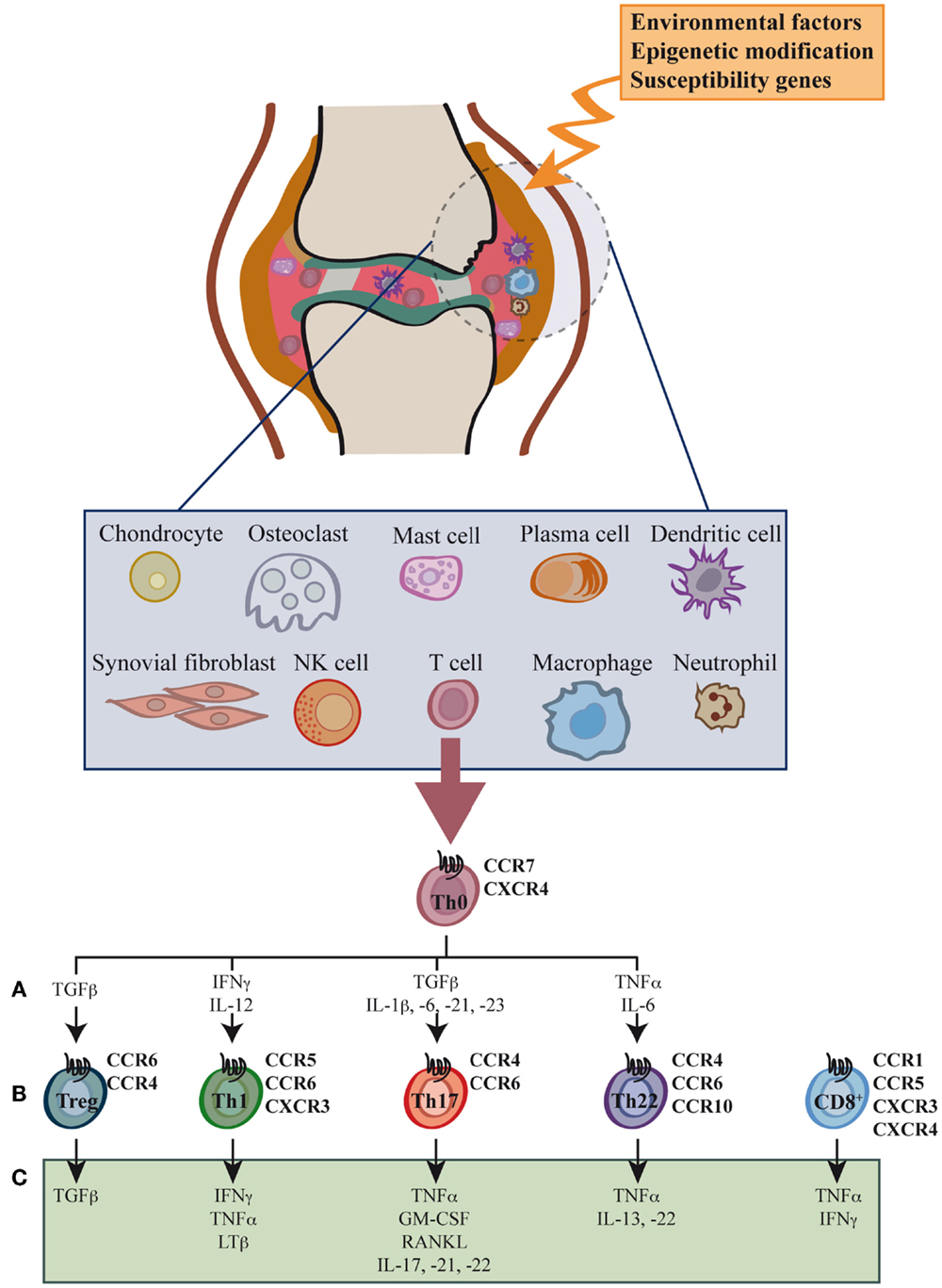
Figure 1. Cell types, cytokines, and chemokine receptors involved in rheumatoid arthritis development. Environmental factors and susceptibility gene interactions promote loss of tolerance to citrullinated self proteins generated by post-translational modifications. Co-stimulation-dependent interactions among DCs, T cells, and B cells generate an autoimmune response to these self proteins. This inflammatory process occurs primarily in the lymph node, but also in the inflamed joint. Adaptive and innate immune cells are attracted to the joint where immune pathways integrate to promote tissue remodeling and damage. Positive feedback loops mediated by interactions among leukocytes, synovial fibroblasts, chondrocytes, and osteoclasts, together with the molecular products of damage, drive the chronic phase in rheumatoid arthritis (RA) pathogenesis. High levels of activated memory CD4+ and CD8+ T cells differentiated through cytokine stimulation of naïve cells infiltrate the synovia (A). RA was classically considered a type 1 T helper (Th1)-mediated disease, but today data indicate that type 17 T helper cells (Th17) are more important in its promotion. Evidence shows that type 22 T helper cells (Th22) also contribute to RA pathogenesis. Function of regulatory T cells (Treg) is also reduced and effector cell resistance to suppression thus helps to alter the immune balance in inflamed joints. The figure shows the chemokine receptor expression pattern (B) and the main secreted cytokines (C) associated with each T cell subtype.
The role of these infiltrating cells is poorly understood. CD4+ T cells, but not CD8+ T cells or B cells, are necessary for disease initiation (21), but not for the inflammatory phase of the disease; hyperactivation of the immune response and the presence of autoantibodies in the synovial microenvironment are sufficient for disease development (22, 23). CD4+ T cell depletion using specific antibodies suppresses autoantibody production and reduces disease severity in the collagen- or antigen-induced arthritis models in rodents (CIA and AIA, respectively) (22, 24–27). Disease can nonetheless be induced in murine models of collagen antibody-induced arthritis (CAIA) in the absence of T cells (28). These differences indicate that murine RA models reflect only partial steps in the human disease and underline the limitations of the in vivo models (29). The limited effectiveness of T cell-depleting strategies (22) in clinical studies is probably due to immunogenicity and poor reconstitution of the immune system and emphasizes the restraints of in vitro testing and in vivo models (29). In contrast, therapies that block T cell co-stimulation are very effective at both early and advanced disease stages (30, 31).
Although RA was generally considered dependent on IFN-γ-producing Th1 cells, recent evidence indicates an important role for Th17 cells in RA development (11, 22) (Figure 1). Cytokines expressed by these cells (IL-17, GM-CSF, IL-22) are associated with synovial inflammation, mainly through their effect on neutrophil activation (10, 32). IFN-γ levels are not high in synovial membranes of RA patients, and RA development is reported in IFNγ-deficient mice (33, 34). In contrast, IL-17 deficiency mitigates arthritis development, as seen in mice that lack IL-17A (35, 36) or those treated with anti-IL-17-blocking antibodies (36, 37); IL-17 overexpression exacerbates disease progression and induces a chronic, erosive form of the disease (38). Although not the site of naïve T cell priming, CD4+ T cell commitment might occur at the inflamed joints that also have larger numbers of activated macrophages and dendritic cells (DCs) (15, 20). In mice, Th17 cell commitment requires IL-6, TGF-β, and IL-23 expression. In human beings, Th17 polarization depends on IL-1β, IL-6, IL-21, and IL-23, but the role of TGF-β is unclear (39). All of these cytokines are produced by tissue-resident macrophages, although the importance of specific DC subsets in T cell priming and polarization is becoming evident. An increase in Th17 cells is induced by monocyte-derived DC and CD1c+ myeloid DC, both found at high frequency in RA patient synovial fluid (40, 41), and by human inflammatory DC (42). DC from RA patient synovial fluid secrete higher levels of CCL17 than DC in peripheral blood; this chemokine could contribute to recruitment of CCR6+ cells, including Th17, to the inflamed joint (41). In mice, disruption of immune homeostasis by mucosal DC depends on the presence of commensal bacteria (43). Triggering of Toll-like receptors by intestinal flora contribute to RA progression by altering the Th17/regulatory T cells (Treg) balance, suggesting a role for the microbiota in Th17 response induction in RA (44, 45).
IL-17 has pleiotropic effects on many cell types, induces migration of innate immune cells, increases production of cytokines, chemokines, and matrix metalloproteases (46, 47), and enhances germinal center formation in animal models (48, 49), all of which contribute to the initiation and inflammatory phases of RA. In addition, IL-17 drives osteoclastogenesis, leading to bone resorption (50). Despite success in other types of arthritic diseases, IL-17-blocking strategies have thus far been less effective than anticipated in RA; this raises questions regarding the contribution of Th17 cells to RA development in human beings (51).
Activated naïve CD4+ T cells differentiate to IL-22-producing Th22 cells in the presence of IL-6 and TNFα. Similar to Th17, Th22 cells express CCR4 and CCR6, as well as CCR10 (52). Th22 cells are implicated in epidermal immunity, although their role in RA is unclear (Figure 1). Th22 cell frequencies are increased in peripheral blood from RA patients (53), and their percentages correlated positively with plasma IL-22 levels in these individuals (54). These observations coincide with reports that link IL-22 with RA activity and bone damage (55, 56). Results in animal models are also debated, whereas IL-22−/− mice show reduced incidence of CIA (57), recombinant IL-22 administration prior to arthritis onset reduces disease severity (58), suggesting a dual role for IL-22 during RA development (59).
The inflammatory environment also induces Treg expansion, and large numbers of proliferating Treg cells can be detected in the inflamed joints of patients (19). Data from murine models indicate that TNFα can boost Treg cell expansion (60), and thus protect mice from induction of autoimmune diseases. In man, however, TNFα has a negative effect on Treg cell expansion in vitro and in vivo (61, 62). The data indicate that Treg, Th1, and Th17 cells are the key T cell subsets in joint inflammation (Figure 1).
T cell plasticity is described in many inflammatory conditions. IL-1β and IL-6 downregulate FoxP3 expression, thus reducing Treg suppressive function (63, 64). In the inflamed synovium, TNFα promotes FoxP3 dephosphorylation and impaired Treg function (65). In these conditions, Treg cell dysfunction correlates with increased IL-17+ and IFN-γ+ CD4+ T cell numbers (65). In these inflammatory conditions, Treg cells differentiate into IL-17- and IFN-γ-producing effector cells (66, 67). Th17 cells in joints also show plasticity, as they co-express IFN-γ and transcription factors characteristic of Th17 (RORC) or Th1 (T-bet) cells; when cultured in synovial fluid, Th17 cells are reported to convert to Th1 cells (68–70). It is thus possible that Th17 cells give rise to a population of Th1 cells in inflamed joints, which could explain the pronounced Th1 responses in the inflamed synovium (68, 71, 72).
With activated macrophages and granulocytes, these T cell subsets contribute to the production of the proinflammatory cytokine cocktail that aggravates RA. TNFα and its receptors are expressed in human rheumatoid joint tissue (73, 74) and have a key role in RA, as they participate in cartilage (75) and bone degradation (76), and also promote IL-1, IL-6, and IL-8 production (77). In the CIA model of RA, administration of a specific anti-TNFα monoclonal antibody (mAb) after disease onset ameliorated inflammation and joint damage (27, 78). This therapy restores Treg cell function in RA patients (65), and antibody-based therapies that target TNFα are widely and successfully used in the clinic (79). IL-1α and IL-1β are also expressed in abundance in the synovial membrane (80), and IL-1Rα-deficient mice develop spontaneous arthritis, mediated in part by amplification of Th17-dependent inflammation (81). IL-18, another member of the IL-1 superfamily, is detected in RA synovium (82); symptoms are reduced for CIA in IL-18-deficient mice, as are those of RA in rodent models when IL-18 is blocked using neutralizing antibodies (83, 84). Given its pleiotropic functions, IL-6 is also important in RA; it regulates the maturation and activation of B and T cells, macrophages, osteoclasts, chondrocytes, and endothelial cells and has broad effects on hematopoiesis in the bone marrow. IL-6 deletion protects DBA/1 mice from CIA, and neutralization of IL-6 reduces the disease (85, 86). IL-12 is the main stimulator of IFN-γ production and of development of Th1 autoimmune responses (87); although the use of neutralizing antibodies in a murine CIA model attenuates symptoms, prolonged treatment worsens the disease (88).
T Cell Trafficking to the Synovium
Synovial inflammation in RA is partially dependent on migration of inflammatory cells, their retention at the inflammation site, and insufficient apoptosis of chronic inflammatory and stromal cells (89). T cell trafficking to the sites of inflammation is enabled by local activation in synovial vessels of the mechanisms necessary for leukocyte recruitment; alterations in these mechanisms can lead to chronic inflammation and autoimmunity (Figure 2).
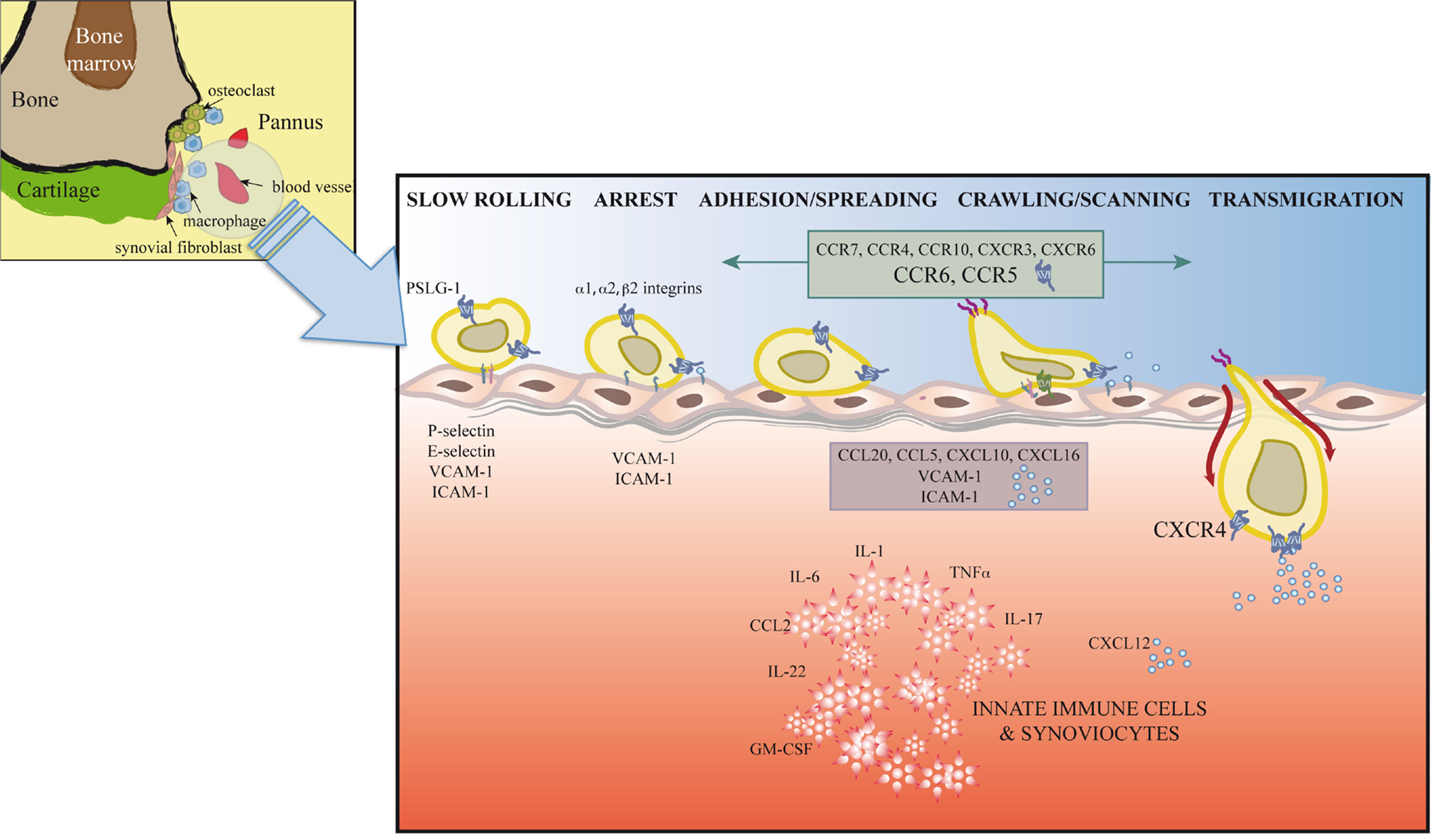
Figure 2. Extravasation model for T cells at the inflamed joint. In response to proinflammatory mediators, leukocytes and vascular cells are activated. Among other immune cells, T cells (Th1, Th17, Treg, and possibly Th22) initiate a serial cascade (rolling, arrest, spreading, crawling, and transmigration) and eventually extravasate from blood vessels to the inflamed joint. The figure shows inflammatory cytokines, selectins, integrins, adhesion molecules, chemokines, and chemokine receptors involved in T cell recruitment to and retention in the joint.
The leukocyte adhesion cascade is a multistep process that requires the coordinated action of rolling, adhesion, and transmigration events. This cascade is currently seen as the result of a chain of events initiated by leukocyte rolling along the endothelium, followed by their activation and adhesion to endothelial cells, and finally, migration to the target tissue (13, 14). Only those leukocyte subsets that express the appropriate set of adhesion molecules and chemoattractants will be recruited to specific sites. Leukocyte rolling is mediated by selectins, which are expressed by most leukocyte populations (L-selectin) and by inflamed endothelial cells (E- and P-selectins) (90) (Figure 2). Rolling involves selectins and P-selectin glycoprotein ligand-1 (PSGL-1), expressed by leukocytes and inflamed endothelial cells, as well as other glycosylated ligands (91). Interaction between PSLG1 and L-selectin is needed for leukocyte–leukocyte interactions that enable leukocyte tethering and adhesion to the inflamed endothelium in conditions of blood flow (91). E-selectin, which is upregulated in the inflamed synovium, is decreased after TNF-α therapy (92). Although serum levels of P- and L-selectin are reported to be increased in RA patients (93), the use of blocking antibodies and selectin-deficient mice only correlate P-selectin levels with disease activity (94, 95).
Integrins also participate in rolling and are responsible for firm leukocyte adhesion and arrest (13, 14) (Figure 2). Unlike circulating leukocytes, in the synovia, these populations express high levels of specific subsets of activated integrins. These integrins interact on the endothelial cell surface with ICAM1 or VCAM1, adhesion molecules that belong to the immunoglobulin superfamily, a prerequisite for cell extravasation. Distinct cell types express specific integrins. Whereas α1 are strongly expressed in activated CD4+ and CD8+ T cells, Th17 cells upregulate α2 integrins, a costimulatory molecule thought to be necessary for IL-17 production (96, 97). This specific integrin upregulation is fostered by proinflammatory cytokines in the synovia such as IL-1 or TNFα (98, 99) and determines cell localization in the inflamed joint. In addition to its effect on cell adhesion, interaction between integrins and their ligands, including fibronectin, collagen, VCAM-1, or degraded cartilage, also induces cell proliferation, cytokine production, and angiogenesis, contributing to disease development (96, 100, 101). Antagonists of integrins and their ligands thus prevent inflammation and angiogenesis in the murine CIA model (97, 102).
Stimulation by IL-1, TNFα, or IFNγ induces high levels of soluble and endothelium-bound ICAM-1, the β2 integrin ligand, in RA patient synovia (98, 103) (Figure 2). The role of ICAM-1 in RA is supported by lower disease activity in a CIA model in ICAM-1-deficient mice and by clinical studies that showed beneficial effects of a blocking anti-ICAM-1 mAb in early RA (104, 105).
Endothelial cells respond to inflammatory conditions by promoting expression of adhesion molecules and chemoattractants that bind directly or indirectly to glycosaminoglycans (GAGs) on the endothelial cell membrane. Integrin activation, initiated through chemoattractant-mediated inside–out signaling, induces the conformational changes responsible for the increased ligand-binding affinity needed for leukocyte arrest. The chemokine-activated signaling pathways responsible for integrin regulation and activation are not yet completely understood. G protein-dependent signaling through small GTPases is involved in rapid LFA-1 activation (106, 107). Recent reports suggest the existence of G protein-independent mechanisms that link Janus kinase (JAK)-mediated chemokine signaling with integrin activation via RhoA, RAC1, and Rap1 (108–110).
Transendothelial migration following chemotactic gradients is the final step in leukocyte migration through paracellular or transcellular pathways into inflamed tissues (Figure 2). Studies using knockout mice indicate a role in leukocyte transmigration of endothelial cell junction molecules such as PECAM1, ICAM2, JAMA, and ESAM (13).
Specificity of this process is achieved through carefully regulated cell signatures, that is, differential expression of the distinct components of this leukocyte adhesion cascade, including selectins, integrins, chemokines, and their respective ligands or receptors. For example, naïve T cells express low LFA-1, α4 integrin, and CCR7 levels, which allow cell recirculation through lymphoid tissues but it is insufficient to permit cells entry into inflamed tissues. In contrast, T effector and memory cells with elevated expression of LFA-1, α-integrins, E- and P-selectin ligands, CCR1, CCR5, and CXCR3 enter these tissues. The role of chemokines in T cell recruitment to the synovia is analyzed in detail in the next section.
These data indicate the potential of T cell migration inhibitors as targets for anti-inflammatory therapy. Whereas the limited number of selectins and integrins raises possible specificity problems of the drugs that target these molecules, the discovery of chemokines suggested the development of small molecules and inhibitors with the desired specificity characteristics. Although selectin, integrin, or chemokine receptor blockade has proved highly effective in animal models of disease, the transfer of these results to human diseases has not yet been successful. Promising therapies have nonetheless been developed that target molecules involved in leukocyte trafficking. This is the case of anti-VLA-4 neutralizing antibodies (natalizumab) for multiple sclerosis therapy (111) and of anti-CCR9 compounds now in phase III clinical trials for treatment of Crohn’s disease and inflammatory bowel disease (IBD) (112, 113). The blockade of signaling pathways involved in leukocyte trafficking is also being explored, with promising results. JAK inhibitors are showing utility in clinic (114), mainly because they regulate cytokine-mediated leukocyte activation, although a potential effect on cell migration should also be considered (115). Other inhibitors that target antigen-mediated B and T cell activation (Syk inhibitors) also show positive results in phase III trials (116). Given their roles in cell proliferation and survival and in macrophage, B cell, mast cell and neutrophil activation, PI3K, and Bruton’s tyrosine kinase (BTK) inhibitors are also candidates for therapy (117, 118).
Chemokines as Target of RA
Due to their central role in the selective recruitment and activation of immune cells at the inflammation site, chemokines and chemokine receptors are currently considered to potential therapeutic targets in several chronic autoimmune disorders. Inducible and homeostatic chemokines are heavily expressed in RA joints, produced mainly by activated synovial tissue and infiltrating leukocytes (119); elevated levels of several chemokines and their receptors are detected in serum and synovial fluid of RA patients (12) (Figure 2). Their relevance in disease progression has been determined in various animal models of the disease. Chemokines are implicated in RA development through recruitment and retention of different leukocyte populations in the inflamed joint (120, 121), but also elicit functions that contribute to pathogenesis. Chemokines can induce cytokine and metalloprotease release by chondrocytes and synovial fibroblasts, which contribute to cartilage destruction (122, 123). Other functions include induction of human chondrocyte death (124), enhanced cell proliferation (12, 125), angiogenesis, and angiostatic activities.
As indicated above, Th17 cells contribute to initiation and inflammatory phases of RA. Although Th17 cells express other chemokine receptors such as CCR4, CCR10, and CXCR3 (126, 127), they are characterized by CCR6 expression. CCL20, the CCR6 ligand, is a selective chemoattractant for T cells, naïve B cells, and immature DCs. CCR6+ Th17 cells have been identified in peripheral blood, synovial fluid, and inflamed tissue (128). CCL20 is expressed strongly at the inflamed joint, which allows Th17 cell activation and migration to the arthritic joint at early stages of the disease. Expression of other chemokine receptors in CCR6+ Th cells is associated with the expression of specific sets of cytokines. CCR4+/CCR6+ Th cells express high IL-17A levels, whereas levels of this interleukin are low in CXCR3+/CCR6+cells, whose IFN-γ levels are high. CCR6+/CCR10+ Th cells express high levels of IL-22, which defines the Th22 cell population. Other chemokine receptors found in CCR6+ Th cells are CCR5, CXCR4, and CXCR6, although they have not been associated with specific cytokine profiles. This cytokine production attracts and activates other cell types to the site of inflammation, including monocytes, neutrophils, synovial, and osteoclasts, which contribute to disease progression (128). Given this cytokine production, the induction of inflammatory chemokines during RA progression is not surprising.
In most cases, IL-1β- and TNF-α-activated cell types in the inflamed joint induce chemokine expression, although other cytokines such as IL-17 and IFNγ were also shown to upregulate expression of several chemokines. IL-1 and TNF-α stimulation of cells induce high CXCL8 levels in synovial tissue and fluid of inflamed joints (129, 130), and anti-CXCL8 treatment prevents neutrophil infiltration and tissue damage in LPS/IL-1-induced arthritis in mice (131). Production of CCL13, a major chemoattractant for eosinophils, T cells, and monocytes, is enhanced in cartilage by IFNγ, IL-1β, and TNF-α stimulation. As anticipated, the expression of these chemokines correlates with the recruitment of cells that express their receptors to the inflamed joint (132) (Figure 2).
CCL2 is also upregulated in synovial tissue of RA patients (130). It is produced by chondrocytes and synovial fibroblasts and can recruit CCR2+ macrophages to the synovia, as well as T cells, NK cells, and basophils (133, 134). Injection of a specific neutralizing anti-CCL2 mAb into rats with CIA reduced ankle swelling associated with decreased macrophage numbers in the joints (135); similar treatment inhibited arthritis in a MRL-lpr mouse model (136). Nonetheless, targeting CCL2 is not always valuable, and anti-CCL2 mAb treatment during the progression phase of a murine CIA model aggravated RA (137). Results were also discouraging in CIA models developed in mice that lacked the CCL2 receptor, CCR2 (138, 139). CCL3 and CCL5 are both expressed by activated T cells stimulated with IL-1β and TNF-α, by fibroblast-like synoviocytes, and by mononuclear cells in RA synovium (140–143); targeting their receptor, CCR5, could be of interest in pathological conditions. Whereas the percentages of CCR1+ and CCR5+ monocytes are lower in RA patient peripheral blood compared with normal controls, synovia of these patients show abundant CCR1 and CCR5 expression, indicating upregulation of these receptors and/or accumulation of CCR1+ and CCR5+ cells in the synovial compartment (144, 145). In mice, subcutaneous treatment with a CCR5 antagonist initiated a few days before clinical signs of arthritis promoted a marked decrease in leukocyte migration to joints, and thus reduced disease incidence and severity (146). Suppression of joint inflammation, reduced joint destruction, and diminished disease development was observed in CIA in rhesus monkeys treated with a CCR5 antagonist (147). These data are in agreement with reports showing that CCR5 density on the T cell surface determines the efficiency of T cell attraction to the joint, which might explain intra-individual variability and resistance of Δ32-CCR5 individuals to RA development (148, 149).
The CXC chemokine also have a role in RA due to their chemotactic effects on cell populations such as neutrophils (CXCL1, CXCL5, CXCL8), monocytes, and T cells (CXCL4, CXCL9, CXCL10, CXCL12, CXCL16), which correlates with the presence of CXCR3+ T cells, recruitment of CXCR6+ Th1 effector cells, and accumulation of CD4+ T cells in the RA synovium (143). There is growing evidence of an important functional role for CXCR4/CXCL12 in T lymphocyte accumulation and positioning within the rheumatoid synovium. CD4+ T cells in the inflamed synovium express high CXCR4 levels, which tallies with the high CXCL12 concentration in RA patient synovia (121) and suggests that CXCR4 is important for T cell retention in RA-affected synovial tissues (120). This is further supported by the observation that Th1 cells are attracted by RA synovial fluid, and that this chemotaxis can be inhibited in vitro by anti-CXCL12 antibodies (150). These studies show that CXCL12 production and CXCR4 expression are responsible for the characteristic pattern of T lymphocyte accumulation seen in the rheumatoid synovium (Figure 2). In accordance with the role of CXCL12/CXCR4 in RA, several CXCR4 antagonists, including the binding site competitor AMD3100, have shown therapeutic activity in arthritic mice (151).
In contrast to CC chemokine, the CXC group can also participate in angiogenic or angiostatic effects in RA patient joints. Synovia from RA patients show increased numbers of blood vessels compared to healthy synovium (12). It is generally thought that the new vessels accommodate the influx of immune cells into the joint and thus contribute indirectly to cell infiltration. RA synovium can show certain histological similarities to lymphoid tissue, including the presence of germinal centers, B cells, T cells, and follicular DCs. This could be due to the induction of newly expressed homeostatic chemokine receptors by the local microenvironment once T cells have entered the synovium, together with local expression of matching chemokines. Consistent with this hypothesis, CCR7 and CXCR4 are expressed by CD4+ memory T cells in RA synovial fluid, whereas circulating CD4+ T cells do not express these receptors (152, 153). Homeostatic chemokines that regulate cell traffic in lymphoid tissues are similarly found in the RA synovia, including CCL19, CXCL12, and CXCL13, and therefore can also participate in this lymphoid-like organization (154).
Although considered an initially promising therapy, results for blockade of chemokines or chemokine receptors in patients have been disappointing (155). A mAb against CCL2, the CCR2 ligand, showed no beneficial effects when administered to RA patients (156). Similarly, anti-CXCL8/IL-8 antibody treatment did not lead to clinical improvement in RA patients (157), and although short-term treatment of active RA patients with a CCR1 antagonist showed a tendency toward clinical improvement compared to controls (158), a phase II clinical study did not demonstrate clinical efficacy after a 6-month treatment (159).
Chemokine biology is more complex than originally anticipated. In addition to their considerable promiscuity and redundancy, the chemokine receptors oligomerize at the cell membrane (160, 161). This oligomerization is not limited to other chemokine receptors, as they can also interact with other GPCR (162, 163) and with other cell surface molecules such as CD4 (164). Chemokine signaling requires preformed receptor dimers (165) that allow G protein coupling to the receptor and activation of G protein-dependent and -independent signaling pathways. Chemokine receptor complexes help to generate diversity in chemokine signaling and function (160, 166, 167). In a complex microenvironment such as that of the inflamed arthritic joint, chemokine receptors are co-expressed, and chemokines and cytokines are upregulated. The lack of drugs that target chemokine receptors efficiently might also reflect greater complexity of the system than initially predicted and indicate that efficient chemokine inhibition could require additional therapeutic approaches that regulate interactions between chemokines, and between chemokines and cytokines that recruit proinflammatory cells to the arthritic joint. In addition, in vivo secreted chemokines bind to GAG, allowing formation of chemotactic gradients that direct leukocytes to inflammation sites. CXCL12 attached to sulfate proteoglycans has been observed on endothelial cells of the RA synovium, a process upregulated by inflammatory cytokines (168). These findings indicate that both chemokine upregulation and the GAG-dependent immobilization of these mediators on endothelial cells are potential targets for intervention.
Conclusion
During the course of RA, T cells and other immune cells are recruited to the synovial tissue, where they produce large amounts of proinflammatory cytokines and interact with synovial fibroblasts and macrophages, all of which contribute to pathogenesis development. These include CD4+ and CD8+ cells, mostly with an activated phenotype. RA was classically considered a Th1-mediated disease, but evidence today indicates clear involvement of Th17, Th22, and Treg cells; it nonetheless remains unclear whether these are truly separate subpopulations or they represent plasticity and heterogeneity within the Th17 lineage. Each of these cell subsets acts at distinct stages in the course of the disease, to participate in the complex network of cell–cell interactions that governs RA initiation and progression, including release of inflammatory mediators, induction of cell proliferation, and angiogenesis. Cell migration into the synovium is controlled by the expression of selectins and their ligands, integrins, adhesion molecules, and chemokines and their receptors; all these molecules define the specific T cell subsets in the inflamed joint. The use of antagonists to and mice deficient in these proteins has been essential for defining their role in different steps of the disease, and prompted the use of inhibitors in clinical studies. The diversity of chemokines and receptors suggested they were ideal targets that only affect specific leukocyte subsets, and over the last two decades, most pharmaceutical and biotechnology companies developed chemokine receptor-targeting reagents that were analyzed for RA therapy. These clinical studies were not as successful as anticipated and dashed the promise of targeting individual chemokine receptors for RA. Alternative strategies aimed at intracellular signaling pathways or interactions between chemokine receptors must thus be considered.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the people at the Chemokine Signaling Group for much of the work that contributed to this review, especially PL, and Ricardo Villares, and to Coral Bastos and Catherine Mark for the secretarial and editorial assistance, respectively. This work was supported in part by grants from the Spanish Ministry of Science and Innovation (SAF 2011-27370 and SAF2014-53416-R), the RETICS Program (RD12/0009/009; RIER), and the Madrid Regional Government (S2010/BMD-2350; RAPHYME).
References
1. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365:2205–19. doi:10.1056/NEJMra1004965
2. Jutley G, Raza K, Buckley CD. New pathogenic insights into rheumatoid arthritis. Curr Opin Rheumatol (2015) 27:249–55. doi:10.1097/BOR.0000000000000174
3. Mohan VK, Ganesan N, Gopalakrishnan R. Association of susceptible genetic markers and autoantibodies in rheumatoid arthritis. J Genet (2014) 93:597–605. doi:10.1007/s12041-014-0380-1
4. Amos CI, Chen WV, Remmers E, Siminovitch KA, Seldin MF, Criswell LA, et al. Data for genetic analysis workshop (GAW) 15 problem 2, genetic causes of rheumatoid arthritis and associated traits. BMC Proc (2007) 1(Suppl 1):S3. doi:10.1186/1753-6561-1-s1-s3
5. Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet (2007) 39:1477–82. doi:10.1038/ng.2007.27
6. Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis – a genomewide study. N Engl J Med (2007) 357:1199–209. doi:10.1056/NEJMoa073491
7. Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med (2007) 357:977–86. doi:10.1056/NEJMoa073003
8. Hughes LB, Reynolds RJ, Brown EE, Kelley JM, Thomson B, Conn DL, et al. Most common single-nucleotide polymorphisms associated with rheumatoid arthritis in persons of European ancestry confer risk of rheumatoid arthritis in African Americans. Arthritis Rheum (2010) 62:3547–53. doi:10.1002/art.27732
9. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature (2014) 506:376–81. doi:10.1038/nature12873
10. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol (2007) 7:429–42. doi:10.1038/nri2094
11. Komatsu N, Takayanagi H. Autoimmune arthritis: the interface between the immune system and joints. Adv Immunol (2012) 115:45–71. doi:10.1016/B978-0-12-394299-9.00002-3
12. Iwamoto T, Okamoto H, Toyama Y, Momohara S. Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. FEBS J (2008) 275:4448–55. doi:10.1111/j.1742-4658.2008.06580.x
13. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7:678–89. doi:10.1038/nri2156
14. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity (2014) 41:694–707. doi:10.1016/j.immuni.2014.10.008
15. Kinne RW, Stuhlmuller B, Burmester GR. Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res Ther (2007) 9:224. doi:10.1186/ar2333
16. Lundy SK, Sarkar S, Tesmer LA, Fox DA. Cells of the synovium in rheumatoid arthritis. T lymphocytes. Arthritis Res Ther (2007) 9:202. doi:10.1186/ar2107
17. Mauri C, Ehrenstein MR. Cells of the synovium in rheumatoid arthritis. B cells. Arthritis Res Ther (2007) 9:205. doi:10.1186/ar2125
18. Smeets TJ, Kraan MC, Galjaard S, Youssef PP, Smith MD, Tak PP. Analysis of the cell infiltrate and expression of matrix metalloproteinases and granzyme B in paired synovial biopsy specimens from the cartilage-pannus junction in patients with RA. Ann Rheum Dis (2001) 60:561–5. doi:10.1136/ard.60.6.561
19. Wehrens EJ, Prakken BJ, van Wijk F. T cells out of control – impaired immune regulation in the inflamed joint. Nat Rev Rheumatol (2013) 9:34–42. doi:10.1038/nrrheum.2012.149
20. Lutzky V, Hannawi S, Thomas R. Cells of the synovium in rheumatoid arthritis. Dendritic cells. Arthritis Res Ther (2007) 9:219. doi:10.1186/ar2200
21. Wong PK, Quinn JM, Sims NA, van Nieuwenhuijze A, Campbell IK, Wicks IP. Interleukin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced osteoclastogenesis. Arthritis Rheum (2006) 54:158–68. doi:10.1002/art.21537
22. Alzabin S, Williams RO. Effector T cells in rheumatoid arthritis: lessons from animal models. FEBS Lett (2011) 585:3649–59. doi:10.1016/j.febslet.2011.04.034
23. Matmati M, Jacques P, Maelfait J, Verheugen E, Kool M, Sze M, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet (2011) 43:908–12. doi:10.1038/ng.874
24. Kollias G, Papadaki P, Apparailly F, Vervoordeldonk MJ, Holmdahl R, Baumans V, et al. Animal models for arthritis: innovative tools for prevention and treatment. Ann Rheum Dis (2011) 70:1357–62. doi:10.1136/ard.2010.148551
25. Kadowaki KM, Matsuno H, Tsuji H, Tunru I. CD4+ T cells from collagen-induced arthritic mice are essential to transfer arthritis into severe combined immunodeficient mice. Clin Exp Immunol (1994) 97:212–8. doi:10.1111/j.1365-2249.1994.tb06070.x
26. Petrow PK, Thoss K, Katenkamp D, Brauer R. Adoptive transfer of susceptibility to antigen-induced arthritis into severe combined immunodeficient (SCID) mice: role of CD4+ and CD8+ T cells. Immunol Invest (1996) 25:341–53. doi:10.3109/08820139609059316
27. Bevaart L, Vervoordeldonk MJ, Tak PP. Evaluation of therapeutic targets in animal models of arthritis: how does it relate to rheumatoid arthritis? Arthritis Rheum (2010) 62:2192–205. doi:10.1002/art.27503
28. Kagari T, Doi H, Shimozato T. The importance of IL-1 beta and TNF-alpha, and the noninvolvement of IL-6, in the development of monoclonal antibody-induced arthritis. J Immunol (2002) 169:1459–66. doi:10.4049/jimmunol.169.3.1459
29. Isaacs JD. Therapeutic T-cell manipulation in rheumatoid arthritis: past, present and future. Rheumatology (Oxford) (2008) 47:1461–8. doi:10.1093/rheumatology/ken163
30. Kremer JM, Dougados M, Emery P, Durez P, Sibilia J, Shergy W, et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: twelve-month results of a phase iib, double-blind, randomized, placebo-controlled trial. Arthritis Rheum (2005) 52:2263–71. doi:10.1002/art.21201
31. Schiff M. Abatacept treatment for rheumatoid arthritis. Rheumatology (2011) 50:437–49. doi:10.1093/rheumatology/keq287
32. van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol (2009) 5:549–53. doi:10.1038/nrrheum.2009.179
33. Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-gamma receptor-deficient mice. J Immunol (1997) 158:5507–13.
34. Matthys P, Vermeire K, Mitera T, Heremans H, Huang S, Billiau A. Anti-IL-12 antibody prevents the development and progression of collagen-induced arthritis in IFN-gamma receptor-deficient mice. Eur J Immunol (1998) 28:2143–51. doi:10.1002/(SICI)1521-4141(199807)28:07<2143::AID-IMMU2143>3.0.CO;2-C
35. Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol (2003) 171:6173–7. doi:10.4049/jimmunol.171.11.6173
36. Zwerina K, Koenders M, Hueber A, Marijnissen RJ, Baum W, Heiland GR, et al. Anti IL-17A therapy inhibits bone loss in TNF-alpha-mediated murine arthritis by modulation of the T-cell balance. Eur J Immunol (2012) 42:413–23. doi:10.1002/eji.201141871
37. Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum (2004) 50:650–9. doi:10.1002/art.20001
38. Lubberts E, Joosten LA, van de Loo FA, Schwarzenberger P, Kolls J, van den Berg WB. Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res (2002) 51:102–4. doi:10.1007/BF02684010
39. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol (2007) 8:942–9. doi:10.1038/ni1496
40. Estrada-Capetillo L, Hernandez-Castro B, Monsivais-Urenda A, Alvarez-Quiroga C, Layseca-Espinosa E, Abud-Mendoza C, et al. Induction of Th17 lymphocytes and Treg cells by monocyte-derived dendritic cells in patients with rheumatoid arthritis and systemic lupus erythematosus. Clin Dev Immunol (2013) 2013:584303. doi:10.1155/2013/584303
41. Moret FM, Hack CE, van der Wurff-Jacobs KM, de Jager W, Radstake TR, Lafeber FP, et al. Intra-articular CD1c-expressing myeloid dendritic cells from rheumatoid arthritis patients express a unique set of T cell-attracting chemokines and spontaneously induce Th1, Th17 and Th2 cell activity. Arthritis Res Ther (2013) 15:R155. doi:10.1186/ar4338
42. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity (2013) 38:336–48. doi:10.1016/j.immuni.2012.10.018
43. Luckey D, Gomez A, Murray J, White B, Taneja V. Bugs & us: the role of the gut in autoimmunity. Indian J Med Res (2013) 138:732–43.
44. Rogier R, Koenders MI, Abdollahi-Roodsaz S. Toll-like receptor mediated modulation of T cell response by commensal intestinal microbiota as a trigger for autoimmune arthritis. J Immunol Res (2015) 2015:527696. doi:10.1155/2015/527696
45. Scher JU, Abramson SB. The microbiome and rheumatoid arthritis. Nat Rev Rheumatol (2011) 7:569–78. doi:10.1038/nrrheum.2011.121
46. Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology (2010) 129:311–21. doi:10.1111/j.1365-2567.2009.03240.x
47. Shen F, Ruddy MJ, Plamondon P, Gaffen SL. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-alpha- induced genes in bone cells. J Leukoc Biol (2005) 77:388–99. doi:10.1189/jlb.0904490
48. Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol (2008) 9:166–75. doi:10.1038/ni1552
49. Hsu HC, Zhou T, Kim H, Barnes S, Yang P, Wu Q, et al. Production of a novel class of polyreactive pathogenic autoantibodies in BXD2 mice causes glomerulonephritis and arthritis. Arthritis Rheum (2006) 54:343–55. doi:10.1002/art.21550
50. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest (1999) 103:1345–52. doi:10.1172/JCI5703
51. Kellner H. Targeting interleukin-17 in patients with active rheumatoid arthritis: rationale and clinical potential. Ther Adv Musculoskelet Dis (2013) 5:141–52. doi:10.1177/1759720X13485328
52. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine (2015) 74:5–17. doi:10.1016/j.cyto.2014.09.011
53. Zhang L, Li YG, Li YH, Qi L, Liu XG, Yuan CZ, et al. Increased frequencies of Th22 cells as well as Th17 cells in the peripheral blood of patients with ankylosing spondylitis and rheumatoid arthritis. PLoS One (2012) 7:e31000. doi:10.1371/journal.pone.0031000
54. Zhao L, Jiang Z, Jiang Y, Ma N, Zhang Y, Feng L, et al. IL-22+ CD4+ T cells in patients with rheumatoid arthritis. Int J Rheum Dis (2013) 16:518–26. doi:10.1111/1756-185X.12099
55. da Rocha LF Jr, Duarte AL, Dantas AT, Mariz HA, Pitta Ida R, Galdino SL, et al. Increased serum interleukin 22 in patients with rheumatoid arthritis and correlation with disease activity. J Rheumatol (2012) 39:1320–5. doi:10.3899/jrheum.111027
56. Kim KW, Kim HR, Park JY, Park JS, Oh HJ, Woo YJ, et al. Interleukin-22 promotes osteoclastogenesis in rheumatoid arthritis through induction of RANKL in human synovial fibroblasts. Arthritis Rheum (2012) 64:1015–23. doi:10.1002/art.33446
57. Geboes L, Dumoutier L, Kelchtermans H, Schurgers E, Mitera T, Renauld JC, et al. Proinflammatory role of the Th17 cytokine interleukin-22 in collagen-induced arthritis in C57BL/6 mice. Arthritis Rheum (2009) 60:390–5. doi:10.1002/art.24220
58. Sarkar S, Zhou X, Justa S, Bommireddy SR. Interleukin-22 reduces the severity of collagen-induced arthritis in association with increased levels of interleukin-10. Arthritis Rheum (2013) 65:960–71. doi:10.1002/art.37849
59. Justa S, Zhou X, Sarkar S. Endogenous IL-22 plays a dual role in arthritis: regulation of established arthritis via IFN-gamma responses. PLoS One (2014) 9:e93279. doi:10.1371/journal.pone.0093279
60. Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol (2007) 179:154–61. doi:10.4049/jimmunol.179.1.154
61. Goldstein I, Ben-Horin S, Koltakov A, Chermoshnuk H, Polevoy V, Berkun Y, et al. alpha1beta1 Integrin+ and regulatory Foxp3+ T cells constitute two functionally distinct human CD4+ T cell subsets oppositely modulated by TNFalpha blockade. J Immunol (2007) 178:201–10. doi:10.4049/jimmunol.178.1.201
62. Herrath J, Muller M, Amoudruz P, Janson P, Michaelsson J, Larsson PT, et al. The inflammatory milieu in the rheumatic joint reduces regulatory T-cell function. Eur J Immunol (2011) 41:2279–90. doi:10.1002/eji.201041004
63. Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood (2009) 113:4240–9. doi:10.1182/blood-2008-10-183251
64. Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood (2008) 112:2340–52. doi:10.1182/blood-2008-01-133967
65. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med (2013) 19:322–8. doi:10.1038/nm.3085
66. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3-T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol (2007) 178:6725–9. doi:10.4049/jimmunol.178.11.6725
67. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity (2008) 29:44–56. doi:10.1016/j.immuni.2008.05.007
68. Cosmi L, Cimaz R, Maggi L, Santarlasci V, Capone M, Borriello F, et al. Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum (2011) 63:2504–15. doi:10.1002/art.30332
69. Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, et al. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci U S A (2010) 107:14751–6. doi:10.1073/pnas.1003852107
70. Nistala K, Moncrieffe H, Newton KR, Varsani H, Hunter P, Wedderburn LR. Interleukin-17-producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheum (2008) 58:875–87. doi:10.1002/art.23291
71. Snir O, Backlund J, Bostrom J, Andersson I, Kihlberg J, Buckner JH, et al. Multifunctional T cell reactivity with native and glycosylated type II collagen in rheumatoid arthritis. Arthritis Rheum (2012) 64:2482–8. doi:10.1002/art.34459
72. Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol (2009) 9:91–105. doi:10.1038/nri2487
73. Chu CQ, Field M, Feldmann M, Maini RN. Localization of tumor necrosis factor alpha in synovial tissues and at the cartilage-pannus junction in patients with rheumatoid arthritis. Arthritis Rheum (1991) 34:1125–32. doi:10.1002/art.1780340908
74. Deleuran BW, Chu CQ, Field M, Brennan FM, Mitchell T, Feldmann M, et al. Localization of tumor necrosis factor receptors in the synovial tissue and cartilage-pannus junction in patients with rheumatoid arthritis. Implications for local actions of tumor necrosis factor alpha. Arthritis Rheum (1992) 35:1170–8. doi:10.1002/art.1780351009
75. Dayer JM, Beutler B, Cerami A. Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J Exp Med (1985) 162:2163–8. doi:10.1084/jem.162.6.2163
76. Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem (2000) 275:4858–64. doi:10.1074/jbc.275.7.4858
77. Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest (2008) 118:3537–45. doi:10.1172/JCI36389
78. Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci U S A (1992) 89:9784–8. doi:10.1073/pnas.89.20.9784
79. Malottki K, Barton P, Tsourapas A, Uthman AO, Liu Z, Routh K, et al. Adalimumab, etanercept, infliximab, rituximab and abatacept for the treatment of rheumatoid arthritis after the failure of a tumour necrosis factor inhibitor: a systematic review and economic evaluation. Health Technol Assess (2011) 15:1–278. doi:10.3310/hta15140
80. Kay J, Calabrese L. The role of interleukin-1 in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) (2004) 43(Suppl 3):iii2–9. doi:10.1093/rheumatology/keh201
81. Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci U S A (2003) 100:5986–90. doi:10.1073/pnas.1035999100
82. McInnes IB, Liew FY, Gracie JA. Interleukin-18: a therapeutic target in rheumatoid arthritis? Arthritis Res Ther (2005) 7:38–41. doi:10.1186/ar1497
83. Plater-Zyberk C, Joosten LA, Helsen MM, Sattonnet-Roche P, Siegfried C, Alouani S, et al. Therapeutic effect of neutralizing endogenous IL-18 activity in the collagen-induced model of arthritis. J Clin Invest (2001) 108:1825–32. doi:10.1172/JCI200112097
84. Wei XQ, Leung BP, Arthur HM, McInnes IB, Liew FY. Reduced incidence and severity of collagen-induced arthritis in mice lacking IL-18. J Immunol (2001) 166:517–21. doi:10.4049/jimmunol.166.1.517
85. Kishimoto T. Interleukin-6: from basic science to medicine – 40 years in immunology. Annu Rev Immunol (2005) 23:1–21. doi:10.1146/annurev.immunol.23.021704.115806
86. Md Yusof MY, Emery P. Targeting interleukin-6 in rheumatoid arthritis. Drugs (2013) 73:341–56. doi:10.1007/s40265-013-0018-2
87. Pope RM, Shahrara S. Possible roles of IL-12-family cytokines in rheumatoid arthritis. Nat Rev Rheumatol (2013) 9:252–6. doi:10.1038/nrrheum.2012.170
88. Joosten LA, Lubberts E, Helsen MM, van den Berg WB. Dual role of IL-12 in early and late stages of murine collagen type II arthritis. J Immunol (1997) 159:4094–102.
89. Pope RM. Apoptosis as a therapeutic tool in rheumatoid arthritis. Nat Rev Immunol (2002) 2:527–35. doi:10.1038/nri846
90. Patel KD, Cuvelier SL, Wiehler S. Selectins: critical mediators of leukocyte recruitment. Semin Immunol (2002) 14:73–81. doi:10.1006/smim.2001.0344
91. Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood (2011) 118:6743–51. doi:10.1182/blood-2011-07-343566
92. Tak PP, Taylor PC, Breedveld FC, Smeets TJ, Daha MR, Kluin PM, et al. Decrease in cellularity and expression of adhesion molecules by anti-tumor necrosis factor alpha monoclonal antibody treatment in patients with rheumatoid arthritis. Arthritis Rheum (1996) 39:1077–81. doi:10.1002/art.1780390702
93. Ates A, Kinikli G, Turgay M, Duman M. Serum-soluble selectin levels in patients with rheumatoid arthritis and systemic sclerosis. Scand J Immunol (2004) 59:315–20. doi:10.1111/j.0300-9475.2004.01389.x
94. Bullard DC, Mobley JM, Justen JM, Sly LM, Chosay JG, Dunn CJ, et al. Acceleration and increased severity of collagen-induced arthritis in P-selectin mutant mice. J Immunol (1999) 163:2844–9.
95. Tarrant TK, Patel DD. Chemokines and leukocyte trafficking in rheumatoid arthritis. Pathophysiology (2006) 13:1–14. doi:10.1016/j.pathophys.2005.11.001
96. Boisvert M, Gendron S, Chetoui N, Aoudjit F. Alpha2 beta1 integrin signaling augments T cell receptor-dependent production of interferon-gamma in human T cells. Mol Immunol (2007) 44:3732–40. doi:10.1016/j.molimm.2007.04.003
97. de Fougerolles AR, Sprague AG, Nickerson-Nutter CL, Chi-Rosso G, Rennert PD, Gardner H, et al. Regulation of inflammation by collagen-binding integrins alpha1beta1 and alpha2beta1 in models of hypersensitivity and arthritis. J Clin Invest (2000) 105:721–9. doi:10.1172/JCI7911
98. Burne MJ, Elghandour A, Haq M, Saba SR, Norman J, Condon T, et al. IL-1 and TNF independent pathways mediate ICAM-1/VCAM-1 up-regulation in ischemia reperfusion injury. J Leukoc Biol (2001) 70:192–8.
99. Lowin T, Straub RH. Integrins and their ligands in rheumatoid arthritis. Arthritis Res Ther (2011) 13:244. doi:10.1186/ar3464
100. Schwartz MA, Assoian RK. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J Cell Sci (2001) 114:2553–60.
101. Stupack DG, Cheresh DA. Integrins and angiogenesis. Curr Top Dev Biol (2004) 64:207–38. doi:10.1016/S0070-2153(04)64009-9
102. Yusuf-Makagiansar H, Anderson ME, Yakovleva TV, Murray JS, Siahaan TJ. Inhibition of LFA-1/ICAM-1 and VLA-4/VCAM-1 as a therapeutic approach to inflammation and autoimmune diseases. Med Res Rev (2002) 22:146–67. doi:10.1002/med.10001
103. Klimiuk PA, Sierakowski S, Latosiewicz R, Cylwik JP, Cylwik B, Skowronski J, et al. Soluble adhesion molecules (ICAM-1, VCAM-1, and E-selectin) and vascular endothelial growth factor (VEGF) in patients with distinct variants of rheumatoid synovitis. Ann Rheum Dis (2002) 61:804–9. doi:10.1136/ard.61.9.804
104. Bullard DC, Hurley LA, Lorenzo I, Sly LM, Beaudet AL, Staite ND. Reduced susceptibility to collagen-induced arthritis in mice deficient in intercellular adhesion molecule-1. J Immunol (1996) 157:3153–8.
105. Kavanaugh AF, Davis LS, Jain RI, Nichols LA, Norris SH, Lipsky PE. A phase I/II open label study of the safety and efficacy of an anti-ICAM-1 (intercellular adhesion molecule-1; CD54) monoclonal antibody in early rheumatoid arthritis. J Rheumatol (1996) 23:1338–44.
106. Contento RL, Campello S, Trovato AE, Magrini E, Anselmi F, Viola A. Adhesion shapes T cells for prompt and sustained T-cell receptor signalling. EMBO J (2010) 29:4035–47. doi:10.1038/emboj.2010.258
107. Montresor A, Toffali L, Constantin G, Laudanna C. Chemokines and the signaling modules regulating integrin affinity. Front Immunol (2012) 3:127. doi:10.3389/fimmu.2012.00127
108. Cascio G, Martin-Cofreces NB, Rodriguez-Frade JM, Lopez-Cotarelo P, Criado G, Pablos JL, et al. CXCL12 regulates through JAK1 and JAK2 formation of productive immunological synapses. J Immunol (2015) 194(11):5509–19. doi:10.4049/jimmunol.1402419
109. Garcia-Bernal D, Parmo-Cabanas M, Dios-Esponera A, Samaniego R, Hernán-P de la Ossa D, Teixido J. Chemokine-induced Zap70 kinase-mediated dissociation of the Vav1-talin complex activates alpha4beta1 integrin for T cell adhesion. Immunity (2009) 31:953–64. doi:10.1016/j.immuni.2009.09.021
110. Perez-Rivero G, Cascio G, Soriano SF, Sanz AG, de Guinoa JS, Rodriguez-Frade JM, et al. Janus kinases 1 and 2 regulate chemokine-mediated integrin activation and naive T-cell homing. Eur J Immunol (2013) 43:1745–57. doi:10.1002/eji.201243178
111. O’Connor P. Natalizumab and the role of alpha 4-integrin antagonism in the treatment of multiple sclerosis. Expert Opin Biol Ther (2007) 7:123–36. doi:10.1517/14712598.7.1.123
112. Eksteen B, Adams DH. GSK-1605786, a selective small-molecule antagonist of the CCR9 chemokine receptor for the treatment of Crohn’s disease. IDrugs (2010) 13:472–81.
113. Walters MJ, Wang Y, Lai N, Baumgart T, Zhao BN, Dairaghi DJ, et al. Characterization of CCX282-B, an orally bioavailable antagonist of the CCR9 chemokine receptor, for treatment of inflammatory bowel disease. J Pharmacol Exp Ther (2010) 335:61–9. doi:10.1124/jpet.110.169714
114. Ni H, Moe S, Myint KT, Htet A. Oral Janus kinase inhibitor for the treatment of rheumatoid arthritis: tofacitinib. ISRN Rheumatol (2013) 2013:357904. doi:10.1155/2013/357904
115. Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med (2012) 367:495–507. doi:10.1056/NEJMoa1109071
116. Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med (2010) 363:1303–12. doi:10.1056/NEJMoa1000500
117. Kelly V, Genovese M. Novel small molecule therapeutics in rheumatoid arthritis. Rheumatology (Oxford) (2013) 52:1155–62. doi:10.1093/rheumatology/kes367
118. Whang JA, Chang BY. Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov Today (2014) 19:1200–4. doi:10.1016/j.drudis.2014.03.028
120. Buckley CD, Amft N, Bradfield PF, Pilling D, Ross E, Arenzana-Seisdedos F, et al. Persistent induction of the chemokine receptor CXCR4 by TGF-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol (2000) 165:3423–9. doi:10.4049/jimmunol.165.6.3423
121. Nanki T, Hayashida K, El-Gabalawy HS, Suson S, Shi K, Girschick HJ, et al. Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J Immunol (2000) 165:6590–8. doi:10.4049/jimmunol.165.11.6590
122. Alaaeddine N, Olee T, Hashimoto S, Creighton-Achermann L, Lotz M. Production of the chemokine RANTES by articular chondrocytes and role in cartilage degradation. Arthritis Rheum (2001) 44:1633–43. doi:10.1002/1529-0131(200107)44:7<1633::AID-ART286>3.0.CO;2-Z
123. Borzi RM, Mazzetti I, Cattini L, Uguccioni M, Baggiolini M, Facchini A. Human chondrocytes express functional chemokine receptors and release matrix-degrading enzymes in response to C-X-C and C-C chemokines. Arthritis Rheum (2000) 43:1734–41. doi:10.1002/1529-0131(200008)43:8<1734::AID-ANR9>3.0.CO;2-B
124. Wei L, Sun X, Kanbe K, Wang Z, Sun C, Terek R, et al. Chondrocyte death induced by pathological concentration of chemokine stromal cell-derived factor-1. J Rheumatol (2006) 33:1818–26.
125. Garcia-Vicuna R, Gomez-Gaviro MV, Dominguez-Luis MJ, Pec MK, Gonzalez-Alvaro I, Alvaro-Gracia JM, et al. CC and CXC chemokine receptors mediate migration, proliferation, and matrix metalloproteinase production by fibroblast-like synoviocytes from rheumatoid arthritis patients. Arthritis Rheum (2004) 50:3866–77. doi:10.1002/art.20615
126. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
127. Lim HW, Lee J, Hillsamer P, Kim CH. Human Th17 cells share major trafficking receptors with both polarized effector T cells and FOXP3+ regulatory T cells. J Immunol (2008) 180:122–9. doi:10.4049/jimmunol.180.1.122
128. Paulissen SM, van Hamburg JP, Dankers W, Lubberts E. The role and modulation of CCR6+ Th17 cell populations in rheumatoid arthritis. Cytokine (2015) 74(1):43–53. doi:10.1016/j.cyto.2015.02.002
129. Tak PP. Chemokine inhibition in inflammatory arthritis. Best Pract Res Clin Rheumatol (2006) 20:929–39. doi:10.1016/j.berh.2006.06.001
130. De Benedetti F, Pignatti P, Bernasconi S, Gerloni V, Matsushima K, Caporali R, et al. Interleukin 8 and monocyte chemoattractant protein-1 in patients with juvenile rheumatoid arthritis. Relation to onset types, disease activity, and synovial fluid leukocytes. J Rheumatol (1999) 26:425–31.
131. Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol (1994) 56:559–64.
132. Iwamoto T, Okamoto H, Kobayashi S, Ikari K, Toyama Y, Tomatsu T, et al. A role of monocyte chemoattractant protein-4 (MCP-4)/CCL13 from chondrocytes in rheumatoid arthritis. FEBS J (2007) 274:4904–12. doi:10.1111/j.1742-4658.2007.06013.x
133. Koch AE, Kunkel SL, Harlow LA, Johnson B, Evanoff HL, Haines GK, et al. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest (1992) 90:772–9. doi:10.1172/JCI115950
134. Luster AD. Chemokines – chemotactic cytokines that mediate inflammation. N Engl J Med (1998) 338:436–45. doi:10.1056/NEJM199802123380706
135. Ogata H, Takeya M, Yoshimura T, Takagi K, Takahashi K. The role of monocyte chemoattractant protein-1 (MCP-1) in the pathogenesis of collagen-induced arthritis in rats. J Pathol (1997) 182:106–14. doi:10.1002/(SICI)1096-9896(199705)182:1<106::AID-PATH816>3.0.CO;2-A
136. Gong JH, Ratkay LG, Waterfield JD, Clark-Lewis I. An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J Exp Med (1997) 186:131–7. doi:10.1084/jem.186.1.131
137. Bruhl H, Cihak J, Schneider MA, Plachy J, Rupp T, Wenzel I, et al. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol (2004) 172:890–8. doi:10.4049/jimmunol.172.2.890
138. Quinones MP, Estrada CA, Kalkonde Y, Ahuja SK, Kuziel WA, Mack M, et al. The complex role of the chemokine receptor CCR2 in collagen-induced arthritis: implications for therapeutic targeting of CCR2 in rheumatoid arthritis. J Mol Med (Berl) (2005) 83:672–81. doi:10.1007/s00109-005-0637-5
139. Zhao Q. Dual targeting of CCR2 and CCR5: therapeutic potential for immunologic and cardiovascular diseases. J Leukoc Biol (2010) 88:41–55. doi:10.1189/jlb.1009671
140. Rathanaswami P, Hachicha M, Sadick M, Schall TJ, McColl SR. Expression of the cytokine RANTES in human rheumatoid synovial fibroblasts. Differential regulation of RANTES and interleukin-8 genes by inflammatory cytokines. J Biol Chem (1993) 268:5834–9.
141. Volin MV, Shah MR, Tokuhira M, Haines GK, Woods JM, Koch AE. RANTES expression and contribution to monocyte chemotaxis in arthritis. Clin Immunol Immunopathol (1998) 89:44–53. doi:10.1006/clin.1998.4590
142. Koch AE, Kunkel SL, Harlow LA, Mazarakis DD, Haines GK, Burdick MD, et al. Macrophage inflammatory protein-1 alpha. A novel chemotactic cytokine for macrophages in rheumatoid arthritis. J Clin Invest (1994) 93:921–8. doi:10.1172/JCI117097
143. Patel DD, Zachariah JP, Whichard LP. CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin Immunol (2001) 98:39–45. doi:10.1006/clim.2000.4957
144. Haringman JJ, Smeets TJ, Reinders-Blankert P, Tak PP. Chemokine and chemokine receptor expression in paired peripheral blood mononuclear cells and synovial tissue of patients with rheumatoid arthritis, osteoarthritis, and reactive arthritis. Ann Rheum Dis (2006) 65:294–300. doi:10.1136/ard.2005.037176
145. Weber C, Weber KS, Klier C, Gu S, Wank R, Horuk R, et al. Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and T(H)1-like/CD45RO(+) T cells. Blood (2001) 97:1144–6. doi:10.1182/blood.V97.4.1144
146. Yang YF, Mukai T, Gao P, Yamaguchi N, Ono S, Iwaki H, et al. A non-peptide CCR5 antagonist inhibits collagen-induced arthritis by modulating T cell migration without affecting anti-collagen T cell responses. Eur J Immunol (2002) 32:2124–32. doi:10.1002/1521-4141(200208)32:8<2124::AID-IMMU2124>3.0.CO;2-S
147. Vierboom MP, Zavodny PJ, Chou CC, Tagat JR, Pugliese-Sivo C, Strizki J, et al. Inhibition of the development of collagen-induced arthritis in rhesus monkeys by a small molecular weight antagonist of CCR5. Arthritis Rheum (2005) 52:627–36. doi:10.1002/art.20850
148. Zapico I, Coto E, Rodriguez A, Alvarez C, Torre JC, Alvarez V. CCR5 (chemokine receptor-5) DNA-polymorphism influences the severity of rheumatoid arthritis. Genes Immun (2000) 1:288–9. doi:10.1038/sj.gene.6363673
149. Desmetz C, Lin YL, Mettling C, Portales P, Noel D, Clot J, et al. Cell surface CCR5 density determines the intensity of T cell migration towards rheumatoid arthritis synoviocytes. Clin Immunol (2007) 123:148–54. doi:10.1016/j.clim.2007.01.004
150. Bradfield PF, Amft N, Vernon-Wilson E, Exley AE, Parsonage G, Rainger GE, et al. Rheumatoid fibroblast-like synoviocytes overexpress the chemokine stromal cell-derived factor 1 (CXCL12), which supports distinct patterns and rates of CD4+ and CD8+ T cell migration within synovial tissue. Arthritis Rheum (2003) 48:2472–82. doi:10.1002/art.11219
151. Matthys P, Hatse S, Vermeire K, Wuyts A, Bridger G, Henson GW, et al. AMD3100, a potent and specific antagonist of the stromal cell-derived factor-1 chemokine receptor CXCR4, inhibits autoimmune joint inflammation in IFN-gamma receptor-deficient mice. J Immunol (2001) 167:4686–92. doi:10.4049/jimmunol.167.8.4686
152. Burman A, Haworth O, Hardie DL, Amft EN, Siewert C, Jackson DG, et al. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J Immunol (2005) 174:1693–700. doi:10.4049/jimmunol.174.3.1693
153. Schmutz C, Hulme A, Burman A, Salmon M, Ashton B, Buckley C, et al. Chemokine receptors in the rheumatoid synovium: upregulation of CXCR5. Arthritis Res Ther (2005) 7:R217–29. doi:10.1186/ar1475
154. Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol (2001) 167:1072–80. doi:10.4049/jimmunol.167.2.1072
155. Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International union of basic and clinical pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev (2014) 66:1–79. doi:10.1124/pr.113.007724
156. Haringman JJ, Gerlag DM, Smeets TJ, Baeten D, van den Bosch F, Bresnihan B, et al. A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum (2006) 54:2387–92. doi:10.1002/art.21975
157. Bizzarri C, Beccari AR, Bertini R, Cavicchia MR, Giorgini S, Allegretti M. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol Ther (2006) 112:139–49. doi:10.1016/j.pharmthera.2006.04.002
158. Haringman JJ, Kraan MC, Smeets TJ, Zwinderman KH, Tak PP. Chemokine blockade and chronic inflammatory disease: proof of concept in patients with rheumatoid arthritis. Ann Rheum Dis (2003) 62:715–21. doi:10.1136/ard.62.8.715
159. Brown MF, Bahnck KB, Blumberg LC, Brissette WH, Burrell SA, Driscoll JP, et al. Piperazinyl CCR1 antagonists – optimization of human liver microsome stability. Bioorg Med Chem Lett (2007) 17:3109–12. doi:10.1016/j.bmcl.2007.03.037
160. Martinez Munoz L, Lucas P, Navarro G, Checa AI, Franco R, Martinez AC, et al. Dynamic regulation of CXCR1 and CXCR2 homo- and heterodimers. J Immunol (2009) 183:7337–46. doi:10.4049/jimmunol.0901802
161. Thelen M, Munoz LM, Rodriguez-Frade JM, Mellado M. Chemokine receptor oligomerization: functional considerations. Curr Opin Pharmacol (2010) 10:38–43. doi:10.1016/j.coph.2009.09.004
162. Barroso R, Martinez Munoz L, Barrondo S, Vega B, Holgado BL, Lucas P, et al. EBI2 regulates CXCL13-mediated responses by heterodimerization with CXCR5. FASEB J (2012) 26:4841–54. doi:10.1096/fj.12-208876
163. Pello OM, Martinez-Munoz L, Parrillas V, Serrano A, Rodriguez-Frade JM, Toro MJ, et al. Ligand stabilization of CXCR4/delta-opioid receptor heterodimers reveals a mechanism for immune response regulation. Eur J Immunol (2008) 38:537–49. doi:10.1002/eji.200737630
164. Martinez-Munoz L, Barroso R, Dyrhaug SY, Navarro G, Lucas P, Soriano SF, et al. CCR5/CD4/CXCR4 oligomerization prevents HIV-1 gp120IIIB binding to the cell surface. Proc Natl Acad Sci U S A (2014) 111:E1960–9. doi:10.1073/pnas.1322887111
165. Rodriguez-Frade JM, Vila-Coro AJ, de Ana AM, Albar JP, Martinez AC, Mellado M. The chemokine monocyte chemoattractant protein-1 induces functional responses through dimerization of its receptor CCR2. Proc Natl Acad Sci U S A (1999) 96:3628–33. doi:10.1073/pnas.96.7.3628
166. Mellado M, Rodriguez-Frade JM, Vila-Coro AJ, Fernandez S, Martin de Ana A, Jones DR, et al. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J (2001) 20:2497–507. doi:10.1093/emboj/20.10.2497
167. Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A (2007) 104:14759–64. doi:10.1073/pnas.0702229104
Keywords: chemokines, cytokines, rheumatoid arthritis, inflammation, cell migration
Citation: Mellado M, Martínez-Muñoz L, Cascio G, Lucas P, Pablos JL and Rodríguez-Frade JM (2015) T cell migration in rheumatoid arthritis. Front. Immunol. 6:384. doi: 10.3389/fimmu.2015.00384
Received: 29 May 2015; Accepted: 13 July 2015;
Published: 27 July 2015
Edited by:
Vinicius Cotta-de-Almeida, Oswaldo Cruz Foundation – FIOCRUZ, BrazilReviewed by:
António Gil Castro, University of Minho, PortugalVasileios Bekiaris, Danish Technical University, Denmark
Copyright: © 2015 Mellado, Martínez-Muñoz, Cascio, Lucas, Pablos and Rodríguez-Frade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario Mellado, Department of Immunology and Oncology, Centro Nacional de Biotecnología/CSIC, Campus Cantoblanco, Darwin, 3, Madrid 28049, Spain, mmellado@cnb.csic.es