 Andrea Gioffredi
Andrea Gioffredi Federica Maritati
Federica Maritati Elena Oliva
Elena Oliva Carlo Buzio
Carlo Buzio- Unit of Nephrology, University Hospital of Parma, Parma, Italy
Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystemic disorder, belonging to the small vessel anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis, defined as an eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract, and necrotizing vasculitis predominantly affecting small to medium-sized vessels, associated with asthma and eosinophilia. EGPA pathogenesis is not well known: HLA-DRB1*04 and *07, HLA-DRB4 and IL10.2 haplotype of the IL-10 promoter gene are the most studied genetic determinants. Among the acquired pathogenetic factors, the exposure to different allergens, infections, vaccinations, drugs, and silica exposure have been involved. Eosinophils are the most characteristic cells in EGPA and different studies have demonstrated their role as effector and immunoregulatory cells. EGPA is considered as a disease with a prevalent activation of the Th-2 cellular-mediated inflammatory response and also humoral immunity plays an important role. A link between B and T inflammatory responses may explain different disease features. EGPA typically develops into three sequential phases: the allergic phase, distinguished by the occurrence of asthma, allergic rhinitis, and sinusitis, the eosinophilic phase, in which the main pathological finding is the eosinophilic organ infiltrations (e.g., lungs, heart, and gastrointestinal system), and the vasculitic phase, characterized by purpura, peripheral neuropathy, and constitutional symptoms. ANCA (especially pANCA anti-myeloperoxidase) are present in 40–60% of the patients. An elevation of IgG4 is frequently found. Corticosteroids and cyclophosphamide are classically used for remission induction, while azathioprine and methotrexate are the therapeutic options for remission maintenance. B-cell depletion with rituximab has shown promising results for remission induction.
Introduction and Epidemiology
Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystemic disorder, belonging to the small vessel anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAVs). According to the 1994 Chapel Hill consensus conference (CHCC), EGPA is defined as an eosinophil-rich and granulomatous inflammation often involving the respiratory tract, and necrotizing vasculitis predominantly affecting small to medium-sized vessels, associated with asthma and eosinophilia. Formerly known as “Churg–Strauss syndrome,” this eponym has been replaced during the 2012 Revised International CHCC, with the aim of focusing on the histopathology of the disease (1). Unlike in the 1990 American College of Rheumatology classification criteria and the former CHCC, the CHCC 2012 has reported for the first time that ANCA are found in EGPA, especially in patients with glomerulonephritis. This reflects some of the newest evidences of the distinction of two EGPA subsets, depending on the presence or the absence of ANCA (Table 1) (2).

Table 1. Diagnostic criteria, classification, and nomenclature of eosinophilic granulomatosis with polyangiitis during the last 20 years.
Eosinophilic granulomatosis with polyangiitis incidence in Europe is 0.5–6.8 new cases/year per million populations, whereas its prevalence is 10.7–13 cases per million populations. It mostly affects subjects between 40 and 60 years old and the mean age at diagnosis is 48 years (3).
Pathogenesis
Eosinophilic granulomatosis with polyangiitis pathogenesis is not well known. The disease is probably the result of a complex interaction in which genetically and environmental factors lead to an inflammatory response whose principal players are eosinophils, T, and B lymphocytes (2) (Figure 1).
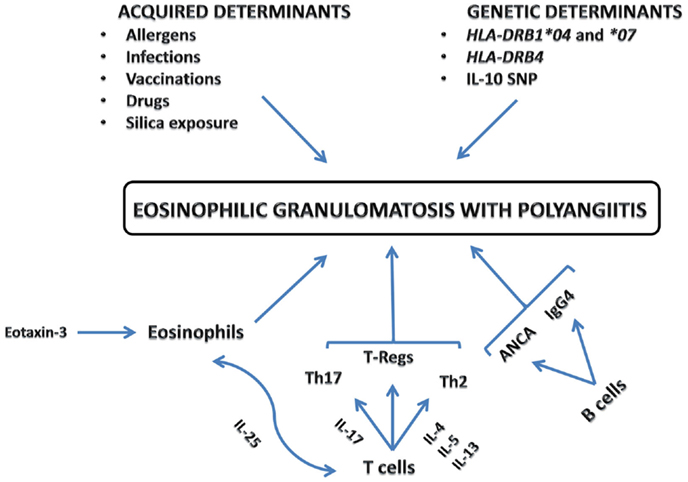
Figure 1. Eosinophilic granulomatosis with polyangiitis pathogenesis.
Genetic Determinants
Eosinophilic granulomatosis with polyangiitis is an HLA-associated disease (4). It has been proven that it is associated with HLA-DRB1*04 and *07 (5) and with HLA-DRB4 (6). This contraction of the class II HLA repertoire suggests a strong CD4+ T lymphocyte activation, possibly triggered by allergens or antigens.
It has been also investigated the presence of single nucleotide polymorphisms (SNP) of the gene, which encodes interleukin (IL)-10, an important molecule for the activation of the Th-2 pathway; EGPA ANCA-negative subset has been associated with the IL10.2 haplotype of the IL-10 promoter gene, a condition, which leads to an increased production of IL-10 (7). This is apparently in line with EGPA pathogenesis, which is characterized by an increased Th-2 response and an increase in IgG4 levels, both of which seem to be mediated by IL-10.
Acquired Determinants
Some environmental triggers have been identified: the exposure to different allergens, infections, vaccinations could trigger the disease. Drugs may also have a pathogenetic role and, among these, the leukotriene receptor antagonists are the most frequently involved more often used as steroid-sparing agents for asthma, their key role in triggering EGPA is still uncertain (8). More recently, also the recombinant anti-IgE monoclonal antibody omalizumab used in patient with asthma has been considered as an EGPA trigger (9–11). According to the most reliable hypotheses, both LTRA and anti-IgE antibody may be involved in EGPA pathogenesis simply unmasking the disease, due to the delayed use of steroids.
A recent review has shown the possible pathogenetic influence of silica exposure in AAVs, including EGPA (12).
Eosinophils
The role of the eosinophils is still uncertain in EGPA but different studies have demonstrated the cytotoxic (13, 14) and pro-coagulant (15, 16) properties of this cell type, which may result in the development of cardiovascular and cerebrovascular complications in patients with any type of hypereosinophilic syndromes including EGPA. Although they are usually considered to be effector cells, they may act as immunoregulatory cells (2): indeed, a cross-talk between T-lymphocytes and eosinophils has been pointed out. In a recent study, high concentrations of IL-25 have been detected in the sera of EGPA patients; eosinophils are the main source of IL-25, which induces T-cells to produce cytokines that stimulate Th-2 and, at the same time, eosinophilic responses (17).
T-Lymphocytes
It has been demonstrated that T-lymphocytes have an important role in the EGPA pathogenesis. T-cells are present in the most of the organ lesions and in some of them, like peripheral neuropathy, they represent the main component. Moreover, serum levels of T-cell activation markers, like IL-2r, are increased during the active phase of the disease (18). T-cells receptors show a restricted repertoire suggesting oligoclonal expansion (19), which is in line with the hypothesis of an antigen-driven disease. Clonal restricted effector CD8+ lymphocytes with a proinflammatory profile have been recently described in patients with EGPA (20).
Specifically, EGPA is considered as a disease with a prevalent activation of the Th-2 pathway. In keeping with this view, it has been demonstrated that tissue infiltrates in patients with EGPA are rich in T-cells with Th-2 makers such as CD294. Furthermore, EGPA patients CD4+ T-cells are able to produce, in vitro, high concentrations of IL-4, IL-5, and IL-13, molecules that hallmark the Th-2 immunoresponse.
High-blood concentrations of IL-17 have been found in patients with EGPA, a finding, which suggests that the involvement of Th17 lymphocytes into EGPA pathogenesis; indeed, these lymphocytes are involved in the pathogenesis of other autoimmune diseases (2).
Finally, reduced levels of regulatory CD4+ T-cells (Tregs) have been discovered in EGPA patients (21, 22). Tregs usually have a protective role toward the development of autoimmune diseases. Lower numbers of Tregs were found in active EGPA patients than in patients with asthma or with chronic eosinophilic pneumonia; additionally, the percentages of circulating Tregs were lower in active than quiescent EGPA (2).
B-Lymphocytes
The role of the humoral immunity in EGPA seems to be less relevant as compared to other autoimmune diseases. Despite this, EGPA patients often show an abnormal humoral response. ANCA are found in about 40% of patients with EGPA, they are characterized by a perinuclear pattern (pANCA) at the immunofluorescence assay and are directed against the neutrophil myeloperoxidase (MPO), as revealed by ELISA. Their pathogenetic role and their potential harmful effect is still matter of debate. Even though animal models and in vitro studies have shown a pathogenic role of the anti-MPO antibodies (23), their role in causing organ damage in EGPA is still unknown.
A substantial number of patients show an increased IgG4 blood levels. In a recent analysis of 46 EGPA patients, IgG4 levels correlated with the number of disease manifestations and the Birmingham vasculitis activity score (BVAS). Furthermore, serum IgG4 levels paralleled the disease course as they normalized during remission. The skewed IgG4 response is likely due to the enhancing effects of the Th-2 cytokines IL-4, IL-5, and IL-13 (24).
Cytokines and Chemokines
Regarding chemotaxis, eotaxin-3 (CCL26), a chemokine, which attracts eosinophils in the sites of inflammation, apparently has a key role into EGPA pathogenesis. Two different studies reported that eotaxin-3 was highly elevated in serum samples of active EGPA patients and correlated highly significantly with eosinophil counts, total immunoglobulin E (IgE) levels, and acute-phase parameters. Immunohistochemical analysis revealed strong expression of eotaxin-3 in endothelial and inflammatory cells in affected tissues of active EGPA patients (25, 26).
Also, CCL17, another Th-2 chemokine, seems to be present into both tissues infiltrates and patients’ sera (27).
Some recent studies have demonstrated that EGPA patients’ T-cells produce, after stimulation in vitro, a large amount of interferon-γ (INF-γ), a cytokine, which boosts Th-1 immune response (28).
The hypothesis of a cross-talk between humoral and cell-mediated immunity and eosinophils is still the object of different pathogenetic studies.
Clinical Features
Eosinophilic granulomatosis with polyangiitis mainly affects patients with asthma (often developed in the adult age), sinusitis, allergic rhinitis, and nasal polyposis (Table 2) (29).
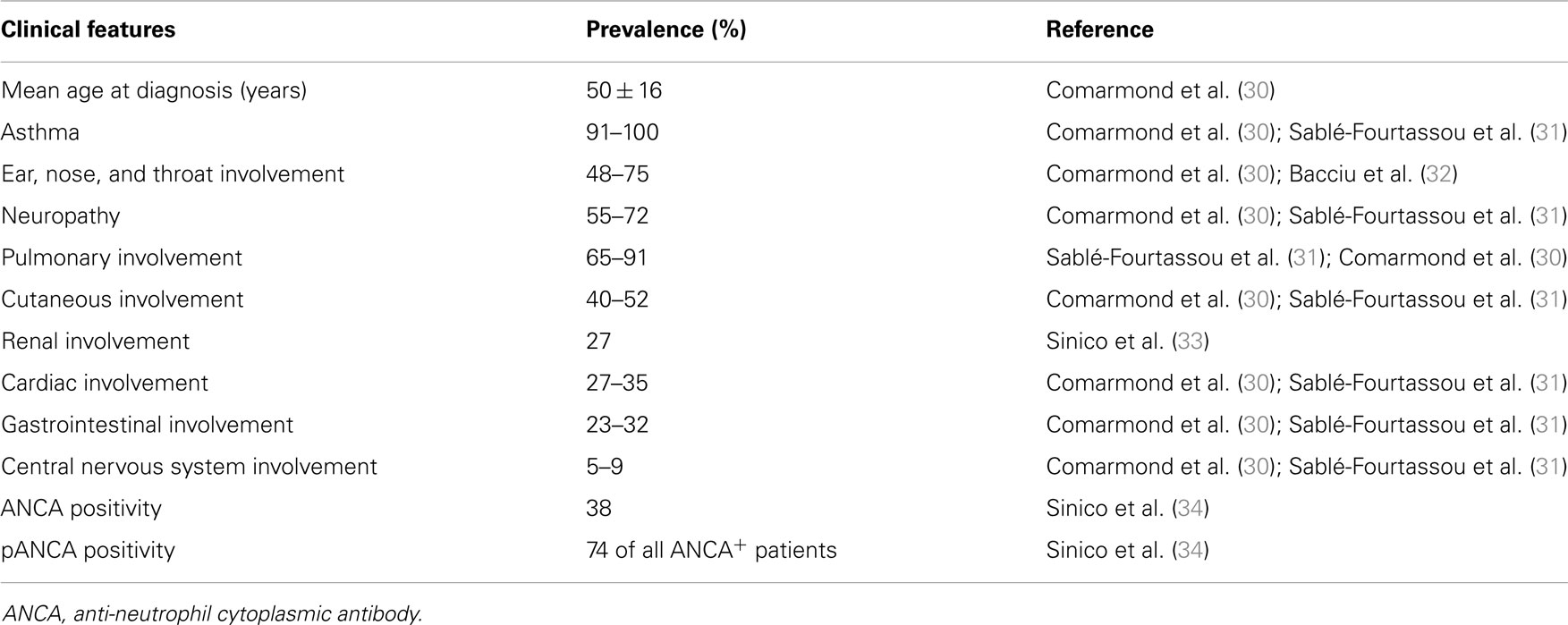
Table 2. Main clinical features in eosinophilic granulomatosis with polyangiitis and their prevalences.
Eosinophilic granulomatosis with polyangiitis typically develops into three sequential phases, marked by a progression of the main symptoms. The first phase, also called prodromic or allergic, is most common in the second or third decade and it is distinguished by the occurrence of asthma, allergic rhinitis, and sinusitis. Subsequently, the eosinophilic phase develops the main pathological findings of this phase are the raise in the peripheral eosinophilic count and the eosinophilic organ infiltrations, especially in lungs, heart, and gastrointestinal system. The third phase is the vasculitic one during this last phase, the patient suffers from the consequences of a necrotizing vasculitis (e.g., purpura, peripheral neuropathy), generally associated with vascular or extravascular granulomatosis and constitutional symptoms like fever, malaise, and weight loss (35).
Eosinophilic granulomatosis with polyangiitis is a multisystemic disease. One of the most frequently involved sites is the respiratory system asthma has a prevalence of about 95% (36). Pulmonary eosinophilic infiltrates may be present and their biopsy is often highly informative for the histopathologic diagnosis (37).
The otorhinolaryngoiatric system is also frequently involved nasal polyposis is one of the conditions, which lead patients to undergo repeat surgery. Allergic rhinosinusitis, epistaxis, and neurosensory hearing loss are other common features (32).
Cardiac involvement may be represents the most harmful manifestation of EGPA characterized by myocardial infarction, pericarditis, or congestive heart failure, it is the main cause of death (30, 36, 38).
Among the most frequent skin manifestations, subcutaneous nodules, and purpura (especially involving the legs) represent a clinical hallmark of the vasculitic phase, a skin biopsy of purpuric lesions generally shows a leukocytoclasic vasculitis (39, 40).
Although less frequent than the other two AAVs, renal involvement occurs in about 25% of the patients and the most typical expression is pauci-immune crescentic glomerulonephritis with a high range of clinical features, from isolated urinary abnormalities (proteinuria, hematuria) to rapidly progressive glomerulonephritis. Kidney involvement is a bad prognosis factor for patients with EGPA (33).
Peripheral neuropathy, either sensory or motor or sensory-motor, affects a large portion of the patients; mononeuritis multiplex, with axonal damage, usually unilateral and asymmetric, is the most characteristic manifestation of peripheral nervous system involvement. Patients report paresthesia and pain in the affected areas (peroneal, tibial, ulnar nerve), especially during the vasculitic stage of the disease (41).
In the gastrointestinal system, the vasculitic phase may be preceded by an eosinophilic gastroenteritis with abdominal pain, diarrhea, and intestinal bleeding (42).
In the 30–40% of the patient, there can be diffuse lymphadenopathy, frequently affecting axillary and cervical lymph nodes (43).
The most frequent laboratory findings in EGPA patients is marked hypereosinophilia, frequently between 5000 and 9000 eosinophils/μL [at least >1500 eosinophils/μL or >10% of the total white blood cells, according to Lanham criteria (44)], this is one of the most common signs of EGPA (36). An increase in non-specific inflammatory markers (ESR, CRP) is often found (36). The role of the complement is still uncertain. ANCA are present approximately in 40–60% of the patients; pANCA (perinuclear) is the prevalent pattern, with antibody specificity for MPO (33, 34, 45).
All these clinical manifestations and laboratory features could be frequently gathered into two patterns: the vasculitic and ANCA-positive phenotype, characterized by manifestations resulting from small and medium-sized vessel vasculitis (e.g., purpura, mononeuritis multiplex, glomerulonephritis) and the eosinophilic, ANCA-negative phenotype, in which the organ is damaged mainly by an eosinophilic infiltration (e.g., pulmonary infiltrates, cardiomyopathy) (2). These findings may have pathogenetic implications, as they suggest that ANCA, as observed in MPO-ANCA mouse models, mediate vasculitis in EGPA as well; however, there are no animal models of EGPA. In addition, the ANCA-positive and ANCA-negative subsets are not clearly separated, as overlapping manifestations occur very frequently.
Histopathology
The main histological findings in EGPA are the extravascular granulomas, small and medium-sized vessels vasculitis, and the eosinophilic infiltrates.
Interstitial and vascular granulomas are composed by eosinophilic necrotic matrix surrounded by giant cells and palisading lymphocytes. The vasculitic process affects mainly small and medium vessels (especially small arteries) and is characterized by fibrinoid necrosis of the vessel wall associated or not with granuloma or eosinophilic infiltrates (46, 47). It is difficult to find all these features together, which makes the histological diagnosis sometimes challenging (48). In addition, specific disease manifestations often show specific histopathological features; for example, purpura is caused by a leucocytoclastic vasculitis (eosinophilic infiltration or fibrinoid necrosis is frequently absent) and alveolar hemorrhage depends on an alveolar capillaritis (without granuloma) (36). Furthermore, glomerulonephritis (33) and peripheral neuropathy frequently lack eosinophilic infiltrates. Gastrointestinal biopsies reveal eosinophilic tissue infiltration and histological signs of mesenteric vessel vasculitis, which may induce bowel ischemia (42).
Cardiac involvement may show coronary vasculitis, myocardial granuloma, eosinophilic endomyocarditis, and pericarditis (36).
Differential Diagnosis
Different conditions have to be considered in the differential diagnosis, mainly eosinophilic and vasculitic diseases.
Parasitic infections as well as hypersensitivity reactions (e.g., to drugs) must be excluded. The hypereosinophilic syndrome (HES) is characterized by persistent eosinophilia and organ involvement without a reason, which can explain hypeosinophilia. Cardiac and pulmonary manifestations are analog to those of EGPA patients but subjects with HES usually do not have asthma or vasculitic complication like purpura or glomerulonephritis; furthermore, ANCA are absent in HES (49). A recent revised classification of HESs has focused on the pathogenesis of many hypereosinophilic disorders: myeloproliferative and lymphocytic forms of HES should be excluded in all patients. Particularly, Fip1-like-1(FIP1L1)/platelet-derived growth factor receptor α (PDGFRA) fusion genes must be investigated (50).
Broncho-pulmonary allergic aspergillosis may mimic pulmonary involvement in EGPA: differential diagnosis is helped by finding Aspergillus spp at bronchoscopy lavage or dosing Aspergillus fumigatus specific serum IgE, which are pathognomonic of allergic aspergillosis (51).
Acute eosinophilic pneumonia is featured by pulmonary infiltrates and bronchoscopy lavage rich in eosinophils but usually originates as an acute illness with fever and dyspnea, without peripheral eosinophilia or other organ involvement.
Chronic eosinophilic pneumonia diagnosis is more insidious. Patients may present with asthma, peripheral eosinophilia, and constitutional symptoms. The absence of other organ manifestations and the negativity of ANCA may help to differentiate chronic eosinophilic pneumonia from EGPA (52).
Eosinophilic granulomatosis with polyangiitis must be distinguished from the other AAVs. Granulomatosis with polyangiitis (GPA) may mimic particular aspects of EGPA, especially in those patients, which present peripheral eosinophilia, the ANCA specificity (cANCA PR3-specific, in GPA) and the presence, in GPA, of pulmonary cavitated nodules associated with nasal crusting and nasal and paranasal sinuses erosion, allow clinicians to differentiate the two vasculitides.
Although microscopic polyangiitis (MPA) could be also characterized by pANCA with MPO specificity, it rarely shows peripheral eosinophilia, nodules, or eosinophilic pulmonary infiltrates (48).
Finally, EGPA must be differentiated from IgG4-related disease (IgG4-RD), which may present with allergic manifestations, blood eosinophilia, pulmonary infiltrates, and sinusitis. However, tissue biopsies in patients with IgG4-RD show fibrosis and obliterative phlebitis, without vasculitis or eosinophilic granulomas (53).
In our center experience, first level examinations include blood tests and, in particular, complete blood cell count, ESR, CRP, immunoglobulins with their subclasses (especially IgG subclasses), rheumatoid factor, ANCA, eosinophil cationic protein (ECP), serum B12 levels (elevated in myeloproliferative neoplasms), and a screening of renal function and urinalysis. Detection of FIP1L1/PDGFRA fusion genes and stool cultures for ova and parasite examination must be done in the early stages of diagnosis. ANCA are thought to be useful in the differential diagnosis between EGPA and other (especially infectious and hematological) eosinophilic disorders. Likewise, finding fusion genes clearly points toward a diagnosis of myeloproliferative HES. The differential diagnosis with lymphocytic forms of HES is more challenging, as most laboratories do not perform clonal analysis of circulating lymphocyte subsets or their intracellular cytokine production, which could be helpful in these conditions.
Second level examinations include imaging studies such as lung and facial computed tomography (CT), as well as functional studies such as electromyography.
Finally, kidney biopsy and a bronchoscopy with bronchoalveolar lavage are reserved for those patients with severe (and often rapidly progressive) clinical manifestations.
Treatment and Outcome
Eosinophilic granulomatosis with polyangiitis treatment is a matter of debate because of the lack of large-scale, randomized controlled trials. The five factors score (FFS) may be a guide for clinicians, this score assigns one point to each of the following items, namely, gastrointestinal involvement, CNS involvement, cardiac involvement, proteinuria >1 g/24 h and serum creatinine >141 μmol/L (35). Patients with poor prognosis factors (FFS ≥1) are often treated with both glucocorticoids (classically prednisone at dosage of 1 mg/kg of total body weight/day with a maximum dosage of 75 mg/day, for 1 month and then tapered) and cyclophosphamide (CYC, 2 mg/kg of total body weight/day), while the typical approach for patients with a better prognosis (e.g., FFS of 0) is glucocorticoid therapy alone (54). Recently, a revised FFS has been proposed an age over 65 years, cardiac symptoms, gastrointestinal involvement, renal insufficiency (serum creatinine >150 μmol/L) and absence of ear, nose, and throat manifestations have been pointed out as predictors of 5-year mortality (55).
Classically, used therapies in EGPA remission maintenance are azathioprine or methotrexate (56).
Although primarily used for GPA, the BVAS, a clinical index of disease activity (57), might be useful to better decide when to stop therapy with CYC and introduce maintenance therapy like azathioprine or methotrexate.
Cyclophosphamide toxicity has long been known (58) and, based on our center experience, we recommend not to exceed the dose of 10–15 g of CYC (including both oral and pulse medications). On the other hand, too-short duration of CYC administration has been associated with more relapses (59).
Azathioprine too requires a constant monitoring of liver function, due to the drug-related hepatotoxicity (60).
B-cell depletion adjunct therapy with rituximab has shown promising results for remission induction (61–67).
Interleukin-5, a major survival factor for eosinophils, has been targeted in patients with EGPA using the monoclonal antibody mepolizumab. Use of mepolizumab in refractory cases (68, 69) and steroid-dependent patients (70) has given positive results but EGPA manifestations recurred on drug cessation.
On the assumption of its inhibitory effects on the eosinophil degranulation, interferon-alpha therapy has been tried with positive results in refractory patients, but the severe drug-related toxicity has greatly limited its use (71, 72).
Plasmapheresis may be an adjunctive therapy particularly in patients with rapidly progressive glomerulonephritis, peripheral neuropathy, or alveolar hemorrhage (2).
Eosinophilic granulomatosis with polyangiitis outcomes are well represented in a retrospective study of 383 EGPA patients in the French Vasculitis Study Group cohort. Vasculitis relapse occurred in 97 patients (25.3%), while 72 additional patients experienced asthma flares, sinusitis, and/or increased eosinophilia. Of the 383 patients, 45 (11.7%) died and the major cause of death was attributed to cardiac events. Five-year and 10-year survival rates were, respectively, 88.9 and 78.6%. Vasculitis relapse-free survival rate at 5 years was 64.8%, while at 10 years was 54.4%. ANCA positivity and cutaneous signs were independent predictors of relapse (30).
Another recent analysis of EGPA patients’ long-term follow up has demonstrated that the outcome of EGPA is good with respect to mortality. According to the analysis of 118 patients with EGPA (enrolled in two prospective trials), 108 (91.5%) patients achieved remission (34 of the 108 achieved long-term remission without relapse) and 12 (10.2%) died (only 5 of them died for EGPA-related causes). During relapses, pulmonary symptoms predominated (81%), followed by ear nose and throat signs (38%) and mononeuritis multiplex (36%) (73).
Finally, in a German cohort of 150 EGPA patients, the analysis of the follow-up of 104 of them has evidenced that 70 patients (67.3%) attained remission after conventional therapies, 21 (14%) suffered from major relapses and 42 (28%) from minor relapses. Twelve patients died 94 ± 16 (mean ± SD) months after diagnosis (74).
Perspective Future
Despite the great levels of knowledge reached, more has to be done to clarify EGPA pathogenesis, a genome-wide association study (GWAS) will probably help to better understand the genetic determinants of the disease. Besides, the environmental factors like silica or any other occupational exposure (e.g., asbestos) must be studied in depth.
In the future, probably, the distinction between ANCA+ and ANCA− small vessels vasculitides will lead to re-define the current classification criteria with a more simplistic view of all the AAVs.
Despite this, clinicians should keep in mind all the distinctive clinical features and differential diagnosis approaches that make EGPA one of the more characteristic and complex AAV.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 Revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum (2013) 65(1):1–11. doi: 10.1002/art.37715
2. Vaglio A, Moosig F, Zwerina J. Churg-Strauss syndrome: update on pathophysiology and treatment. Curr Opin Rheumatol (2012) 24(1):24–30. doi:10.1097/BOR.0b013e32834d85ce
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Mouthon L, Dunogue B, Guillevin L. Diagnosis and classification of eosinophilic granulomatosis with polyangiitis (formerly named Churg-Strauss syndrome). J Autoimmun (2014) 48-49:99–103. doi:10.1016/j.jaut.2014.01.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Alberici F, Martorana D, Bonatti F, Gioffredi A, Lyons PA, Vaglio A. Genetics of ANCA-associated vasculitides: HLA and beyond. Clin Exp Rheumatol (2014) 32(3 Suppl 82):90–7.
5. Wieczorek S, Hellmich B, Gross WL, Epplen JT. Associations of Churg-Strauss syndrome with the HLA-DRB1 locus, and relationship to the genetics of antineutrophil cytoplasmic antibody-associated vasculitides: comment on the article by Vaglio et al. Arthritis Rheum (2008) 58(1):329–30. doi:10.1002/art.23209
6. Vaglio A, Martorana D, Maggiore U, Grasselli C, Zanetti A, Pesci A, et al. HLA-DRB4 as a genetic risk factor for Churg-Strauss syndrome. Arthritis Rheum (2007) 56(9):3159–66. doi:10.1002/art.22834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Wieczorek S, Hellmich B, Arning L, Moosig F, Lamprecht P, Gross WL, et al. Functionally relevant variations of the interleukin-10 gene associated with antineutrophil cytoplasmic antibody-negative Churg-Strauss syndrome, but not with Wegener’s granulomatosis. Arthritis Rheum (2008) 58(6):1839–48. doi:10.1002/art.23496
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Bibby S, Healy B, Steele R, Kumareswaran K, Nelson H, Beasley R. Association between leukotriene receptor antagonist therapy and Churg-Strauss syndrome: an analysis of the FDA AERS database. Thorax (2010) 65(2):132–8. doi:10.1136/thx.2009.120972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Wechsler ME, Wong DA, Miller MK, Lawrence-Miyasaki L. Churg-Strauss syndrome in patients treated with omalizumab. Chest (2009) 136(2):507–18. doi:10.1378/chest.08-2990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Giavina-Bianchi P, Giavina-Bianchi M, Agondi R, Kalil J. Administration of anti-IgE to a Churg-Strauss syndrome patient. Int Arch Allergy Immunol (2007) 144(2):155–8. doi:10.1159/000103228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Puechal X, Rivereau P, Vinchon F. Churg-Strauss syndrome associated with omalizumab. Eur J Intern Med (2008) 19(5):364–6. doi:10.1016/j.ejim.2007.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Gomez-Puerta JA, Gedmintas L, Costenbader KH. The association between silica exposure and development of ANCA-associated vasculitis: systematic review and meta-analysis. Autoimmun Rev (2013) 12(12):1129–35. doi:10.1016/j.autrev.2013.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Young JD, Peterson CG, Venge P, Cohn ZA. Mechanism of membrane damage mediated by human eosinophil cationic protein. Nature (1986) 321(6070):613–6. doi:10.1038/321613a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Shah AM, Brutsaert DL, Meulemans AL, Andries LJ, Capron M. Eosinophils from hypereosinophilic patients damage endocardium of isolated feline heart muscle preparations. Circulation (1990) 81(3):1081–8. doi:10.1161/01.CIR.81.3.1081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Maino A, Rossio R, Cugno M, Marzano AV, Tedeschi A. Hypereosinophilic syndrome, Churg-Strauss syndrome and parasitic diseases: possible links between eosinophilia and thrombosis. Curr Vasc Pharmacol (2012) 10(5):670–5. doi:10.2174/157016112801784594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Slungaard A, Vercellotti GM, Tran T, Gleich GJ, Key NS. Eosinophil cationic granule proteins impair thrombomodulin function. A potential mechanism for thromboembolism in hypereosinophilic heart disease. J Clin Invest (1993) 91(4):1721–30. doi:10.1172/JCI116382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Terrier B, Bièche I, Maisonobe T, Laurendeau I, Rosenzwajg M, Kahn JE, et al. Interleukin-25: a cytokine linking eosinophils and adaptive immunity in Churg-Strauss syndrome. Blood (2010) 116(22):4523–31. doi:10.1182/blood-2010-02-267542
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Schmitt WH, Csernok E, Kobayashi S, Klinkenborg A, Reinhold-Keller E, Gross WL. Churg-Strauss syndrome: serum markers of lymphocyte activation and endothelial damage. Arthritis Rheum (1998) 41(3):445–52. doi:10.1002/1529-0131(199803)41:3<445::AID-ART10>3.3.CO;2-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Guida G, Vallario A, Stella S, Boita M, Circosta P, Mariani S, et al. Clonal CD8+ TCR-Vbeta expanded populations with effector memory phenotype in Churg Strauss syndrome. Clin Immunol (2008) 128(1):94–102. doi:10.1016/j.clim.2008.03.505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Boita M, Guida G, Circosta P, Elia AR, Stella S, Heffler E, et al. The molecular and functional characterization of clonally expanded CD8+ TCR BV T cells in eosinophilic granulomatosis with polyangiitis (EGPA). Clin Immunol (2014) 152(1–2):152–63. doi:10.1016/j.clim.2014.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Lepse N, Abdulahad WH, Kallenberg CG, Heeringa P. Immune regulatory mechanisms in ANCA-associated vasculitides. Autoimmun Rev (2011) 11(2):77–83. doi:10.1016/j.autrev.2011.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Free ME, Bunch DO, McGregor JA, Jones BE, Berg EA, Hogan SL, et al. Patients with antineutrophil cytoplasmic antibody-associated vasculitis have defective Treg cell function exacerbated by the presence of a suppression-resistant effector cell population. Arthritis Rheum (2013) 65(7):1922–33. doi:10.1002/art.37959
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Jennette JC, Xiao H, Falk R, Gasim AM. Experimental models of vasculitis and glomerulonephritis induced by antineutrophil cytoplasmic autoantibodies. Contrib Nephrol (2011) 169:211–20. doi:10.1159/000314776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Vaglio A, Strehl JD, Manger B, Maritati F, Alberici F, Beyer C, et al. IgG4 immune response in Churg-Strauss syndrome. Ann Rheum Dis (2012) 71(3):390–3. doi:10.1136/ard.2011.155382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Zwerina J, Bach C, Martorana D, Jatzwauk M, Hegasy G, Moosig F, et al. Eotaxin-3 in Churg-Strauss syndrome: a clinical and immunogenetic study. Rheumatology (Oxford) (2011) 50(10):1823–7. doi:10.1093/rheumatology/keq445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Polzer K, Karonitsch T, Neumann T, Eger G, Haberler C, Soleiman A, et al. Eotaxin-3 is involved in Churg-Strauss syndrome – a serum marker closely correlating with disease activity. Rheumatology (Oxford) (2008) 47(6):804–8. doi:10.1093/rheumatology/ken033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Jakiela B, Sanak M, Szczeklik W, Sokolowska B, Plutecka H, Mastalerz L, et al. Both Th2 and Th17 responses are involved in the pathogenesis of Churg-Strauss syndrome. Clin Exp Rheumatol (2011) 29(1 Suppl 64):S23–34.
28. Kiene M, Csernok E, Müller A, Metzler C, Trabandt A, Gross WL. Elevated interleukin-4 and interleukin-13 production by T cell lines from patients with Churg-Strauss syndrome. Arthritis Rheum (2001) 44(2):469–73. doi:10.1002/1529-0131(200102)44:2<469::AID-ANR66>3.0.CO;2-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Vaglio A, Buzio C, Zwerina J. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): state of the art. Allergy (2013) 68(3):261–73. doi:10.1111/all.12088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum (2013) 65(1):270–81. doi:10.1002/art.37721
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Sablé-Fourtassou R, Cohen P, Mahr A, Pagnoux C, Mouthon L, Jayne D, et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med (2005) 143(9):632–8. doi:10.7326/0003-4819-143-9-200511010-00006
32. Bacciu A, Bacciu S, Mercante G, Ingegnoli F, Grasselli C, Vaglio A, et al. Ear, nose and throat manifestations of Churg-Strauss syndrome. Acta Otolaryngol (2006) 126(5):503–9. doi:10.1080/00016480500437435
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Sinico RA, Di Toma L, Maggiore U, Tosoni C, Bottero P, Sabadini E, et al. Renal involvement in Churg-Strauss syndrome. Am J Kidney Dis (2006) 47(5):770–9. doi:10.1053/j.ajkd.2006.01.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Sinico RA, Di Toma L, Maggiore U, Bottero P, Radice A, Tosoni C, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum (2005) 52(9):2926–35. doi:10.1002/art.21250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Pagnoux C, Guilpain P, Guillevin L. Churg-Strauss syndrome. Curr Opin Rheumatol (2007) 19(1):25–32. doi:10.1097/BOR.0b013e3280119854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) (1999) 78(1):26–37. doi:10.1097/00005792-199901000-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Allen JN, Davis WB. Eosinophilic lung diseases. Am J Respir Crit Care Med (1994) 150(5 Pt 1):1423–38. doi:10.1164/ajrccm.150.5.7952571
38. Neumann T, Manger B, Schmid M, Kroegel C, Hansch A, Kaiser WA, et al. Cardiac involvement in Churg-Strauss syndrome: impact of endomyocarditis. Medicine (Baltimore) (2009) 88(4):236–43. doi:10.1097/MD.0b013e3181af35a5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Schwartz RA, Churg J. Churg-Strauss syndrome. Br J Dermatol (1992) 127(3):199–204. doi:10.1111/j.1365-2133.1992.tb00114.x
40. Bosco L, Peroni A, Schena D, Colato C, Girolomoni G. Cutaneous manifestations of Churg-Strauss syndrome: report of two cases and review of the literature. Clin Rheumatol (2011) 30(4):573–80. doi:10.1007/s10067-010-1593-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Cattaneo L, Chierici E, Pavone L, Grasselli C, Manganelli P, Buzio C, et al. Peripheral neuropathy in Wegener’s granulomatosis, Churg-Strauss syndrome and microscopic polyangiitis. J Neurol Neurosurg Psychiatry (2007) 78(10):1119–23. doi:10.1136/jnnp.2006.111013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Vaglio A, Corradi D, Ronda N, Garini G, Buzio C. Large bowel obstruction heralding Churg-Strauss syndrome. Am J Gastroenterol (2004) 99(3):562–3. doi:10.1111/j.1572-0241.2004.04105.x
43. Churg A, Brallas M, Cronin SR, Churg J. Formes frustes of Churg-Strauss syndrome. Chest (1995) 108(2):320–3. doi:10.1378/chest.108.2.320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore) (1984) 63(2):65–81. doi:10.1097/00005792-198403000-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Keogh KA, Specks U. Churg-Strauss syndrome: clinical presentation, antineutrophil cytoplasmic antibodies, and leukotriene receptor antagonists. Am J Med (2003) 115(4):284–90. doi:10.1016/S0002-9343(03)00359-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum (1990) 33(8):1094–100. doi:10.1002/art.1780330806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Lie JT. Illustrated histopathologic classification criteria for selected vasculitis syndromes. American College of Rheumatology Subcommittee on Classification of Vasculitis. Arthritis Rheum (1990) 33(8):1074–87. doi:10.1002/art.1780330804
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Vaglio A, Casazza I, Grasselli C, Corradi D, Sinico RA, Buzio C. Churg-Strauss syndrome. Kidney Int (2009) 76(9):1006–11. doi:10.1038/ki.2009.210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Corradi D, Vaglio A, Maestri R, Legname V, Leonardi G, Bartoloni G, et al. Eosinophilic myocarditis in a patient with idiopathic hypereosinophilic syndrome: insights into mechanisms of myocardial cell death. Hum Pathol (2004) 35(9):1160–3. doi:10.1016/j.humpath.2004.05.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Simon HU, Rothenberg ME, Bochner BS, Weller PF, Wardlaw AJ, Wechsler ME, et al. Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol (2010) 126(1):45–9. doi:10.1016/j.jaci.2010.03.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Sinico RA, Bottero P. Churg-Strauss angiitis. Best Pract Res Clin Rheumatol (2009) 23(3):355–66. doi:10.1016/j.berh.2009.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Wechsler ME. Pulmonary eosinophilic syndromes. Immunol Allergy Clin North Am (2007) 27(3):477–92. doi:10.1016/j.iac.2007.07.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Vaglio A, Zwerina J. IgG4-related disease. N Engl J Med (2012) 366(17):1643–47. doi:10.1056/NEJMc1202768$SA5
54. Mahr A, Moosig F, Neumann T, Szczeklik W, Taillé C, Vaglio A, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): evolutions in classification, etiopathogenesis, assessment and management. Curr Opin Rheumatol (2014) 26(1):16–23. doi:10.1097/BOR.0000000000000015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P, et al. The five-factor score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) (2011) 90(1):19–27. doi:10.1097/MD.0b013e318205a4c6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Jayne D. New-generation therapy for ANCA-associated vasculitis. Clin Exp Nephrol (2013) 17(5):694–6. doi:10.1007/s10157-013-0855-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, Emery P, et al. Birmingham vasculitis activity score (BVAS) in systemic necrotizing vasculitis. QJM (1994) 87(11):671–8.
58. Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med (1992) 116(6):488–98. doi:10.7326/0003-4819-116-6-488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Cohen P, Pagnoux C, Mahr A, Arène JP, Mouthon L, Le Guern V, et al. Churg-Strauss syndrome with poor-prognosis factors: a prospective multicenter trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in forty-eight patients. Arthritis Rheum (2007) 57(4):686–93. doi:10.1002/art.22679
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. de Boer NK, van Bodegraven AA, Jharap B, de Graaf P, Mulder CJ. Drug Insight: pharmacology and toxicity of thiopurine therapy in patients with IBD. Nat Clin Pract Gastroenterol Hepatol (2007) 4(12):686–94. doi:10.1038/ncpgasthep1000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Pepper RJ, Fabre MA, Pavesio C, Gaskin G, Jones RB, Jayne D, et al. Rituximab is effective in the treatment of refractory Churg-Strauss syndrome and is associated with diminished T-cell interleukin-5 production. Rheumatology (Oxford) (2008) 47(7):1104–5. doi:10.1093/rheumatology/ken175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Jones RB, Ferraro AJ, Chaudhry AN, Brogan P, Salama AD, Smith KG, et al. A multicenter survey of rituximab therapy for refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum (2009) 60(7):2156–68. doi:10.1002/art.24637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Cartin-Ceba R, Keogh KA, Specks U, Sethi S, Fervenza FC. Rituximab for the treatment of Churg-Strauss syndrome with renal involvement. Nephrol Dial Transplant (2011) 26(9):2865–71. doi:10.1093/ndt/gfq852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Saech J, Owczarczyk K, Roesgen S, Hallek M, Rubbert A. Successful use of rituximab in a patient with Churg-Strauss syndrome and refractory central nervous system involvement. Ann Rheum Dis (2010) 69(6):1254–5. doi:10.1136/ard.2009.109850
65. Kaushik VV, Reddy HV, Bucknall RC. Successful use of rituximab in a patient with recalcitrant Churg-Strauss syndrome. Ann Rheum Dis (2006) 65(8):1116–7. doi:10.1136/ard.2005.047308
66. Koukoulaki M, Smith KG, Jayne DR. Rituximab in Churg-Strauss syndrome. Ann Rheum Dis (2006) 65(4):557–9. doi:10.1136/ard.2005.042937
67. Thiel J, Hässler F, Salzer U, Voll RE, Venhoff N. Rituximab in the treatment of refractory or relapsing eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). Arthritis Res Ther (2013) 15(5):R133. doi:10.1186/ar4313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Kahn JE, Grandpeix-Guyodo C, Marroun I, Catherinot E, Mellot F, Roufosse F, et al. Sustained response to mepolizumab in refractory Churg-Strauss syndrome. J Allergy Clin Immunol (2010) 125(1):267–70. doi:10.1016/j.jaci.2009.10.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Moosig F, Gross WL, Herrmann K, Bremer JP, Hellmich B. Targeting interleukin-5 in refractory and relapsing Churg-Strauss syndrome. Ann Intern Med (2011) 155(5):341–3. doi:10.7326/0003-4819-155-5-201109060-00026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Kim S, Marigowda G, Oren E, Israel E, Wechsler ME. Mepolizumab as a steroid-sparing treatment option in patients with Churg-Strauss syndrome. J Allergy Clin Immunol (2010) 125(6):1336–43. doi:10.1016/j.jaci.2010.03.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Metzler C, Schnabel A, Gross WL, Hellmich B. A phase II study of interferon-alpha for the treatment of refractory Churg-Strauss syndrome. Clin Exp Rheumatol (2008) 26(3 Suppl 49):S35–40.
72. Tatsis E, Schnabel A, Gross WL. Interferon-alpha treatment of four patients with the Churg-Strauss syndrome. Ann Intern Med (1998) 129(5):370–4. doi:10.7326/0003-4819-129-5-199809010-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Samson M, Puéchal X, Devilliers H, Ribi C, Cohen P, Stern M, et al. Long-term outcomes of 118 patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) enrolled in two prospective trials. J Autoimmun (2013) 43:60–9. doi:10.1016/j.jaut.2013.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Moosig F, Bremer JP, Hellmich B, Holle JU, Holl-Ulrich K, Laudien M, et al. A vasculitis centre based management strategy leads to improved outcome in eosinophilic granulomatosis and polyangiitis (Churg-Strauss, EGPA): monocentric experiences in 150 patients. Ann Rheum Dis (2013) 72(6):1011–7. doi:10.1136/annrheumdis-2012-201531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: eosinophilic granulomatosis with polyangiitis, vasculitis, eosinophils, vascular diseases, ANCA-associated vasculitis
Citation: Gioffredi A, Maritati F, Oliva E and Buzio C (2014) Eosinophilic granulomatosis with polyangiitis: an overview. Front. Immunol. 5:549. doi: 10.3389/fimmu.2014.00549
Received: 04 July 2014; Accepted: 15 October 2014;
Published online: 03 November 2014.
Edited by:
Cornelia Weyand, Stanford University, USAReviewed by:
Matthew Cook, Australian National University, AustraliaMohey Eldin El Shikh, Queen Mary University of London, UK
Eric Matteson, Mayo Clinic, USA
Bhaskar Dasgupta, Southend University Hospital, UK
Copyright: © 2014 Gioffredi, Maritati, Oliva and Buzio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Gioffredi, Unit of Nephrology, University Hospital of Parma, Via Gramsci 14, Parma 43126, Italy e-mail: gioffredi.andrea@virgilio.it