 Marie-Therese Haider
Marie-Therese Haider Hanna Taipaleenmäki
Hanna Taipaleenmäki- Molecular Skeletal Biology Laboratory, Department of Trauma, Hand and Reconstructive Surgery, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Bone metastases are a common and devastating feature of late-stage breast cancer. Metastatic bone disease is a consequence of disturbed bone remodeling due to pathological interactions between cancer cells and the bone microenvironment (BME). In the BME, breast cancer cells severely alter the balanced bone formation and bone resorption driven by osteoblasts and osteoclasts. The complex cellular cross talk in the BME is governed by secreted molecules, signaling pathways and epigenetic cues including non-coding RNAs. MicroRNAs (miRNAs) are small non-coding RNAs that reduce protein abundance and regulate several biological processes, including bone remodeling. Under pathological conditions, abnormal miRNA signaling contributes to the progression of diseases, such as bone metastasis. Recently miRNAs have been demonstrated to regulate several key drivers of bone metastasis. Furthermore, miRNAs are implicated as important regulators of cellular interactions within the metastatic BME. As a consequence, targeting the BME by miRNA delivery or antagonism has been reported to limit disease progression in experimental and preclinical conditions positioning miRNAs as emerging novel therapeutic tools in metastatic bone disease. This review will summarize our current understanding on the composition and function of the metastatic BME and discuss the recent advances how miRNAs can modulate pathological interactions in the bone environment.
Introduction
Breast cancer is one of the most common malignancies in the world. Approximately 12% of women are diagnosed with breast cancer during their lifetime (1). After successful treatment of the primary tumor that often comprises surgery, adjuvant chemo- and radiation therapy, and the administration of anti-hormonal drugs, patients frequently suffer from distant metastases even decades after a disease-free interval (2). Bone is the most common site for breast cancer metastases, and approximately 70% of patients with advanced breast cancer suffer from osteolytic bone metastases (3). Osteolytic metastases are frequently associated with skeletal-related events (SREs), including fractures and spinal cord compression, that are often accompanied by severe pain and hypercalcemia (4).
In a physiological context, bone mass is maintained by the balanced activities of matrix-producing osteoblasts (OBs) that originate from mesenchymal cells and can become matrix-entrapped osteocytes (OCYs), and bone-resorbing osteoclasts (OCs) that arise from the hematopoietic lineage (5). OC function and bone resorption is stimulated by the receptor activator of NFκB ligand (RANKL) that is expressed in membrane-bound and soluble forms by OBs and OCYs (Figure 1). The activity is restricted by osteoprotegerin, which is a soluble decoy receptor against RANKL (6). Under pathological conditions, for instance in the context of metastatic breast cancer disease, breast cancer cells colonize the bone marrow microenvironment and severely disturb the balance between bone formation and bone resorption (7). This multi-directional process termed “vicious cycle” perpetuates metastatic bone destruction (8).
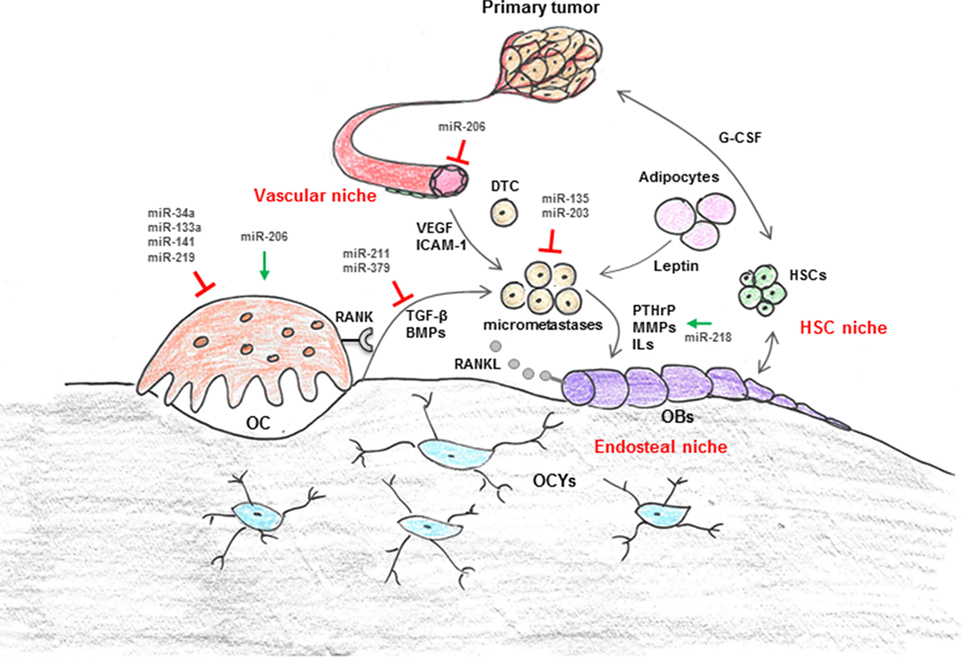
Figure 1. Regulation of cellular interaction in breast cancer bone metastasis by microRNA (miRNAs). The bone microenvironment (BME) is composed of cellular entities, including hematopoietic stem cells (HSC niche), osteoblasts (OBs), osteoclasts (OCs), and adipocytes (endosteal niche) as well as vascular endothelial cells and pericytes (vascular niche). These niches are suggested to control survival, dormancy, and growth of disseminated tumor cells (DTCs) through production of cytokines (i.e., leptin, G-CSF, VEGF, etc.) and intracellular signals in addition to cell-to-cell contact. In a physiological context, the highly coordinated cross talk between bone-forming OBs and bone-resorbing OCs maintains bone mass. OC function is regulated via OB and osteocyte (OCYs) derived RANKL. In the context of metastatic breast cancer disease, breast cancer cells severely disturb the balance between bone formation and resorption through secretion of various growth factors and cytokines [i.e., interleukins (ILs), parathyroid hormone-related protein (PTHrP), matrix metalloproteinases (MMPs), RANKL]. Recently, it has also been suggested that cells from the primary tumor themselves modify the distant microenvironment, for example through systemic factors (i.e., VEGF, TGF-β, G-CSF, miRNAs), in order to make it more attractive for DTCs. Several components of the BME are negatively (red blocks) or positively (green arrows) regulated by miRNAs.
MicroRNAs (miRNAs) are small non-coding RNAs and key regulators of various biological processes including bone remodeling and cancer progression (9, 10). miRNAs bind to the 3′UTR of their target mRNAs, and depending on the degree of complementarity interfere with the mRNA stability and/or by block protein translation (9). Abnormal miRNA expression has been implicated in the pathology of osteoporosis, primary bone tumors, and bone metastases of various cancers (11–14). Furthermore, in vivo delivery of miRNA mimics or miRNA antagonists has been established as an attractive therapeutic approach to reverse bone degeneration, or to prevent cancer-induced bone diseases (15, 16). Thus, miRNAs can be used as therapeutic targets and may provide a novel tool to treat breast cancer-induced osteolytic disease.
Several miRNAs have been identified to regulate breast cancer cell-intrinsic oncogenic properties, such as proliferation, migration, and invasion (17–19). However, how miRNAs regulate non-cell autonomous interactions in the bone microenvironment (BME) remains largely unknown. This review highlights the recent understanding of the role of miRNAs in the metastatic BME and their potential use as therapeutic targets to modulate the pathological environment and limit disease progression.
Metastatic Bone Disease
Bone is the most prevalent metastatic site for breast cancer cell colonization and growth. Bone metastasis is a complex multistep process starting from the dissemination of malignant cells into bloodstream, survival of these circulating tumor cells (CTCs) in the circulation, homing to distant organs and eventually metastases formation in the distant site (2). Disseminated tumor cells (DTCs) can be detected in the bone marrow of approximately 30% of breast cancer patients and predict for poor overall survival, breast cancer-specific survival, and disease-free survival compared to patients without DTCs (20).
Once bone metastases occur, the disease is incurable, and treatment remains palliative (21). The standard of care for patients with bone metastases comprises anti-resorptive drugs that reduce the progression of bone destruction and increase survival (22). For instance, bisphosphonates are well established in the treatment of osteolytic disease. Bisphosphonates are incorporated into the bone matrix and taken up by OCs during bone resorption, leading to OC apoptosis and a consecutive reduction of bone resorption (22). An alternative therapeutic approach is the use of the human monoclonal antibody Denosumab (Xgeva®) that inhibits RANKL and has been shown to delay the time to first and subsequent SRE in breast cancer patients (23). Although breast cancer patients greatly benefit from the use of bisphosphonates and Denosumab, a better understanding of the control of the “vicious cycle” in the BME and the underlying cellular and molecular mechanisms is needed as it is likely to help identifying novel therapeutic concepts to restrict SREs.
Tumor Microenvironment (TME)—The BME in Breast Cancer Bone Metastasis
Over the last decade, a variety of preclinical studies have emphasized the contribution of the TME to disease progression (24–28). The TME comprises the cellular environment in which the tumor exists, the surrounding extracellular matrix, and signaling molecules. Several aspects of how the TME impacts cancer growth are well established such as the role of endothelial cells in tumor angiogenesis (29, 30). However, others including the role of the TME in mediating tumor cell invasion, dissemination, and metastasis remain poorly defined (31).
Circulating tumor cells have a high affinity for bone, in particular areas of active bone remodeling (32). The highly balanced cross talk between OBs and OCs, the presence of various other bone marrow-derived cell populations and soluble factors including osteopontin (OPN) and matrix metalloproteinases (MMPs) make bone an attractive site (“soil”) for DTCs (“seeds”). Nearly almost a century ago Steven Pagets’ “seed and soil theory” proposed that therapies to modify the TME might be of equal importance as therapies targeted against the tumor cells themselves (33). Hence, cells of the BME are becoming increasingly recognized as potential therapeutic targets for breast cancer bone metastasis (24–27, 34, 35).
Upon their arrival in bone, DTCs encounter a heterogeneous BME, which is composed of various cells originating from either hematopoietic or mesenchymal stem cells (HSCs, MSCs, respectively) (Figure 1). These include lymphoid and myeloid lineage cells (e.g., immune cells, megakaryocytes, erythrocytes, and macrophages such as OCs) as well as adipocytes and bone and connective tissue-forming cells (e.g., chondrocytes and OBs). In addition, the BME contains a dense, interconnected vascular system which maintains hematopoiesis and osteogenesis (36, 37). Within bone, these various cellular entities form supporting microenvironments, “niches,” which are thought to regulate tumor cell homing, survival, and dormancy (28, 38–41) (Figure 1). The most well-studied niches are the HSC-, endosteal- (OBs, OCs), and vascular niche (vascular endothelial cells, pericytes). Both, the endosteal and the vascular niche control self-renewal, differentiation, and proliferation of HSCs through cell-to-cell contacts as well as by producing a variety of cytokines and intracellular signals (42–45). It is thought that tumor cells respond, similar like HSCs, to these signals. Among the most well studied pathways is the CXCL12/CXCR4 axis. CXCL12 or stromal cell-derived factor-1 (SDF-1) is produced and secreted by bone marrow stromal cells, primarily the OB, endothelial, and epithelial cells (46) (Figure 1). Its cognate receptor CXCR4 is expressed in high levels on various cancer cell lines, including MDA-MB-231 (47). Overexpression of CXCR4 in MDA-MB-231 cells increases bone metastasis, and very recently, it has been demonstrated that both newly and established metastases were anchored in the bone marrow by CXCR4/CXCL12 interactions (48, 49). Further niche signals are suggested to include OPN, vascular adhesion molecule-1, intercellular adhesion molecule-1, chemokines such as Interleukins (ILs) and various growth factors, including bone morphogenetic proteins, Transforming growth factor-β1 (TGF-β), and Vascular endothelial growth factor (VEGF) (50–52) (Figure 1). Emerging data also implicate the importance of the immune- and bone marrow adipocyte niche in bone metastasis (28, 53, 54). Studies by Templeton et al. highlighted the role of adipocytes, one of the most abundant stromal components in the BME, in breast cancer cell osteotropism and early colonization by demonstrating that adipokines, including leptin, promote the migration of MDA-MB-231 breast cancer cells to human bone tissue fragments in vitro (28). Nevertheless, exact mechanisms that guide DTCs toward the metastatic site in bone remain to be established. Recently, it was proposed that cells from the primary tumor themselves modify the distant (bone) microenvironment, for example through systemic factors (i.e., VEGF, TGF-β, LOX, G-CSF, miRNAs), in order to make it more attractive for DTCs (27, 55, 56).
Given the heterogeneity of the BME, the fate of DTCs might be determined by the nature of their arrival site within bone. A recent review by Croucher et al. suggests that long-term dormancy might be supported when tumor cells face quiescent/static microenvironments (e.g., endosteal surfaces covered by bone lining cells or stable vasculature), whereas active, dynamic BMEs including areas of osteoclastic bone resorption and sprouting vasculature foster proliferation and/or reactivation of dormant tumor cells (26, 57).
Once activated, breast cancer cells secrete growth factors, such as Parathyroid hormone-related protein (PTHrP), ILs, and MMPs, which stimulate OBs to produce excessive amounts of RANKL and other cytokines (58–60). RANKL increases OCs activity and subsequent bone degradation. During bone resorption, matrix-derived growth factors, e.g., TGF-β1 are released into the metastatic microenvironment and further stimulate cancer cell proliferation (7). This “vicious cycle” perpetuates metastatic bone destruction leading to osteolytic disease (8). Therefore, targeting the BME, for instance by miRNAs, to disable this cycle is not only scientifically interesting but also clinically relevant approach.
Tumor-Derived miRNAs Influencing the BME
MicroRNAs have been recently recognized as key regulators of various biological processes, including cancer progression and metastasis. miRNAs are small (20–22 nucleotides in length), endogenous non-coding RNAs which posttranscriptionally regulate mRNA stability and protein translation (9). More than 1,800 miRNAs are expressed in humans and according to prediction tools, each miRNA regulates numerous target genes (61–63). An important feature of miRNAs is that miRNAs can be encapsulated in extracellular vehicles and released to bloodstream (64–66), which makes them attractive minimal or non-invasive source of biomarkers of various diseases, including bone disorders (67, 68).
MicroRNAs expressed by tumor cells can act as master regulators of bone metastases formation by targeting metastasis-driving factors and consequently altering cancer cell behavior (17–19). In addition, tumor-derived miRNAs can exert their oncogenic or tumor-suppressive action by altering the BME. A specific feature of bone metastatic breast cancer cells is that they exhibit pathologically elevated expression of bone-related genes [e.g., Runt-related transcription factor 2 (Runx2)] and signaling pathways, including the Wnt pathway (69–71). Runx2 is necessary for normal bone formation but often dysregulated in bone metastatic breast cancer cells due to a downregulation of Runx2-targeting miRNAs, including miR-135 and miR-203 (72). Runx2 promotes tumor growth in bone and knocking down Runx2 in cancer cells protects from breast cancer-induced osteolytic disease, positioning Runx2 as an attractive target to reduce bone metastatic burden (73). Indeed, pharmacological delivery of synthetic miR-135 and miR-203 mimics into metastatic breast cancer cells reduces Runx2 protein abundance and consequently, diminishes tumor growth and spontaneous metastasis to bone (72). Furthermore, reconstitution of miR-135 and miR-203 greatly impairs tumor growth in the BME and alleviates osteolytic disease. The bone protecting effect occurred through downregulation of several metastasis-promoting Runx2 target genes, including IL-11, MMP-13, and PTHrP, and subsequent inhibition of OC activity (72).
Similarly, Wnt signaling promotes OB differentiation and function under physiological conditions but a hyper activation of the signaling pathway is implicated in numerous cancers, including metastatic breast cancer (70, 74). In bone metastatic breast cancer cells, Wnt signaling induces the expression of PTHrP thus aggravating the vicious cycle (75). In OBs, Wnt signaling and miR-218 create a positive feed-forward loop through targeting of Wnt inhibitors, such as Dkk1 and sFRP1 by miR-218 (76). Similarly, miR-218 activates Wnt signaling in metastatic breast cancer cells (76). Consequently, miR-218 enhances MDA-MB-231 cell proliferation and increases the expression of Wnt target genes in a Wnt-dependent manner (76, 77). Furthermore, miR-218 promotes PTHrP secretion in cancer cells and subsequent activation of RANKL in OBs, leading to an enhanced OB-mediated OC differentiation. Importantly, antagonizing miR-218 reversed these effects in vitro and prevented the formation of cancer-induced osteolytic lesions in vivo (77). Tumor-derived osteolytic cytokines are also regulated by miR-211 and miR-379 (78). Both miRNAs prevented TGF-β-induced upregulation of IL-11 and downregulated several genes involved in TGF-β pathway (78). Thus, miR-211 and miR-379 block the vicious cycle by preventing breast cancer cells from receiving signals from the metastatic BME.
Besides regulating the vicious cycle of bone metastasis, tumor-derived miRNAs, including miR-126, have been established in pathological angiogenesis in the BME (79). miR-126, which is silenced in breast cancer cells with bone metastatic potential, suppresses endothelial recruitment and metastatic angiogenesis in a non-cell autonomous manner and, importantly, inhibits bone metastatic colonization of breast cancer cells. The underlying mechanism involves a coordinated targeting of two newly identified pro-metastatic genes; insulin-like growth factor binding protein 2 (IGFPB2) and c-Mer tyrosine kinase (MERTK). Metastatic breast cancer cells secrete IGFPB2 that acts on insulin-like growth factor (IGF1) type I receptor on endothelial cells and modulates IGF1 activation and subsequently endothelial recruitment. In addition, endothelial recruitment is promoted upon cleavage of cMERTK receptor from the breast cancer cells, which antagonizes the binding of GAS6 to endothelial MERTK receptors. A series of elegant loss-of-function and replacement experiments revealed individual components of the pro-angiogenic IGFPB2/IGF1/IGF1R and GAS6-MERTK signaling pathways as direct targets of miR-126 and establish miR-126 as a crucial factor regulating endothelial interactions in the metastatic BME (79).
Recently, miRNAs released from cancer cells in microvesicles or exosomes have been shown to directly control cell–cell interactions in the BME. For instance, miR-192, which is highly abundant in metastatic lung cancer, can be secreted from the cancer cells in extracellular vesicles and transferred to endothelial cells (80). Cancer cell-derived miR-192 is efficiently taken up by endothelial cells in vitro and in vivo, and inhibits tumor-induced angiogenesis leading to reduced metastatic burden and decreased osteolytic disease in mice.
Targeting the BME by miRNAs
Since OC activity is a hallmark of metastatic bone disease, the current treatment as well as the majority of basic research is focusing on restricting OC activity and attenuating pathological bone resorption. Along these lines, several studies have established miRNAs as crucial regulators of pathological OC differentiation. Especially miRNAs that suppress bone resorption provide an attractive approach to limit disease progression (81, 82). For instance, miR-34a was recently reported to inhibit physiological and pathological OC differentiation and to block osteoporosis and cancer-induced bone destruction (83). Using several genetic mouse models, Krzezinski et al. demonstrated that OC-targeted overexpression of miR-34a impairs bone resorption resulting in resistance of bone metastases. Conversely, deletion of miR-34a activated OCs leading to reduced bone mass and exacerbated bone metastasis burden. Mechanistically, miR-34a targets a homeodomain protein TG-interacting factor 2, a novel positive regulator of osteoclast differentiation and function. In a therapeutically relevant setting, systemic delivery of miR-34a mimic oligonucleotides via chitosan nanoparticles diminished bone metastatic burden and osteolysis (83). Since miR-34a had no direct effect of cancer cell proliferation these effects are likely mediated by osteoclasts and possibly other cells in the BME emphasizing the importance of the TME in disease progression.
In another comprehensive study, a group of miRNAs was shown to regulate tumor-induced osteoclast differentiation. Five miRNAs, miR-33a, miR-133a, miR-141, miR-190, and miR-219 were downregulated during osteoclast differentiation under physiological and pathophysiological conditions (84). Among them, miR-133a, miR-141, and miR-219 impaired osteoclast differentiation in vitro by targeting important osteoclast-promoting factors Mitf, Mmp14, Calcitonin receptor, and Traf5. In vivo administration of synthetic miR-141 and miR-219 oligonucleotides reduced physiological bone resorption, impaired tumor growth in bone and prevented pathological bone destruction. In this study, two miRNAs, miR-16 and miR-378 secreted in exosomes by osteoclasts were found to be increased in patients with bone metastases compared to healthy controls and the expression correlated with bone metastasis burden (84). Interestingly, miR-378 promotes tumor growth, angiogenesis, and tumor cell survival through the repression of tumor suppressors SuFu and Fus-1 (85). Although beyond this review, it is important to emphasize that miRNA signatures are being pursued as novel clinical diagnostic targets for predicting metastasis or therapeutic resistance (1, 4).
miR-214 is strongly increased in bone specimen of breast cancer patients with osteolytic bone metastases compared to healthy controls and patients without bone involvement (86). Consistently, osteoclasts isolated from mice with bone metastases express significantly higher levels of miR-214 compared to controls. In addition, miR-214 is elevated in bone tissue and serum of aged patients with fractures and miR-214 expression is accompanied with increased osteoclast activity and bone resorption, indicating that miR-214 regulates bone remodeling in health and disease (87, 88). Indeed, miR-214 is expressed and has a functional role in both OBs and osteoclasts. In OBs, miR-214 inhibits differentiation in vitro and bone formation in vivo by targeting the activating transcription factor 4. As a consequence, delivery of OB-targeted antagomiR-214 in osteoporotic mice increased bone formation and restored bone mass (87). In contrast, in osteoclasts, miR-214 promotes osteoclast function and bone resorption through inhibition of the phosphatase and tensin homolog and Traf3, a negative regulator of NFkB signaling and osteoclast differentiation (86, 89). As a consequence, osteoclast-targeted deletion of miR-214 reduced bone resorption and prevented the development of osteolytic lesions in mice (86). The number and size of non-bone metastases was not changed in mice lacking miR-214 in the osteoclast lineage indicating that the tumor-suppressive effect of bone metastases is mediated by the BME. Importantly, pharmacological delivery of osteoclast targeted (d-Asp8)-liposome conjucated antimiR-214 oligonucleotides reduced physiological and pathological bone resorption and protected from osteolytic bone metastases, suggesting that inhibition of miR-214 could provide an attractive therapeutic strategy to prevent pathological bone destruction (86).
Intriguingly, miR-214 is secreted from osteoclasts in exosomes into circulation and acts on local and distant OBs (88). Treatment of wild-type mice with exosomes isolated from mice with osteoclast-targeted overexpression of miR-214 reduced bone formation demonstrating that the osteoclast-derived miR-214 is fully functional in OBs. This was further supported by an increased bone formation after systemic administration of antagomiR-214 encapsulated in osteoclast-targeted (d-Asp)8-liposomes (88). Given the dual bone anabolic and anti-catabolic effect of antago-miR-214, a systemic delivery of antago-miR-214 might provide a potent strategy to not only prevent osteolytic disease but also reverse existing lesions.
Perspectives
MicroRNAs play a pivotal role in tissue development and homeostasis. Therefore, dysregulation of individual miRNAs is implicated in several pathological conditions, including the onset and progression of metastatic bone disease. While the role of miRNAs regulating oncogenic properties of tumor cells is relatively well established, the direct or indirect regulation of TME by miRNAs has only recently started to be uncovered. In particular, miRNAs that mediate cell–cell interactions in the BME provide a novel therapeutic approach to disable the pathological cross talk in the bone marrow. For instance, identifying and targeting miRNAs that are pathologically elevated in osteoclasts and promote the vicious cycle could offer novel strategies for diagnosis and treatment of bone metastases. Although the evidence is thus far exclusively based on preclinical data, the applications of using miRNAs as adjuvant tools in bone metastases targets are very promising. Therefore, a better understanding of the complex miRNA-mediated cellular interactions is not only scientifically interesting but also critical in transmitting the knowledge from the bench to bedside.
Author Contributions
M-TH and HT reviewed literature and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
M-TH is supported by a post-doctoral fellowship from the Humboldt Foundation. HT received funding from the Deutsche Forschungsgemeinschaft (TA1154/1-1).
References
1. McGuire A, Brown JAL, Kerin MJ. Metastatic breast cancer: the potential of miRNA for diagnosis and treatment monitoring. Cancer Metastasis Rev (2015) 34:145–55. doi:10.1007/s10555-015-9551-7
2. Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer (2004) 4:448–56. doi:10.1038/nrc1370
4. D’Oronzo S, Brown J, Coleman R. The role of biomarkers in the management of bone-homing malignancies. J Bone Oncol (2017) 9:1–9. doi:10.1016/j.jbo.2017.09.001
5. Baron R, Hesse E. Update on bone anabolics in osteoporosis treatment: rationale, current status, and perspectives. J Clin Endocrinol Metab (2012) 97:311–25. doi:10.1210/jc.2011-2332
6. O’Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone (2013) 54:258–63. doi:10.1016/j.bone.2012.08.121
7. Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer (2011) 11:411–25. doi:10.1038/nrc3055
8. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer (2002) 2:584–93. doi:10.1038/nrc867
9. Lian JB, Stein GS, van Wijnen AJ, Stein JL, Hassan MQ, Gaur T, et al. MicroRNA control of bone formation and homeostasis. Nat Rev Endocrinol (2012) 8:212–27. doi:10.1038/nrendo.2011.234
10. Drusco A, Croce CM. MicroRNAs and cancer: a long story for short RNAs. Adv Cancer Res (2017) 135:1–24. doi:10.1016/bs.acr.2017.06.005
11. Taipaleenmäki H. Regulation of bone metabolism by microRNAs. Curr Osteoporos Rep (2018) 16(1):1–12. doi:10.1007/s11914-018-0417-0
12. Browne G, Taipaleenmäki H, Stein GS, Stein JL, Lian JB. MicroRNAs in the control of metastatic bone disease. Trends Endocrinol Metab (2014) 25:320–7. doi:10.1016/j.tem.2014.03.014
13. van der Deen M, Taipaleenmäki H, Zhang Y, Teplyuk NM, Gupta A, Cinghu S, et al. MicroRNA-34c inversely couples the biological functions of the runt-related transcription factor RUNX2 and the tumor suppressor p53 in osteosarcoma. J Biol Chem (2013) 288:21307–19. doi:10.1074/jbc.M112.445890
14. Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med (2012) 4:143–59. doi:10.1002/emmm.201100209
15. Taipaleenmäki H, Bjerre Hokland L, Chen L, Kauppinen S, Kassem M. Mechanisms in endocrinology: micro-RNAs: targets for enhancing osteoblast differentiation and bone formation. Eur J Endocrinol (2012) 166:359–71. doi:10.1530/EJE-11-0646
16. Krzeszinski JY, Wan Y. New therapeutic targets for cancer bone metastasis. Trends Pharmacol Sci (2015) 36:360–73. doi:10.1016/j.tips.2015.04.006
17. Zoni E, van der Pluijm G. The role of microRNAs in bone metastasis. J Bone Oncol (2016) 5:104–8. doi:10.1016/j.jbo.2016.04.002
18. Zhao Q, Luo F, Ma J, Yu X. Bone metastasis-related microRNAs: new targets for treatment? Curr Cancer Drug Targets (2015) 15:716–25. doi:10.2174/1568009615666150629102408
19. Croset M, Kan C, Clézardin P. Tumour-derived miRNAs and bone metastasis. Bonekey Rep (2015) 4:688. doi:10.1038/bonekey.2015.56
20. Fontanella C, Fanotto V, Rihawi K, Aprile G, Puglisi F. Skeletal metastases from breast cancer: pathogenesis of bone tropism and treatment strategy. Clin Exp Metastasis (2015) 32:819–33. doi:10.1007/s10585-015-9743-0
21. Coleman R. Bone targeted treatments in cancer – the story so far. J Bone Oncol (2016) 5:90–2. doi:10.1016/j.jbo.2016.03.002
22. Coleman RE. Impact of bone-targeted treatments on skeletal morbidity and survival in breast cancer. Oncology (Williston Park) (2016) 30:695–702.
23. Stopeck AT, Lipton A, Body J-J, Steger GG, Tonkin K, de Boer RH, et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol (2010) 28:5132–9. doi:10.1200/JCO.2010.29.7101
24. Haider M-T, Holen I, Dear TN, Hunter K, Brown HK. Modifying the osteoblastic niche with zoledronic acid in vivo-potential implications for breast cancer bone metastasis. Bone (2014) 66:240–50. doi:10.1016/j.bone.2014.06.023
25. Ubellacker JM, Haider M-T, DeCristo MJ, Allocca G, Brown NJ, Silver DP, et al. Zoledronic acid alters hematopoiesis and generates breast tumor-suppressive bone marrow cells. Breast Cancer Res (2017) 19:23. doi:10.1186/s13058-017-0815-8
26. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol (2013) 15:807–17. doi:10.1038/ncb2767
27. Cox TR, Rumney RMH, Schoof EM, Perryman L, Høye AM, Agrawal A, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature (2015) 522:106–10. doi:10.1038/nature14492
28. Templeton ZS, Lie W-R, Wang W, Rosenberg-Hasson Y, Alluri RV, Tamaresis JS, et al. Breast cancer cell colonization of the human bone marrow adipose tissue niche. Neoplasia (2015) 17:849–61. doi:10.1016/j.neo.2015.11.005
29. Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer (2001) 1:46–54. doi:10.1038/35094059
30. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol (2002) 29:asonc02906q0015. doi:10.1053/sonc.2002.37263
31. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
32. Brown HK, Ottewell PD, Evans CA, Holen I. Location matters: osteoblast and osteoclast distribution is modified by the presence and proximity to breast cancer cells in vivo. Clin Exp Metastasis (2012) 29:927–38. doi:10.1007/s10585-012-9481-5
33. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev (1989) 8:98–101.
34. Zheng H, Bae Y, Kasimir-Bauer S, Tang R, Chen J, Ren G, et al. Therapeutic antibody targeting tumor- and osteoblastic niche-derived jagged1 sensitizes bone metastasis to chemotherapy. Cancer Cell (2017) 32:731–47.e6. doi:10.1016/j.ccell.2017.11.002
35. Haider M-T, Hunter KD, Robinson SP, Graham TJ, Corey E, Dear TN, et al. Rapid modification of the bone microenvironment following short-term treatment with cabozantinib in vivo. Bone (2015) 81:581–92. doi:10.1016/j.bone.2015.08.003
36. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature (2014) 507:323–8. doi:10.1038/nature13145
37. Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature (2016) 532:323–8. doi:10.1038/nature17624
38. Zalucha JL, Jung Y, Joseph J, Wang J, Berry JE, Shiozawa Y, et al. The role of osteoclasts in early dissemination of prostate cancer tumor cells. J Cancer Stem Cell Res (2015) 3:e1005. doi:10.14343/JCSCR.2015.3e1005
39. Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell (2015) 27:193–210. doi:10.1016/j.ccell.2014.11.017
40. Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest (2011) 121:1298–312. doi:10.1172/JCI43414
41. Shiozawa Y, Havens AM, Pienta KJ, Taichman RS. The bone marrow niche: habitat to hematopoietic and mesenchymal stem cells, and unwitting host to molecular parasites. Leukemia (2008) 22:941–50. doi:10.1038/leu.2008.48
42. Taichman RS. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood (2005) 105:2631–9. doi:10.1182/blood-2004-06-2480
43. Psaila B, Lyden D, Roberts I. Megakaryocytes, malignancy and bone marrow vascular niches. J Thromb Haemost (2012) 10:177–88. doi:10.1111/j.1538-7836.2011.04571.x
44. Calvi LM. Osteoblastic activation in the hematopoietic stem cell niche. Ann N Y Acad Sci (2006) 1068:477–88. doi:10.1196/annals.1346.021
45. Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature (2006) 439:599–603. doi:10.1038/nature04247
46. Ponomaryov T, Peled A, Petit I, Taichman RS, Habler L, Sandbank J, et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J Clin Invest (2000) 106:1331–9. doi:10.1172/JCI10329
47. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature (2001) 410:50–6. doi:10.1038/35065016
48. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell (2003) 3:537–49. doi:10.1016/S1535-6108(03)00132-6
49. Price TT, Burness ML, Sivan A, Warner MJ, Cheng R, Lee CH, et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci Transl Med (2016) 8:340ra73. doi:10.1126/scitranslmed.aad4059
50. McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell (2008) 133:994–1005. doi:10.1016/j.cell.2008.04.045
51. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell (2011) 20:701–14. doi:10.1016/j.ccr.2011.11.002
52. Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer (2010) 10:138–46. doi:10.1038/nrc2791
53. Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell (2016) 165:45–60. doi:10.1016/j.cell.2016.02.025
54. Monteiro AC, Leal AC, Gonçalves-Silva T, Mercadante ACT, Kestelman F, Chaves SB, et al. T cells induce pre-metastatic osteolytic disease and help bone metastases establishment in a mouse model of metastatic breast cancer. PLoS One (2013) 8:e68171. doi:10.1371/journal.pone.0068171
55. Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the “pre-metastatic niche”: within bone and beyond. Cancer Metastasis Rev (2007) 25:521–9. doi:10.1007/s10555-006-9036-9
56. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol (2015) 17:816–26. doi:10.1038/ncb3169
57. Croucher PI, McDonald MM, Martin TJ. Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer (2016) 16:373–86. doi:10.1038/nrc.2016.44
58. Powell GJ, Southby J, Danks JA, Stillwell RG, Hayman JA, Henderson MA, et al. Localization of parathyroid hormone-related protein in breast cancer metastases: increased incidence in bone compared with other sites. Cancer Res (1991) 51:3059–61.
59. Martin TJ. Manipulating the environment of cancer cells in bone: a novel therapeutic approach. J Clin Invest (2002) 110:1399–401. doi:10.1172/JCI0217124
60. Li J, Karaplis AC, Huang DC, Siegel PM, Camirand A, Yang XF, et al. PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J Clin Invest (2011) 121:4655–69. doi:10.1172/JCI46134
61. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell (2005) 120:15–20. doi:10.1016/j.cell.2004.12.035
62. Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature (2005) 433:769–73. doi:10.1038/nature03315
63. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res (2014) 42:D68–73. doi:10.1093/nar/gkt1181
64. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol (2007) 9:654–9. doi:10.1038/ncb1596
65. Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol (2011) 13:423–33. doi:10.1038/ncb2210
66. Wang K, Zhang S, Weber J, Baxter D, Galas DJ. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res (2010) 38:7248–59. doi:10.1093/nar/gkq601
67. Weber JA, Baxter DH, Zhang S, Huang DY, Huang KH, Lee MJ, et al. The microRNA spectrum in 12 body fluids. Clin Chem (2010) 56:1733–41. doi:10.1373/clinchem.2010.147405
68. Hackl M, Heilmeier U, Weilner S, Grillari J. Circulating microRNAs as novel biomarkers for bone diseases – complex signatures for multifactorial diseases? Mol Cell Endocrinol (2016) 432:83–95. doi:10.1016/j.mce.2015.10.015
69. Sethi N, Kang Y. Dysregulation of developmental pathways in bone metastasis. Bone (2011) 48:16–22. doi:10.1016/j.bone.2010.07.005
70. Matsuda Y, Schlange T, Oakeley EJ, Boulay A, Hynes NE. WNT signaling enhances breast cancer cell motility and blockade of the WNT pathway by sFRP1 suppresses MDA-MB-231 xenograft growth. Breast Cancer Res (2009) 11:R32. doi:10.1186/bcr2317
71. Pratap J, Lian JB, Stein GS. Metastatic bone disease: role of transcription factors and future targets. Bone (2011) 48:30–6. doi:10.1016/j.bone.2010.05.035
72. Taipaleenmäki H, Browne G, Akech J, Zustin J, van Wijnen AJ, Stein JL, et al. Targeting of Runx2 by miR-135 and miR-203 impairs progression of breast cancer and metastatic bone disease. Cancer Res (2015) 75:1433–44. doi:10.1158/0008-5472.CAN-14-1026
73. Pratap J, Lian JB, Javed A, Barnes GL, van Wijnen AJ, Stein JL, et al. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev (2006) 25:589–600. doi:10.1007/s10555-006-9032-0
74. Blagodatski A, Poteryaev D, Katanaev VL. Targeting the Wnt pathways for therapies. Mol Cell Ther (2014) 2:28. doi:10.1186/2052-8426-2-28
75. Johnson RW, Merkel AR, Page JM, Ruppender NS, Guelcher SA, Sterling JA. Wnt signaling induces gene expression of factors associated with bone destruction in lung and breast cancer. Clin Exp Metastasis (2014) 31:945–59. doi:10.1007/s10585-014-9682-1
76. Hassan MQ, Maeda Y, Taipaleenmaki H, Zhang W, Jafferji M, Gordon JAR, et al. miR-218 directs a Wnt signaling circuit to promote differentiation of osteoblasts and osteomimicry of metastatic cancer cells. J Biol Chem (2012) 287:42084–92. doi:10.1074/jbc.M112.377515
77. Taipaleenmäki H, Farina NH, van Wijnen AJ, Stein JL, Hesse E, Stein GS, et al. Antagonizing miR-218-5p attenuates Wnt signaling and reduces metastatic bone disease of triple negative breast cancer cells. Oncotarget (2016) 7:79032–46. doi:10.18632/oncotarget.12593
78. Pollari S, Leivonen S-K, Perälä M, Fey V, Käkönen S-M, Kallioniemi O. Identification of microRNAs inhibiting TGF-β-induced IL-11 production in bone metastatic breast cancer cells. PLoS One (2012) 7:e37361. doi:10.1371/journal.pone.0037361
79. Png KJ, Halberg N, Yoshida M, Tavazoie SF. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature (2012) 481:190–4. doi:10.1038/nature10661
80. Valencia K, Luis-Ravelo D, Bovy N, Antón I, Martínez-Canarias S, Zandueta C, et al. miRNA cargo within exosome-like vesicle transfer influences metastatic bone colonization. Mol Oncol (2014) 8:689–703. doi:10.1016/j.molonc.2014.01.012
81. Kagiya T. MicroRNAs and osteolytic bone metastasis: the roles of microRNAs in tumor-induced osteoclast differentiation. J Clin Med (2015) 4:1741–52. doi:10.3390/jcm4091741
82. Ell B, Kang Y. MicroRNAs as regulators of tumor-associated stromal cells. Oncotarget (2013) 4:2166–7. doi:10.18632/oncotarget.1625
83. Krzeszinski JY, Wei W, Huynh H, Jin Z, Wang X, Chang T-C, et al. miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature (2014) 512:431–5. doi:10.1038/nature13375
84. Ell B, Mercatali L, Ibrahim T, Campbell N, Schwarzenbach H, Pantel K, et al. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell (2013) 24:542–56. doi:10.1016/j.ccr.2013.09.008
85. Lee DY, Deng Z, Wang C-H, Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci U S A (2007) 104:20350–5. doi:10.1073/pnas.0706901104
86. Liu J, Li D, Dang L, Liang C, Guo B, Lu C, et al. Osteoclastic miR-214 targets TRAF3 to contribute to osteolytic bone metastasis of breast cancer. Sci Rep (2017) 7:40487. doi:10.1038/srep40487
87. Wang X, Guo B, Li Q, Peng J, Yang Z, Wang A, et al. miR-214 targets ATF4 to inhibit bone formation. Nat Med (2012) 19:93–100. doi:10.1038/nm.3026
88. Li D, Liu J, Guo B, Liang C, Dang L, Lu C, et al. Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. Nat Commun (2016) 7:10872. doi:10.1038/ncomms10872
Keywords: breast cancer, bone metastases, microRNA, bone microenvironment, osteoclast, osteoblast
Citation: Haider M-T and Taipaleenmäki H (2018) Targeting the Metastatic Bone Microenvironment by MicroRNAs. Front. Endocrinol. 9:202. doi: 10.3389/fendo.2018.00202
Received: 07 February 2018; Accepted: 11 April 2018;
Published: 27 April 2018
Edited by:
Julie A. Sterling, Vanderbilt University, United StatesReviewed by:
Graziana Colaianni, University of Bari, ItalyJennifer Tickner, University of Western Australia, Australia
Copyright: © 2018 Haider and Taipaleenmäki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanna Taipaleenmäki, h.taipaleenmaeki@uke.de