 Jason H. Kurzer
Jason H. Kurzer Olga K. Weinberg
Olga K. Weinberg- 1Department of Pathology, Stanford University School of Medicine, Stanford, CA, United States
- 2Department of Pathology, UT Southwestern, Dallas, TX, United States
Next generation sequencing has uncovered several genes with associated mutations in hematologic malignancies that can serve as potential biomarkers of disease. Keeping abreast of these genes is therefore of paramount importance in the field of hematology. This review focuses on PHF6, a highly conserved epigenetic transcriptional regulator that is important for neurodevelopment and hematopoiesis. PHF6 serves as a tumor suppressor protein, with PHF6 mutations and deletions often implicated in the development of T-lymphoblastic leukemia and less frequently in acute myeloid leukemia and other myeloid neoplasms. PHF6 inactivation appears to be an early event in T-lymphoblastic leukemogenesis, requiring cooperating events, including NOTCH1 mutations or overexpression of TLX1 and TLX3 for full disease development. In contrast, PHF6 mutations tend to occur later in myeloid malignancies, are frequently accompanied by RUNX1 mutations, and are often associated with disease progression. Moreover, PHF6 appears to play a role in lineage plasticity within hematopoietic malignancies, with PHF6 mutations commonly present in mixed phenotype acute leukemias with a predilection for T-lineage marker expression. Due to conflicting data, the prognostic significance of PHF6 mutations remains unclear, with a subset of studies showing no significant difference in outcomes compared to malignancies with wild-type PHF6, and other studies showing inferior outcomes in certain patients with mutated PHF6. Future studies are necessary to elucidate the role PHF6 plays in development of T-lymphoblastic leukemia, progression of myeloid malignancies, and its overall prognostic significance in hematopoietic neoplasms.
Introduction
Plant homeodomain (PHD) finger proteins consist of a family of epigenetic regulators that bind to a variety of targets, including both post-translationally modified and unmodified histones (1). The PHD finger protein, homeodomain finger protein 6 (PHF6), is a highly conserved, 365 amino acid, 41kDa protein, that was first identified in the X-linked neurodevelopmental disorder, Börjeson-Forssman-Lehmann syndrome (BFLS) (2, 3). PHF6 contains two imperfect PHD-like zinc finger domains, two nuclear localization signals as well as a nucleolar localization sequence (Figure 1) (2, 4–6). Expression of PHF6 is found in almost all tissues, with particularly high expression in the brain/developing central nervous system as well as in all hematopoietic subpopulations (high levels in CD34+ precursor cells and B-cells; low levels in NK-cells and monocytes) implicating a role for PHF6 in a variety of functions including neurogenesis and hematopoiesis (2, 7, 8).
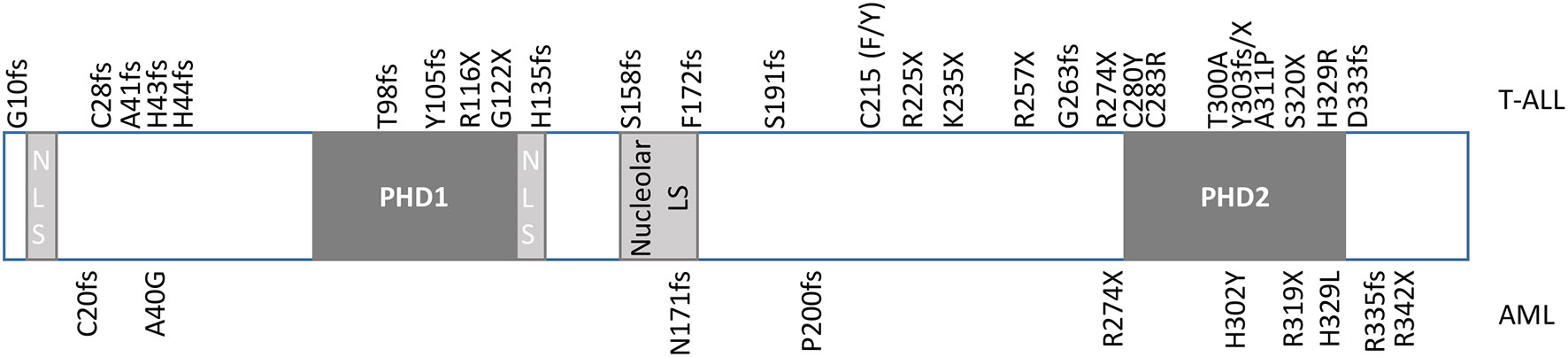
Figure 1 Diagram of PHF6. PHF6 has two imperfect PHD-like zinc finger domains (PHD1 and PHD2), two nuclear localization signals (NLS), and one nucleolar localization signal. Mutations originally identified in T-ALL by Van Vlierberghe et al. (4) are identified at the top of the diagram, whereas mutations originally identified in AML by Van Vliergerghe et al. (21) are identified at the bottom of the diagram.
Function of PHF6
Supporting a fundamental role for PHF6 in general development, PHF6-homozygous knockout mice die perinatally (9). However, scientists have also developed various knockout models in mice to explore the role of PHF6 in hematopoiesis (8, 10–13). McRae et al. showed that germline deletion of murine PHF6 is lethal in males whereas heterozygous females survived to adulthood (10). Using Phf6lox/Y;Tie-creTg/+ male mice with a Phf6-null mutation in hematopoietic and endothelial cells, McRae et al. showed an increase in Lin-c-Kit+Sca-1+ (LSK) stem and progenitor cell-enriched populations, specifically the heterogeneous progenitor cell population (HPC-1), with an enrichment in cycling forms (10). Inactivation of PHF6 at various stages of development results in increased embryologic proliferative properties of hematopoietic stem cells, but also an increased ability for PHF6-deficient neonatal and adult HSCs to repopulate the bone marrow in serial transplant assays (10–13). Studies suggest that this enhanced proliferative ability of PHF6-deleted HSCs results, at least in part, from IFN-α signaling, the inhibition of TNFα-associated growth suppressing genes, and perhaps upregulation of JAK1 signaling (10–12).
Wendorff et al. similarly showed that conditional knockout of PHF6 displayed expansions of total immature hematopoietic LSK cells, but also showed increased multipotent MPP2, and MPP3 populations in 8-week-old mice, and no differences in the numbers of more mature myeloid progenitors, uncommitted lymphoid progenitors, or B-cell precursors (11). However, conditional knockout of PHF6 does show a mild reduction in double-negative DN2 and DN3 thymic progenitors and decreased numbers of peripheral blood CD4 and CD8 positive T-cells in 8-week-old mice (10, 11, 13). Moreover, knockdown studies in cord blood and thymus-derived hematopoietic precursors showed that PHF6 loss fosters a preferential differentiation of B-lymphocytes, reduced erythroid development, and accelerated T-cell development via downregulation of NOTCH1 (8).
Like other PHD finger proteins, PHF6 functions in chromatin-mediated regulation of gene expression. It is capable of binding double-stranded DNA, but not histones, in vitro via its atypical PHD2 domain (14). Co-immunoprecipitation experiments using antibodies to PHF6, reveal it interacts with constituents of the nucleosome remodeling deacetylase (NuRD) complex, including CHD4, HDAC1, and Rbb4 (6, 15). The NuRD complex is a major ATP-dependent chromatin remodeling complex, implicated in nucleosome positioning and both repressing and activating genes involved in embryonic development (16). PHF6 has additionally been shown to bind to the PAF1 transcription elongation complex, regulating Neuroglycan C/Chondroitn sulfate proteoglycan 5 (NGC/CSPG5) and ultimately neuronal migration in murine cerebral cortex development (17). One consequence of PHF6-mediated transcription regulation includes the modulation of levels of the RNA Pol I preinitiation complex activator, upstream binding factor (UBF), thereby suppressing UBF-mediated rRNA transcription (5, 18).
Knockdown studies of PHF6 suggest a tumor suppressor role for the protein, as PHF6 deficiency in HeLa cell lines resulted in increased UBF protein levels, and increased DNA damage at the rDNA locus (5). Deficiency of PHF6 results in the formation of DNA-RNA hybrid (R-loops) and increased R-loop-dependent rDNA damage (5). Moreover, knockdown of PHF6 interferes with the G2 checkpoint recovery of U2OS cells, leading to decreased DNA repair in response to ionizing radiation (19). These data implicate a tumor suppressor role for PHF6 resulting from regulating DNA damage response.
PHF6 and Hematologic Disease
Despite the discovery of PHF6 via its role in BFL syndrome and its prominent role in neurogenesis, mutations of PHF6 have only been identified to date in hematologic malignancies (20). The first and most well-documented hematologic malignancy harboring mutations of PHF6 is T-lymphoblastic leukemia (T-ALL), with fewer cases identified in acute myeloid leukemia (AML), and rarer cases identified in pre-malignant clonal hematopoiesis (4, 21–23). Mutations in hematologic malignancies include deletion, missense, frameshift, and nonsense mutations, which span the whole coding region, with missense mutations concentrated in the PHD2 domain (Figure 1) (4, 21, 24). The ultimate effect of these mutations is to inhibit PHF6 function or deplete its levels, and as such, provide supporting evidence that this protein serves as a tumor suppressor. Indeed, a patient with BFLS was noted to develop T-ALL (25).
T-Lymphoblastic Leukemia
The PHF6 locus is one of the most frequently mutated genes in T-lymphoblastic leukemia (T-ALL). Inactivating mutations of PHF6 have been identified in 5-16% of pediatric and 19-40% of adult patients with T-ALL and ~25% of adults with T-lymphoblastic lymphoma (T-LBL) with some groups identifying an association with NOTCH1 mutations (67%-84.6% of PHF6 mutated/deleted T-ALL with NOTCH1 mutations versus 39.8% PHF6 WT) (4, 26–38). Copy number alterations of PHF6 in pediatric T-ALL has been reported to be between 13-14% (39, 40). The location of PHF6 on chrX26.2 lead researches to speculate that such mutations may at least partially explain the 2-3 fold increased incidence of T-ALL in males, as PHF6 mutations were originally found predominantly in male T-ALL patients (31.5% vs 2.6%) (4). Subsequent studies, however, have failed to show a gender preference (26–28, 33). Recently, PHF6 mutations have additionally been found in up to 25% (3/12) of early T-cell precursor subtype of T-ALL (ETP), a form of T-ALL that frequently expresses myeloid-associated markers (41, 42).
The role PHF6 plays in leukemogenesis is actively under investigation. Analyzing whole exome sequencing data from diagnostic and relapse leukemias, Wendorff et al. showed that somatic mutations of PHF6 occur early in leukeomogenesis (11). Nevertheless, animal models have revealed that while PHF6 mutations/deletions may be initial events, they are insufficient for tumor initiation without additional driver mutations. For example, Miyagi et al. did not observe leukemia development subsequent to serial transplantation of PHF6-deleted HSCs in mice (12). Likewise, no T-ALL tumors developed in a PHF6+/- zebrafish model (43).
Studies show that PHF6 dysfunction cooperates with several other driver mutations. For example, inactivation of PHF6 in hematopoietic progenitors has been reported to facilitate NOTCH1-induced T-ALL, potentially through increasing leukemia-initiating cells and development of a “leukemia stem cell transcriptional program” in lymphoblasts (11, 13). A study of 102 pediatric T-ALL cases in Taiwan showed that PHF6 mutations frequently cooperate with HOX11L2 overexpression and/or WT1 mutations (44). In addition, PHF6 mutations have been shown to be associated (at times in conjunction with DNM2) with T-ALLs that overexpress homeobox transcription factors, TLX1 and TLX3 (4, 32, 45). Indeed, ectopic expression of TLX3 in PHF6-deleted mice facilitated early onset leukemia and a hTLX1;PHF6+/- zebrafish model demonstrated fully penetrant early-onset leukemia development, underscoring the role of cooperation between these mutations in leukemogenesis (10, 43). Other associations include those with mutations in X-linked genes, USP9X and MED1 as well as with IL7R-JAK pathway genes, WT1, PTPN2 deletions, and HOX11L2 overexpression (33, 34, 44, 46). In the setting of ETP, PHF6 mutations frequently occur with mutations in EZH2, EED, and SUZ12 (41). Finally, a network of miRNAs, including miR-19b, miR-20a/93, miR-26a, miR-92, and miR-223, have been shown to target multiple PHF6 and other T-ALL-associated tumor suppressors, and thus promote leukemia (47).
PHF6 has additionally been shown to interact with LMO2 to bind DNA via the LMO2/TAL1/LDB/GATA2 complex in T-ALL cell lines (48). The PHF6/LMO2/TAL1/LDB/GATA2 complex was shown to bind at DNA segments associated with hematopoietic or lymphoid organ development, hematopoiesis, as well as T-cell activation and differentiation (48).
While sample sizes are small, patients with T-ALL harboring PHF6 mutations tend to be older, have been demonstrated to have lower white blood cell counts than other T-ALL patients, as well as lower hemoglobin and platelet levels, splenomegaly/lymphadenopathy, and have blasts with a tendency to express CD13 (26, 29, 37). Genomic analysis of matched diagnosis, germline (remission) and relapse DNA samples from 46 T-ALL cases reveals that PHF6 alterations are found commonly at diagnosis, and persist at relapse (49). While mutations in PHF6 have been implicated in increased resistance to prednisolone in T-ALL cell lines, the majority of studies have shown no correlation with PHF6 mutations and overall survival in patients with T-ALL, and a potential favorable prognosis associated with T-LBL (4, 26, 27, 29, 50, 51). Nevertheless, one study of pediatric T-ALL found PHF6 mutations/deletions predict an inferior overall survival upon multivariate analysis (44). Another study of Chinese adults found that the co-existence of PHF6 and NOTCH1 mutations in T-ALL conferred a shorter event-free survival and a poor prognosis (37). Further study is therefore required to assess the true prognostic significance of PHF6 mutations in T-ALL.
Myeloid Neoplasms
The largest study of PHF6 mutations in myeloid malignancies involved targeted sequencing of 1760 cases with myeloid neoplasms (24). This study revealed 54 patients with 62 somatic mutations of PHF6 (24). With regard to disease burden, the percentage of blasts in the bone marrow tended to be higher in patients with myeloid neoplasms harboring PHF6 mutations (24). As for cytogenetics, abnormal karyotypes showed no significant predilection for PHF6 mutations, although +8, t(8;21), and complex karyotypes were abnormalities most often identified (24). At the molecular level, co-mutated genes associated with PHF6 mutations included RUNX1, U2AF1, SMC1A, ZRSR2, EZH2, and ASXL1, whereas PHF6 was found to be mutually exclusive with SF3B1 (24). In contrast to T-ALL, mutations of PHF6 tend to occur later in disease evolution, sometimes with different mutations in parallel clones (24).
Acute Myeloid Leukemia
Inactivating somatic PHF6 mutations have been found in ~2-3% of AMLs (20, 21, 24, 28, 52, 53). Similar to T-ALL, a male predominance was initially reported but not further substantiated for AML identified with PHF6 mutations (21, 24, 53). With respect to AML subtypes, PHF6 mutations are found in 15% of AML with inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and 15.4% of cases of AML with myelodysplasia related changes (MRC) (24, 54). Interestingly, a case report of AML with MRC harboring a P2RY8-CRLF2 fusion was found to have gained a PHF6 mutation upon transformation to AML, suggesting a potential role for PHF6 in the transition of MDS to AML (55). Further evidence for PHF6 mutations acquired secondarily and leading to progression of myeloid neoplasms was found in patients with germline mutations of RUNX1, where PHF6 mutations were implicated in the transition to MDS in one patient and the transition to AML in the other (56, 57). Interestingly, PHF6 has been shown to frequently co-occur with RUNX1 in AML (58).
Despite PHF6 mutations leading to inactivation of the protein, an analysis of PHF6 expression levels in AML regardless of mutation status revealed that PHF6 protein levels are higher in patients with AML than normal controls, a finding seemingly at odds with its role as a tumor suppressor (59). Moreover, increased PHF6 levels correlated with an increased percentage of blasts, with a possible correlation with CD34 positivity (59). Decreased PHF6 protein expression correlated with longer overall survival than those with high expression levels (2 years versus 6 months) (59).
A study of 318 pediatric patients with de novo AML identified PHF6 mutations in 6 (2%) cases with FAB subtypes of M0, M1, and M2 (53). The median age for this group was 12.6 years (versus 9.5 in wild type PHF6 AML), with 4 of 6 succumbing to the disease. Co-genetic abnormalities included RUNX1/RUNX1T1 translocations, NUP98/KDM5A translocations, and mutations in WT1, RAS, ETV6, TET2, IDH1, and BCORL1 (53). Measuring the expression level of PHF6 showed decreased PHF6 levels in patients with mutations compared to M0, M1, and M2 AML subtypes with wild-type PHF6, again supporting a tumor suppressor role for PHF6 and providing at least some genetic context to the results found by Mousa et al. above (53, 59).
A study of 398 patients with AML younger than 60 years of age revealed PHF6 to be associated with decreased overall survival in patients with intermediate-risk AML with wild-type FLT3-ITD (60). This implicates PHF6 mutations as a potential prognostic marker to be used in intermediate-risk AML, although it should be noted that this finding has not been replicated by others (24). Interestingly, subdividing AML with MRC cases that are associated with complex karyotypes into typical (those harboring 5q, 7q and/or 17p abnormalities) and atypical (those without these abnormalities) shows that AML with atypical complex karyotypes tend to have PHF6 mutations more frequently, TP53 mutations less frequently, be younger, have a higher WBC and blast percentage, and higher complete remission and overall survival rates (61).
Myelodysplastic Syndrome
Animal models suggest a possible role for PHF6 mutations in myelodysplastic syndrome (MDS), as aged mice with knocked-out PHF6 exhibit megakaryocytic dysplasia and associated decreased platelet counts as well as extramedullary hematopoiesis (13). Nevertheless, PHF6 mutations are relatively rare (~3%) in MDS (24, 62–64). The limited data indicate that PHF6 mutations are found most frequently (5.3% of MDS cases) in the high-grade subtypes (MDS with excess blasts) (24). PHF6 mutations tended to show low variant allele frequencies and acquisition in sub-clonal populations (24). The most frequent co-mutations were seen in ASXL1, RUNX1, TET2, and DNMT3A (64). A study of 21 MDS patients harboring PHF6 mutations revealed 61.9% had normal karyotypes and no patients had complex karyotypes (64).
Myeloproliferative Neoplasms
PHF6 mutations are rarely identified in myeloproliferative neoplasms (MPN) (0.7%), occurring in only 1.6% of chronic myelogenous leukemia (24). A screen of 81patients with CML in myeloid blast crisis identified 2 male patients with PHF6 mutations, with at least one patient showing no PHF6 mutations in the preceding chronic phase (65). This finding raises the possibility that, similar to MDS, the accumulation of PHF6 mutations might mediate progression of the disease. Relatedly, a review of 22 patients with PHF6 mutations in myeloproliferative neoplasms at three institutions revealed an enrichment in cases with increased fibrosis and/or blast crisis (66). Other than JAK2, the most common co-mutations in these MPNs were ASXL1, and TET2 with a median of 2.5 non JAK2 co-mutated genes (66).
With respect to mixed myelodysplastic syndrome/myeloproliferative neoplasm cases, PHF6 mutations were seen in 4.7% of CMML patients (24). To date, no effect on survival has been seen in PHF6-mutation associated CMML patients (64).
B-Lymphoblastic Leukemia
Despite their prevalence in T-ALL, PHF6 mutations have only rarely been identified in B-lymphoblastic leukemia (4, 41, 67). 50% of the rare MEF2D-rearranged B-ALL was found to have PHF6 mutations (67). Intriguingly, use of a retrovirally-expressed shRNA screening library into a B-lymphobastic leukemia cell line revealed that knockdown of PHF6 levels inhibited cell growth and leukemia growth in transplanted models (68). Indeed, CRISPR-Cas9-mediated deletion of PHF6 in a murine BCR-ABL1+; p19-/-; mCherry+ B-ALL cell line resulted in delayed tumor formation after injection in immunocompetent mice compared to wild-type B-ALL cells (69). Moreover, PHF6 KO B-ALL cells induced a malignancy closer in presentation to lymphoma than leukemia, with tumor cells showing reduced expression of CD19 and B220 and B-cell development genes (e.g., Cd74, IL4ra, Lyn, Ly86, and BLK), and upregulation of CD4 and T-cell signal transduction genes (69). These data therefore implicate PHF6 mutations as lineage specific with respect to tumorigenesis, and even implicate mutation of PHF6 as a potential mediator of lineage plasticity in hematopoietic neoplasms.
Acute Leukemia of Ambiguous Lineage
The association of PHF6 mutations with leukemias of ambiguous lineage further supports a role for these mutations in lineage plasticity. An analysis of 29 mixed phenotype acute leukemia cases at Memorial Sloan Kettering Cancer Center revealed PHF6 (23%) and DNMT3A (23%) as the most common recurrent mutations (70). Mutations in PHF6 and DNMT3A are mutually exclusive in MPAL, correlate with T-lineage marker expression (83% and 100%, respectively), and have higher relapse at 2 years (58% and 79%, respectively) compared to MPAL lacking these mutations (70).
In addition to T/M cases, Xiao et al. identified PHF6 mutations in patients with T/B/M and T/B phenotypes (70). Similarly, Getta et al. found PHF6 mutations in 3 of 16 MPAL patients with at least 2 of 3 patients of the MPAL, NOS subtype (B/T or B/T/M) (71). Furthermore, a review of 9 patients from multiple institutions identified PHF6 mutations in 5 patients with B/T MPAL (56%) (72).
Similar to T-ALL, PHF6 mutations are believed to be early events in MPAL, as Xiao et al. found every blast population isolated from selected cases showed nearly 100% VAF (70). Interestingly, PHF6-associated MPAL correlates with younger patients, higher hemoglobin and higher platelet values (70, 72).
Bond et al. performed an analysis of the transcriptional program of AMLs and T-ALLs and identified an expression program at the interface of these two diseases (73). Comparing T-ALL to T-ALL-like AMLs and AML-like T-ALLs, it was found that all T-ALLs with PHF6 mutations were accompanied by NOTCH1 mutations, whereas 3/5 PHF6 mutated interface cases lacked NOTCH1 mutations (73).
Interestingly, a study of acute undifferentiated leukemias found PHF6 mutations in 7/16 cases of AUL, whereas only 1/25 cases of minimally differentiated AML harbored these mutations (74). Nevertheless, in the limited study, no clinical outcome differences were seen between the two groups (74).
Discussion
The last two decades have elucidated a role for PHF6 in neurodevelopment and hematopoiesis, and revealed it as a potent tumor suppressor with an exclusive tendency for hematologic malignancies (Figure 2). PHF6 mutations are most common in T-ALL, and appear early in the disease course yet appear insufficient for leukemia development. Given the conflicting data regarding the prognostic significance of PHF6 mutations in T-ALL, additional studies are necessary to clarify the role of PHF6 inactivation in the disease course. It is likely that appropriately powered studies will need to assess its significance in specific patient cohorts.
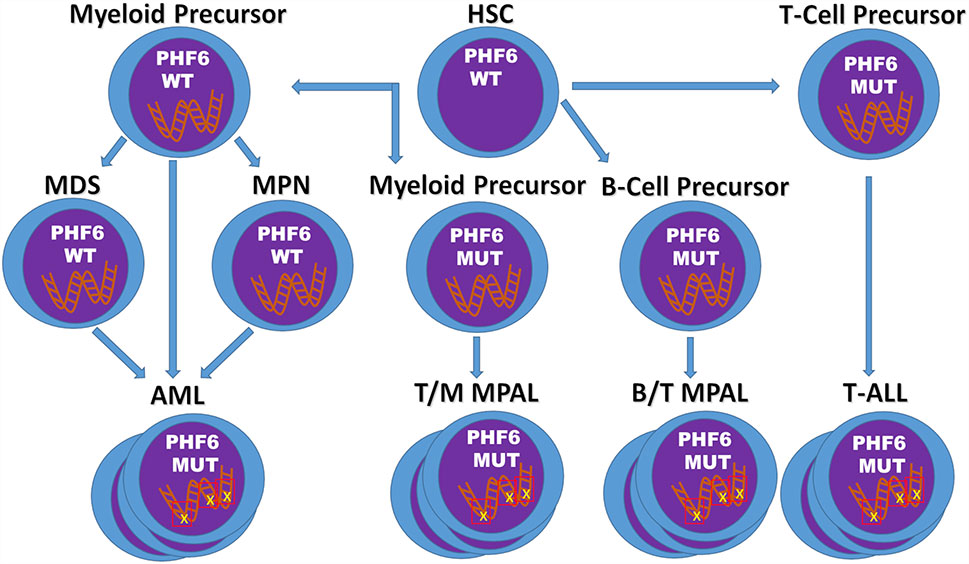
Figure 2 Model of PHF6 in Hematopoietic Malignancies. The timing and context of the acquisition of PHF6 deletions/mutations appear to determine the fate of the resulting malignancy. In T-ALL, PHF6 deletions/mutations arise early, but are insufficient for transformation into leukemia. In contrast, in myeloid neoplasms, PHF6 deletions/mutations tend to develop later possibly leading to disease progression. Finally, acquisition of inactivating mutations of PHF6 in myeloid or B-cell precursors may promote T-cell gene expression and eventual development of mixed phenotype acute leukemias.
In contrast to T-ALL, PHF6 mutations are less frequent in myeloid malignancies. Of interest, these mutations are more frequently found later in the disease course at points of disease progression. Further work is therefore required to determine the mechanism by which PHF6 pushes these neoplasms to a more aggressive disease, as well as to determine the overall prognostic significance of PHF6 mutations in myeloid malignancies in general. If additional studies continue to show a role for PHF6 mutations in disease progression, laboratories may wish to offer specific targeted analysis of PHF6 for the monitoring of myeloid disease.
Finally, it is becoming clear that PHF6 plays a role in lineage plasticity of hematopoietic malignancies, as PHF6 mutations exist in T/Myeloid MPAL as well as MPAL, NOS (particularly B/T MPAL) and is frequently associated with early T-ALL which frequently shows myeloid marker expression. The underlying contextual factors, including cell of origin and cooperative gene mutations, remain to be elucidated to understand what drives a PHF6-associated malignancy to be T-lineage, myeloid lineage, or a combination of both.
Author Contributions
OW conceived of the review subject matter and provided editorial review while JK primarily composed the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Musselman CA, Kutateladze TG. Handpicking Epigenetic Marks With PHD Fingers. Nucleic Acids Res (2011) 39:9061–71. doi: 10.1093/nar/gkr613
2. Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, Gedeon AK, et al. Mutations in PHF6 Are Associated With Börjeson-Forssman-Lehmann Syndrome. Nat Genet (2002) 32:661–5. doi: 10.1038/ng1040
3. Todd MAM, Ivanochko D, Picketts DJ. PHF6 Degrees of Separation: the Multifaceted Roles of a Chromatin Adaptor Protein. Genes (2015) 6:325–52. doi: 10.3390/genes6020325
4. Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, et al. PHF6 Mutations in T-Cell Acute Lymphoblastic Leukemia. Nat Genet (2010) 42:338–42. doi: 10.1038/ng.542
5. Wang J, Leung JW, Gong Z, Feng L, Shi X, Chen J. PHF6 Regulates Cell Cycle Progression by Suppressing Ribosomal RNA Synthesis. J Biol Chem (2013) 288:3174–83. doi: 10.1074/jbc.M112.414839
6. Todd MAM, Picketts DJ. PHF6 Interacts With the Nucleosome Remodeling and Deacetylation (Nurd) Complex. J Proteome Res (2012) 11:4326–37. doi: 10.1021/pr3004369
7. Voss AK, Gamble R, Collin C, Shoubridge C, Corbett M, Gécz J, et al. Protein and Gene Expression Analysis of Phf6, the Gene Mutated in the Börjeson-Forssman-Lehmann Syndrome of Intellectual Disability and Obesity. Gene Expr. Patterns GEP (2007) 7:858–71. doi: 10.1016/j.modgep.2007.06.007
8. Loontiens S, Dolens A-C, Strubbe S, Van de Walle I, Moore FE, Depestel L, et al. Phf6 Expression Levels Impact Human Hematopoietic Stem Cell Differentiation. Front Cell Dev Biol (2020) 8:599472. doi: 10.3389/fcell.2020.599472
9. Cheng C, Deng P-Y, Ikeuchi Y, Yuede C, Li D, Rensing N, et al. Characterization of a Mouse Model of Börjeson-Forssman-Lehmann Syndrome. Cell Rep (2018) 25:1404–14.e6. doi: 10.1016/j.celrep.2018.10.043
10. McRae HM, Garnham AL, Hu Y, Witkowski MT, Corbett MA, Dixon MP, et al. PHF6 Regulates Hematopoietic Stem and Progenitor Cells and its Loss Synergizes With Expression of TLX3 to Cause Leukemia. Blood (2019) 133:1729–41. doi: 10.1182/blood-2018-07-860726
11. Wendorff AA, Quinn SA, Rashkovan M, Madubata CJ, Ambesi-Impiombato A, Litzow MR, et al. Phf6 Loss Enhances HSC Self-Renewal Driving Tumor Initiation and Leukemia Stem Cell Activity in T-ALL. Cancer Discov (2019) 9:436–51. doi: 10.1158/2159-8290.CD-18-1005
12. Miyagi S, Sroczynska P, Kato Y, Nakajima-Takagi Y, Oshima M, Rizq O, et al. The Chromatin-Binding Protein Phf6 Restricts the Self-Renewal of Hematopoietic Stem Cells. Blood (2019) 133:2495–506. doi: 10.1182/blood.2019000468
13. Hsu Y-C, Chen T-C, Lin C-C, Yuan C-T, Hsu C-L, Hou H-A, et al. Phf6-Null Hematopoietic Stem Cells Have Enhanced Self-Renewal Capacity and Oncogenic Potentials. Blood Adv (2019) 3:2355–67. doi: 10.1182/bloodadvances.2019000391
14. Liu Z, Li F, Ruan K, Zhang J, Mei Y, Wu J, et al. Structural and Functional Insights Into the Human Börjeson-Forssman-Lehmann Syndrome-Associated Protein PHF6 *. J Biol Chem (2014) 289:10069–83. doi: 10.1074/jbc.M113.535351
15. Liu Z, Li F, Zhang B, Li S, Wu J, Shi Y. Structural Basis of Plant Homeodomain Finger 6 (PHF6) Recognition by the Retinoblastoma Binding Protein 4 (RBBP4) Component of the Nucleosome Remodeling and Deacetylase (Nurd) Complex. J Biol Chem (2015) 290:6630–8. doi: 10.1074/jbc.M114.610196
16. Basta J, Rauchman M. The Nucleosome Remodeling and Deacetylase (Nurd) Complex in Development and Disease. Transl Res J Lab Clin Med (2015) 165:36–47. doi: 10.1016/j.trsl.2014.05.003
17. Zhang C, Mejia LA, Huang J, Valnegri P, Bennett EJ, Anckar J, et al. The X-Linked Intellectual Disability Protein PHF6 Associates With the PAF1 Complex and Regulates Neuronal Migration in the Mammalian Brain. Neuron (2013) 78:986–93. doi: 10.1016/j.neuron.2013.04.021
18. Todd MAM, Huh MS, Picketts DJ. The Sub-Nucleolar Localization of PHF6 Defines its Role in Rdna Transcription and Early Processing Events. Eur J Hum Genet EJHG (2016) 24:1453–9. doi: 10.1038/ejhg.2016.40
19. Warmerdam DO, Alonso-de Vega I, Wiegant WW, van den Broek B, Rother MB, Wolthuis RM, et al. PHF6 Promotes non-Homologous End Joining and G2 Checkpoint Recovery. EMBO Rep (2020) 21:e48460. doi: 10.15252/embr.201948460
20. Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, et al. The Landscape of Somatic Mutations in Epigenetic Regulators Across 1,000 Paediatric Cancer Genomes. Nat Commun (2014) 5:3630. doi: 10.1038/ncomms4630
21. Van Vlierberghe P, Patel J, Abdel-Wahab O, Lobry C, Hedvat CV, Balbin M, et al. PHF6 Mutations in Adult Acute Myeloid Leukemia. Leukemia (2011) 25:130–4. doi: 10.1038/leu.2010.247
22. Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. N Engl J Med (2015) 373:35–47. doi: 10.1056/NEJMoa1414799
23. Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, Barda N, et al. Prediction of Acute Myeloid Leukaemia Risk in Healthy Individuals. Nature (2018) 559:400–4. doi: 10.1038/s41586-018-0317-6
24. Mori T, Nagata Y, Makishima H, Sanada M, Shiozawa Y, Kon A, et al. Somatic PHF6 Mutations in 1760 Cases With Various Myeloid Neoplasms. Leukemia (2016) 30:2270–3. doi: 10.1038/leu.2016.212
25. Chao MM, Todd MA, Kontny U, Neas K, Sullivan MJ, Hunter AG, et al. T-Cell Acute Lymphoblastic Leukemia in Association With Börjeson-Forssman-Lehmann Syndrome Due to a Mutation in PHF6. Pediatr Blood Cancer (2010) 55:722–4. doi: 10.1002/pbc.22574
26. Grossmann V, Haferlach C, Weissmann S, Roller A, Schindela S, Poetzinger F, et al. The Molecular Profile of Adult T-Cell Acute Lymphoblastic Leukemia: Mutations in RUNX1 and DNMT3A Are Associated With Poor Prognosis in T-ALL. Genes Chromosomes Cancer (2013) 52:410–22. doi: 10.1002/gcc.22039
27. Wang Q, Qiu H, Jiang H, Wu L, Dong S, Pan J, et al. Mutations of PHF6 are Associated With Mutations of NOTCH1, JAK1 and Rearrangement of SET-NUP214 in T-Cell Acute Lymphoblastic Leukemia. Haematologica (2011) 96:1808–14. doi: 10.3324/haematol.2011.043083
28. Yoo NJ, Kim YR, Lee SH. Somatic Mutation of PHF6 Gene in T-Cell Acute Lymphoblatic Leukemia, Acute Myelogenous Leukemia and Hepatocellular Carcinoma. Acta Oncol Stockh Swed (2012) 51:107–11. doi: 10.3109/0284186X.2011.592148
29. Huh HJ, Lee SH, Yoo KH, Sung KW, Koo HH, Jang JH, et al. Gene Mutation Profiles and Prognostic Implications in Korean Patients With T-Lymphoblastic Leukemia. Ann Hematol (2013) 92:635–44. doi: 10.1007/s00277-012-1664-2
30. Chang Y-H, Yu C-H, Jou S-T, Lin C-Y, Lin K-H, Lu M-Y, et al. Targeted Sequencing to Identify Genetic Alterations and Prognostic Markers in Pediatric T-Cell Acute Lymphoblastic Leukemia. Sci Rep (2021) 11:769. doi: 10.1038/s41598-020-80613-6
31. Sentís I, Gonzalez S, Genescà E, García-Hernández V, Muiños F, Gonzalez C, et al. The Evolution of Relapse of Adult T Cell Acute Lymphoblastic Leukemia. Genome Biol (2020) 21:284. doi: 10.1186/s13059-020-02192-z
32. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The Genomic Landscape of Pediatric and Young Adult T-Lineage Acute Lymphoblastic Leukemia. Nat Genet (2017) 49:1211–8. doi: 10.1038/ng.3909
33. Spinella J-F, Cassart P, Richer C, Saillour V, Ouimet M, Langlois S, et al. Genomic Characterization of Pediatric T-Cell Acute Lymphoblastic Leukemia Reveals Novel Recurrent Driver Mutations. Oncotarget (2016) 7:65485–503. doi: 10.18632/oncotarget.11796
34. Vicente C, Schwab C, Broux M, Geerdens E, Degryse S, Demeyer S, et al. Targeted Sequencing Identifies Associations Between IL7R-JAK Mutations and Epigenetic Modulators in T-Cell Acute Lymphoblastic Leukemia. Haematologica (2015) 100:1301–10. doi: 10.3324/haematol.2015.130179
35. Zhang H-H, Wang H-S, Qian X-W, Fan C-Q, Li J, Miao H, et al. Genetic Variants and Clinical Significance of Pediatric Acute Lymphoblastic Leukemia. Ann Transl Med (2019) 7:296. doi: 10.21037/atm.2019.04.80
36. Richter-Pechańska P, Kunz JB, Hof J, Zimmermann M, Rausch T, Bandapalli OR, et al. Identification of a Genetically Defined Ultra-High-Risk Group in Relapsed Pediatric T-Lymphoblastic Leukemia. Blood Cancer J (2017) 7:e523. doi: 10.1038/bcj.2017.3
37. Li M, Xiao L, Xu J, Zhang R, Guo J, Olson J, et al. Co-Existence of PHF6 and NOTCH1 Mutations in Adult T-Cell Acute Lymphoblastic Leukemia. Oncol Lett (2016) 12:16–22. doi: 10.3892/ol.2016.4581
38. De Keersmaecker K, Atak ZK, Li N, Vicente C, Patchett S, Girardi T, et al. Exome Sequencing Identifies Mutation in CNOT3 and Ribosomal Genes RPL5 and RPL10 in T-Cell Acute Lymphoblastic Leukemia. Nat Genet (2013) 45:186–90. doi: 10.1038/ng.2508
39. Teachey DT, Pui C-H. Comparative Features and Outcomes Between Paediatric T-Cell and B-Cell Acute Lymphoblastic Leukaemia. Lancet Oncol (2019) 20:e142–54. doi: 10.1016/S1470-2045(19)30031-2
40. Lejman M, Włodarczyk M, Styka B, Pastorczak A, Zawitkowska J, Taha J, et al. Advantages and Limitations of SNP Array in the Molecular Characterization of Pediatric T-Cell Acute Lymphoblastic Leukemia. Front Oncol (2020) 10:1184. doi: 10.3389/fonc.2020.01184
41. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The Genetic Basis of Early T-Cell Precursor Acute Lymphoblastic Leukaemia. Nature (2012) 481:157–63. doi: 10.1038/nature10725
42. Noronha EP, Marques LVC, Andrade FG, Thuler LCS, Terra-Granado E, Pombo-de-Oliveira MS, et al. The Profile of Immunophenotype and Genotype Aberrations in Subsets of Pediatric T-Cell Acute Lymphoblastic Leukemia. Front Oncol (2019) 9:316. doi: 10.3389/fonc.2019.00316
43. Loontiens S, Vanhauwaert S, Depestel L, Dewyn G, Van Loocke W, Moore FE, et al. A Novel TLX1-Driven T-ALL Zebrafish Model: Comparative Genomic Analysis With Other Leukemia Models. Leukemia (2020) 34:3398–403. doi: 10.1038/s41375-020-0938-2
44. Yeh T-C, Liang D-C, Liu H-C, Jaing T-H, Chen S-H, Hou J-Y, et al. Clinical and Biological Relevance of Genetic Alterations in Pediatric T-Cell Acute Lymphoblastic Leukemia in Taiwan. Pediatr Blood Cancer (2019) 66:e27496. doi: 10.1002/pbc.27496
45. Seki M, Kimura S, Isobe T, Yoshida K, Ueno H, Nakajima-Takagi Y, et al. Recurrent SPI1 (PU.1) Fusions in High-Risk Pediatric T Cell Acute Lymphoblastic Leukemia. Nat Genet (2017) 49:1274–81. doi: 10.1038/ng.3900
46. Alcantara M, Simonin M, Lhermitte L, Touzart A, Dourthe ME, Latiri M, et al. Clinical and Biological Features of PTPN2-Deleted Adult and Pediatric T-Cell Acute Lymphoblastic Leukemia. Blood Adv (2019) 3:1981–8. doi: 10.1182/bloodadvances.2018028993
47. Mavrakis KJ, Van Der Meulen J, Wolfe AL, Liu X, Mets E, Taghon T, et al. A Cooperative Microrna-Tumor Suppressor Gene Network in Acute T-Cell Lymphoblastic Leukemia (T-ALL). Nat Genet (2011) 43:673–8. doi: 10.1038/ng.858
48. Stanulović VS, Binhassan S, Dorrington I, Ward DG, Hoogenkamp M. PHF6 Interacts With LMO2 During Normal Haematopoiesis and in Leukaemia and Regulates Gene Expression and Genome Integrity. bioRxiv (2020), 2020.08.18.255471. doi: 10.1101/2020.08.18.255471
49. Oshima K, Zhao J, Pérez-Durán P, Brown JA, Patiño-Galindo JA, Chu T, et al. Mutational and Functional Genetics Mapping of Chemotherapy Resistance Mechanisms in Relapsed Acute Lymphoblastic Leukemia. Nat Cancer (2020) 1:1113–27. doi: 10.1038/s43018-020-00124-1
50. Xiang J, Wang G, Xia T, Chen Z. The Depletion of PHF6 Decreases the Drug Sensitivity of T-Cell Acute Lymphoblastic Leukemia to Prednisolone. Biomed Pharmacother Biomed Pharmacother (2019) 109:2210–7. doi: 10.1016/j.biopha.2018.11.083
51. Kroeze E, Loeffen JLC, Poort VM, Meijerink JPP. T-Cell Lymphoblastic Lymphoma and Leukemia: Different Diseases From a Common Premalignant Progenitor? Blood Adv (2020) 4:3466–73. doi: 10.1182/bloodadvances.2020001822
52. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal Evolution in Relapsed Acute Myeloid Leukaemia Revealed by Whole-Genome Sequencing. Nature (2012) 481:506–10. doi: 10.1038/nature10738
53. Rooij JDE de, van den Heuvel‐Eibrink MM, van de Rijdt NKAM, Verboon LJ, de Haas V, Trka J, et al. PHF6 Mutations in Paediatric Acute Myeloid Leukaemia. Br J Haematol (2016) 175:967–71. doi: 10.1111/bjh.13891
54. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
55. Aypar U, Taylor J, Garcia JS, Momeni-Boroujeni A, Gao Q, Baik J, et al. P2RY8-CRLF2Fusion-Positive Acute Myeloid Leukemia With Myelodysplasia-Related Changes: Response to Novel Therapy. JCO Precis Oncol (2020) 4:152–60. doi: 10.1200/PO.19.00294
56. Ng IK, Lee J, Ng C, Kosmo B, Chiu L, Seah E, et al. Preleukemic and Second-Hit Mutational Events in an Acute Myeloid Leukemia Patient With a Novel Germline RUNX1 Mutation. Biomark Res (2018) 6:16. doi: 10.1186/s40364-018-0130-2
57. Staňo Kozubík K, Radová L, Pešová M, Réblová K, Trizuljak J, Plevová K, et al. C-Terminal RUNX1 Mutation in Familial Platelet Disorder With Predisposition to Myeloid Malignancies. Int J Hematol (2018) 108:652–7. doi: 10.1007/s12185-018-2514-3
58. Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, et al. Somatic Mutations Precede Acute Myeloid Leukemia Years Before Diagnosis. Nat Med (2018) 24:1015–23. doi: 10.1038/s41591-018-0081-z
59. Mousa NO, Gado M, Assem MM, Dawood KM, Osman A, Mousa NO, et al. Expression Profiling of Some Acute Myeloid Leukemia - Associated Markers to Assess Their Diagnostic/Prognostic Potential. Genet Mol Biol (2021) 44(1). doi: 10.1590/1678-4685-gmb-2019-0268
60. Patel JP, Gado M, Assem MM, Dawood KM, Osman A, Mousa NO, et al. Prognostic Relevance of Integrated Genetic Profiling in Acute Myeloid Leukemia. N Engl J Med (2012) 366:1079–89. doi: 10.1016/j.yonc.2012.07.030
61. Mrózek K, Eisfeld A-K, Kohlschmidt J, Carroll AJ, Walker CJ, Nicolet D, et al. Complex Karyotype in De Novo Acute Myeloid Leukemia: Typical and Atypical Subtypes Differ Molecularly and Clinically. Leukemia (2019) 33:1620–34. doi: 10.1038/s41375-019-0390-3
62. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of Genetic Lesions in 944 Patients With Myelodysplastic Syndromes. Leukemia (2014) 28:241–7. doi: 10.1038/leu.2013.336
63. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature (2011) 478:64–9. doi: 10.1038/nature10496
64. Chien KS, Kanagal-Shamanna R, Naqvi K, Sasaki K, Alvarado Y, Takahashi K, et al. The Impact of PHF6 Mutations in Myelodysplastic Syndromes, Chronic Myelomonocytic Leukemia, and Acute Myeloid Leukemia. Blood (2019) 134:1436–6. doi: 10.1182/blood-2019-131188
65. Li X, Yao H, Chen Z, Wang Q, Zhao Y, Chen S. Somatic Mutations of PHF6 in Patients With Chronic Myeloid Leukemia in Blast Crisis. Leuk Lymphoma (2013) 54:671–2. doi: 10.3109/10428194.2012.725203
66. Kurzer JH, Nardi V, Ouseph M, Weinberg OK. Clinical and Genomic Characterization of Myeloproliferative Neoplasms Harboring PHF6 Mutations, Abstract 1411. Abstr USCAP 2020 Hematop 1316-1502 Mod Pathol (2020) 33:1328–9. doi: 10.1038/s41379-020-0475-6
67. Ohki K, Kiyokawa N, Saito Y, Hirabayashi S, Nakabayashi K, Ichikawa H, et al. Clinical and Molecular Characteristics of MEF2D Fusion-Positive B-Cell Precursor Acute Lymphoblastic Leukemia in Childhood, Including a Novel Translocation Resulting in MEF2D-HNRNPH1 Gene Fusion. Haematologica (2019) 104:128–37. doi: 10.3324/haematol.2017.186320
68. Meacham CE, Lawton LN, Soto-Feliciano YM, Pritchard JR, Joughin BA, Ehrenberger T, et al. A Genome-Scale In Vivo Loss-of-Function Screen Identifies Phf6 as a Lineage-Specific Regulator of Leukemia Cell Growth. Genes Dev (2015) 29:483–8. doi: 10.1101/gad.254151.114
69. Soto-Feliciano YM, Bartlebaugh JME, Liu Y, Sánchez-Rivera FJ, Bhutkar A, Weintraub AS, et al. PHF6 Regulates Phenotypic Plasticity Through Chromatin Organization Within Lineage-Specific Genes. Genes Dev (2017) 31:973–89. doi: 10.1101/gad.295857.117
70. Xiao W, Bharadwaj M, Levine M, Farnhoud N, Pastore F, Getta BM, et al. PHF6 and DNMT3A Mutations Are Enriched in Distinct Subgroups of Mixed Phenotype Acute Leukemia With T-Lineage Differentiation. Blood Adv (2018) 2:3526–39. doi: 10.1182/bloodadvances.2018023531
71. Getta BM, Roshal M, Zheng J, Park JH, Stein EM, Levine R, et al. Allogeneic Hematopoietic Stem Cell Transplantation With Myeloablative Conditioning Is Associated With Favorable Outcomes in Mixed Phenotype Acute Leukemia. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant (2017) 23:1879–86. doi: 10.1016/j.bbmt.2017.06.026
72. Mi X, Griffin G, Lee W, Patel S, Ohgami R, Ok CY, et al. Genomic and Clinical Characterization of B/T Mixed Phenotype Acute Leukemia Reveals Recurrent Features and T-ALL Like Mutations. Am J Hematol (2018) 93:1358–67. doi: 10.1002/ajh.25256
73. Bond J, Krzywon A, Lhermitte L, Roumier C, Roggy A, Belhocine M, et al. A Transcriptomic Continuum of Differentiation Arrest Identifies Myeloid Interface Acute Leukemias With Poor Prognosis. Leukemia (2021) 35:724–36. doi: 10.1038/s41375-020-0965-z
74. Weinberg OK, Hasserjian RP, Baraban E, Ok CY, Geyer JT, Philip JKSS, et al. Clinical, Immunophenotypic, and Genomic Findings of Acute Undifferentiated Leukemia and Comparison to Acute Myeloid Leukemia With Minimal Differentiation: a Study From the Bone Marrow Pathology Group. Mod Pathol Off J U S Can Acad Pathol Inc (2019) 32:1373–85. doi: 10.1038/s41379-019-0263-3
Keywords: PHF6, leukemia, tumor suppressor, T-ALL, AML
Citation: Kurzer JH and Weinberg OK (2021) PHF6 Mutations in Hematologic Malignancies. Front. Oncol. 11:704471. doi: 10.3389/fonc.2021.704471
Received: 03 May 2021; Accepted: 28 June 2021;
Published: 26 July 2021.
Edited by:
Julia T. Geyer, Weill Cornell Medical Center, United StatesReviewed by:
Yi Miao, Nanjing Medical University, ChinaRanganatha R. Somasagara, North Carolina Central University, United States
Copyright © 2021 Kurzer and Weinberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olga K. Weinberg, Olga.Weinberg@UTSouthwestern.edu