 Mar Pacheco-Herrero1*†
Mar Pacheco-Herrero1*† Luis O. Soto-Rojas2†
Luis O. Soto-Rojas2† Charles R. Harrington3
Charles R. Harrington3 Yazmin M. Flores-Martinez4Marcos M. Villegas-Rojas5
Yazmin M. Flores-Martinez4Marcos M. Villegas-Rojas5 Alfredo M. León-Aguilar5Paola A. Martínez-Gómez2
Alfredo M. León-Aguilar5Paola A. Martínez-Gómez2 B. Berenice Campa-Córdoba6,7Ricardo Apátiga-Pérez6,7
B. Berenice Campa-Córdoba6,7Ricardo Apátiga-Pérez6,7 Carolin N. Corniel-Taveras1
Carolin N. Corniel-Taveras1 Jesabelle de J. Dominguez-García1Víctor Manuel Blanco-Alvarez8
Jesabelle de J. Dominguez-García1Víctor Manuel Blanco-Alvarez8 José Luna-Muñoz7,9*
José Luna-Muñoz7,9*- 1Neuroscience Research Laboratory, Faculty of Health Sciences, Pontificia Universidad Católica Madre y Maestra, Santiago de los Caballeros, Dominican Republic
- 2Facultad de Estudios Superiores Iztacala, Universidad Nacional Autónoma de México, Mexico City, Mexico
- 3School of Medicine, Medical Sciences and Nutrition, University of Aberdeen, Aberdeen, United Kingdom
- 4Programa Institucional de Biomedicina Molecular, Escuela Nacional de Medicina y Homeopatía, Instituto Politécnico Nacional, Mexico City, Mexico
- 5Unidad Profesional Interdisciplinaria de Biotecnología del Instituto Politécnico Nacional (UPIBI- IPN), Mexico City, Mexico
- 6Departamento de Fisiología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Mexico City, Mexico
- 7National Dementia BioBank, Ciencias Biológicas, Facultad de Estudios Superiores, Cuautitlán, Mexico
- 8Facultad de Enfermeria, Benemérita Universidad Autónoma de Puebla, Puebla, Mexico
- 9Banco Estado de Cerebros-UNPHU, Universidad Nacional Pedro Henriquez Ureña, Santo Domingo, Dominican Republic
The current pandemic caused by the new severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has become a public health emergency. To date, March 1, 2021, coronavirus disease 2019 (COVID-19) has caused about 114 million accumulated cases and 2.53 million deaths worldwide. Previous pieces of evidence suggest that SARS-CoV-2 may affect the central nervous system (CNS) and cause neurological symptoms in COVID-19 patients. It is also known that angiotensin-converting enzyme-2 (ACE2), the primary receptor for SARS-CoV-2 infection, is expressed in different brain areas and cell types. Thus, it is hypothesized that infection by this virus could generate or exacerbate neuropathological alterations. However, the molecular mechanisms that link COVID-19 disease and nerve damage are unclear. In this review, we describe the routes of SARS-CoV-2 invasion into the central nervous system. We also analyze the neuropathologic mechanisms underlying this viral infection, and their potential relationship with the neurological manifestations described in patients with COVID-19, and the appearance or exacerbation of some neurodegenerative diseases.
Introduction
Coronavirus disease 2019 (COVID-19) was first reported in December 2019 in Wuhan, in Hubei province, China (1). In March 2020, the world health organization (WHO) declared the COVID-19 a pandemic. Almost a year later, on March 1, 2021, more than 114 million cases and 2.53 million deaths have been reported (2–4). Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19, is 100 nm with an oval shape and covered with crown-shaped glycoprotein spikes (5). It is transmitted through respiratory droplets from infected individuals or contact with fomites. Once SARS-CoV-2 enters the body, the onset of symptoms ranges from 2 to 14 days. Patients can manifest clinically from asymptomatic or mild symptoms to moderate or severe symptoms (6, 7). Mild to moderate symptoms manifest as fever, dry cough, nasal congestion, sore throat, runny nose, fatigue, myalgias, diarrhea, anosmia, and ageusia as main symptoms (6, 7). The severe condition is characterized by atypical pneumonia, which can be observed as “ground-glass opacification” with bilateral multi-lobular consolidations by imaging studies (8). Between 5 and 30% of patients develop acute respiratory distress syndrome, characterized by rapid onset and with generalized inflammation in the lungs, requiring invasive life support therapy, such as mechanical ventilation (6, 7). There is increasing evidence that these critically ill COVID-19 patients suffer a so-called “cytokine storm syndrome,” characterized by the release of many pro-inflammatory cytokines [interleukin (IL)-1β and IL-6] and a low number of T cells into the bloodstream (9). The mean period from the onset of the symptoms to death is around 13 days [interquartile range (IQR) 11–18 days], and this depends on advanced age (>65 years) and comorbidities such as diabetes mellitus (DM), hypertension, cardiovascular disease, or chronic obstructive pulmonary disease (COPD) (10). These comorbidities are also risk factors for severity and transfer to intensive care unit (ICU), endotracheal intubation, and death in patients with COVID-19 (11). However, there may be bias in the epidemiological data due to the in-hospital stay of the patient, as well as the human development index of each country. SARS-CoV-2 also may be able to invade multiple organs, including the nervous system, and thus cause multiple organ dysfunction syndrome (MODS) (12). The neurological manifestations are beginning to take on unquestionable importance, mainly in the critical patient (13, 14). Neurological manifestations of COVID-19 and other coronavirus infections involve febrile seizures, disorientation, difficulty in speaking, encephalitis, and stroke (15–18). The mechanisms by which SARS-CoV-2 can spread, infect, cause damage to nerve cells and finally affect both the central (CNS) and peripheral (PNS) nervous system, are not yet understood. This review will analyze the potential mechanisms by which SARS-CoV-2 can invade the CNS and PNS and generate a neurotoxic environment that may trigger or worsen neurological disorders.
SARS-CoV-2: Structure and Mechanism of Infection
The coronaviruses (CoVs) belong to the Orthocoronaviridae subfamily; order: Nidovirales; subordination: Cornidovirineae; family: Coronaviridae (19). They can be grouped into four genera, including α/β/γ/δ-CoV: α and β infect mammals and γ/δ infect birds (20). CoVs are large, positive-stranded RNA viruses, and they are enveloped with a lipid membrane derived from a host cell. The protein protruding from the virus membrane is the spike (S) protein, giving the virus the appearance of a solar corona (1) (Figure 1A). Coronaviruses have single-stranded RNA of between 26.4 and 31.7 kilobases, making them the largest of RNA viruses (21). CoVs have several main structural proteins (Figure 1A): nucleocapsid (N) proteins, which surround the RNA genome; membrane (M) proteins (also known as E1 membrane glycoprotein or matrix protein) (20); envelope (E) proteins, involved in virus assembly, and S protein, which mediates virus entry into host cells. Some CoVs also encode an envelope-associated hemaglutinin-esterase protein (HE) used as an invading mechanism (22).
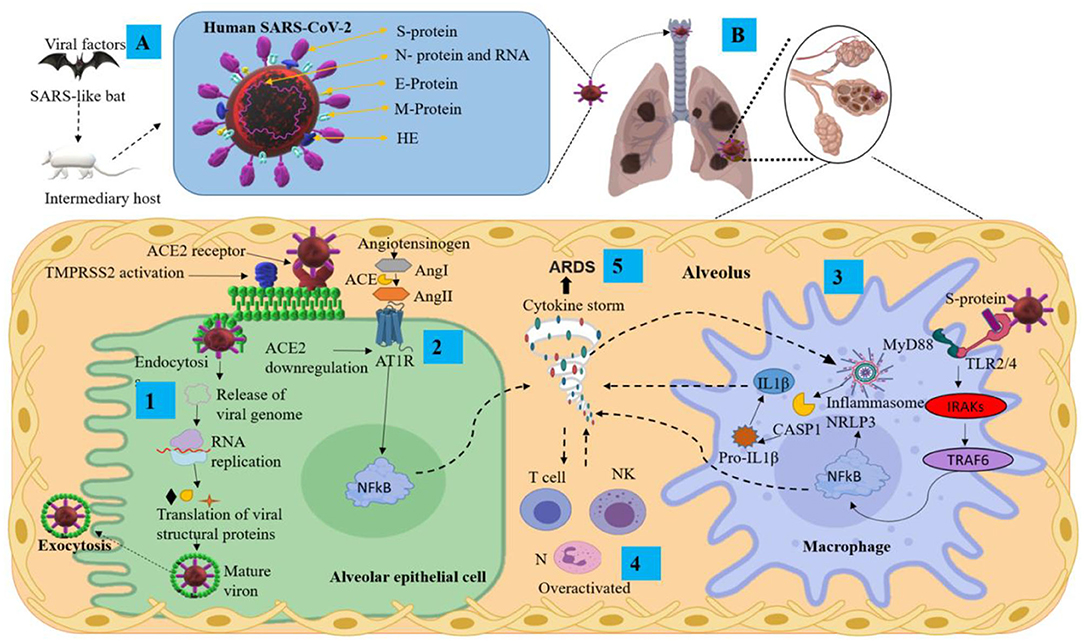
Figure 1. Pathological mechanisms of SARS-CoV-2 in the pulmonary alveolus. (A) Mode of transmission and main structural proteins of SARS-CoV-2. (B) Mechanisms of SARS-CoV-2 infection and pulmonary inflammatory immune response. ACE2, angiotensin-converting enzyme 2; Ang, angiotensin; ARDS, acute respiratory distress syndrome; AT1R, angiotensin II type I receptor; CASP1, aaspase 1; E protein, envelope small membrane protein; HE, hemagglutinin esterase; IL1β, interleukin 1 beta; IRAKs, interleukin-1 receptor-associated kinases; M protein, membrane protein; MyD88, myeloid differentiation primary response 88; N protein, nucleoprotein; N, neutrophils; NF-κB, nuclear factor Kappa B; NK, natural killer cells; NLRP3, nucleotide-binding domain-, leucine-rich repeat-containing receptor, pyrin domain-containing 3; RNA, ribonucleic acid; S protein, spike protein; TMPRSS2, transmembrane serine protease 2; TRAF6, tumor necrosis factor receptor-associated factor 6.
The S protein is the main antigenic component of SARS-CoV-2 structural proteins and is comprised of two subunits, S1 and S2 (23). This protein is multifunctional, contributing to host receptor binding, pathogenesis, and cell tropism. The S protein binds to host receptors on target cells, inducing virion particle endocytosis, and then catalyzes the fusion between host and viral membranes, allowing the virus genome penetration into the host cytoplasm (24). The S1 domain has a high-affinity association with the host receptor angiotensin-converting enzyme 2 (ACE2) (25). The receptor-binding domain (RBD) of the S protein binds to the extracellular peptidase domain of ACE2, mediating cell entry (25, 26). SARS-CoV-2 uses the SARS-CoV receptor ACE2 for entry and the transmembrane serine protease 2 (TMPRSS2) for S protein priming. The endosomal cysteine proteases cathepsin B and L (CatB/L) can be used to mature the S protein (27, 28). However, while TMPRSS2 is indispensable for viral spread and pathogenesis, the CatB/L activity is not essential (Figure 1B step 1). Once the virus enters the host cell, viral replication begins with translation of the replicase-polymerase gene and assembly of the replication-transcription complex. This complex also transcribes the genomic regions to structural proteins. New virions are assembled in the endoplasmic reticulum and Golgi apparatus released from the cell (Figure 1B step 1) (29). Finally, the newly assembled SARS-CoV-2 virions possess protein S on the surface and are ready to infect any cell that expresses the ACE2 receptor with no further requirement for TMPRSS2 activity (30).
SARS-CoV2 Pathophysiology
The SARS-CoV-2 pathophysiology is not yet clear. It has been suggested that it can be similar to SARS-CoV (31, 32) with two possible responses (32):
1) After the viral infection occurs, active viral replication and dissemination through ACE2 receptors occurs with the associated host antiviral responses. SARS-CoV-2 downregulates ACE2 receptors, with loss of their catalytic effect at the membrane surface. Inflammation and thrombosis have been related to enhanced and unimpeded angiotensin II effects through the ACE-Angiotensin II-AT1 receptor axis (33) (Figure 1B step 2). The SARS-CoV-2 infection can lead to an acute immune response. This response is driven by inflammatory alveolar and monocyte-derived macrophages that can be activated by pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) released by infected pneumocytes (34–36). Subsequently, several pro-inflammatory mediators such as tumor necrosis factor-alpha (TNF-α) and IL-1β, secreted by alveolar macrophages, initiate the acute inflammatory cascade that triggers cell death and damage. Aside from PAMP/DAMP production, the recruitment of immune cells and activation of the nucleotide-binding domain leucine-rich repeat-containing receptor, pyrin domain-containing 3 (NLRP3), establish a pro-inflammatory positive feedback cascade (32, 34, 35) (Figure 1B steps 3 and 4). This localized inflammatory cell death could lead to a hyper-inflammatory microenvironment and spread to the vasculature, inducing leakage, edema, and pneumonia in COVID-19 patients (35, 37). Serum of COVID-19 patients is characterized by increased levels of the following: IL-2, IL-7, IL-10, TNF-α, protein monocyte chemoattractant-1 (MCP1; also known as C-C motif chemokine ligand 2 CCL2), granulocyte colony-stimulating factor (G-CSF), macrophage inflammatory protein 1 alpha (MIP1α; also known as CCL3), C-X-C motif chemokine ligand 10 (CXCL10), C-reactive protein (CRP), D-dimers and ferritin (19, 38–40).
2) The SARS-CoV-2 infection can also lead to the generation of adaptive immunity and neutralizing antibody (NAb). The virus-NAb complex can trigger Fc receptor (FcR)-mediated inflammatory response and acute lung injury. SARS-CoV-2 can infect cells that have FcRs, which provide the ability for antibody-mediated internalization. This mechanism can occur in macrophages, monocytes, or B cells even without ACE2 and TMPRSS2 expression, and especially during infection (41). The internalization of the virus–antibody immune complexes can also promote tissue damage and inflammation by activating myeloid cells via FcRs (42). Both primary and secondary responses culminate in the postulated pathogenesis of SARS-CoV-2 infection (32). Interestingly, it has been suggested that fatal COVID-19 is characterized as a cytokine release syndrome (CRS) induced by a cytokine storm and associated with adverse outcomes of acute respiratory distress syndrome (ARDS) (Figure 1B step 5) and high mortality rate (43, 44).
For mechanistic insights into the life cycle of SARS-CoV-2, the mouse hepatitis virus (MHV) represents a suitable comparator. MHV is a βCoV very similar to SARS-CoV, MERS-CoV, and SARS-CoV-2 (45). Therefore, an MHV animal model could contribute to the elucidation of the neuropathological mechanisms of SARS-CoV-2 in the following aspects: (1) MHV can invade and replicate in the CNS, triggering lesions in the white matter (45); (2) Infection with MHV induces meningoencephalitis in an acute stage and subsequently subacute chronic inflammatory demyelination in the brain and spinal cord (46); (3) CD4 and CD8 T lymphocytes, especially γδ T cells, play an important role in the MHV-induced demyelination process (47); (4) MHV can be translocated from the initial inoculation brain area to the spinal cord through the transit of viral particles in glial and neural cells, as well as by mechanisms that involve the fusion of lipid membranes (48); (5) After intranasal MHV-CoV inoculation in mice, the virus can access the CNS through the olfactory nerve and spread from this area to neuroanatomically interconnected structures such as the limbic system and the brainstem (49).
Potential Neuroinvasive Pathways of SARS-CoV-2
There are four possible routes by which SARS-CoV-2 could enter the CNS: (1) the hematopoietic pathway and subsequent rupture of the blood-brain barrier (BBB); (2) via blood-cerebrospinal fluid (B-CSF); (3) transsynaptic viral spreading; (4) through the entry to circumventricular organs (CVO). In this section, we will discuss the four routes in greater detail.
1) Coronaviruses access the bloodstream via the airway and infect immune cells, which may cross BBB facilitated by pro-inflammatory cytokines and chemokines (Figure 2B step 1). The mechanism by which infected immune cells cross the BBB may occur via intercellular adhesion molecule 1 (ICAM-1) mediated transport that is upregulated by TNF-α, followed by activation of matrix metalloproteinases (MMPs) such as MMP9, which specifically influences cellular leakage and membrane degradation (50). Besides, SARS-CoV-2 tropism, toward the CNS endothelial cells (BECs) favors BBB disruption; by entering the cytosol of the astrocyte via the receptor; the virus increases the release of pro-inflammatory cytokines, such as IL-2, IL-6, IL-7, IL-8, TNFα, CCL2, CCL3, CCL7, and CXCL10. The reactive astrocyte could lead to activation of microglia and the peripheral immune infiltrate such as macrophages, neutrophils, and lymphocytes (Figure 2B step 2) (51–54), which could end in neurotoxicity. DM, hypertension, and metabolic syndrome are risk factors for both contracting COVID-19 and a poor prognosis in patients. These comorbidities also contribute to vascular and BBB alteration (55), increase neuroinflammation, and exacerbate neuropathology (56).
2) The CSF circulation comprises both a directional CSF flow and a pulsatile to and from movement throughout the entire brain and which involves a local fluid exchange between blood, interstitial fluid, and CSF (57). It has been suggested that viral infection may occur via B-CSF and alter gene expression in the choroid plexus. This process activates the nuclear factor kappa (NF-kB), upregulates MMP9, and affects B-CSF permeability and immune cell trafficking (MMP8, TNFα, IL6, IL1B, MCP1, intercellular adhesion molecule 1 (ICAM1) (58), leading to a neuroinflammatory environment.
3) Another entry route to the CNS for SARS-CoV-2 could be through axonal transport and transneuronal spread from olfactory, gustatory, trigeminal, and vagal nerves, allowing the virus to infect the brainstem in the early stages of infection (Figures 3A–D) (52, 59). The transneuronal pathway is one of the potential routes that would allow SARS-CoV-2 to enter through the primary sensory neurons, which communicate with the mitral cells. Mitral cells have projections toward the ventricle and the medulla, and this favors the transfer of the virus from the cerebrospinal fluid toward the lymphatic system within the CNS and toward the PNS (60). The virus could also enter the CNS following the transneuronal olfactory bulb pathway and is reflected by changes at the level of the olfactory nerve, bulb, and cortex (61–63). It has been proposed that SARS-CoV-2 could spread retrogradely through transsynaptic transfer, using an exocytosis/endocytosis mechanism or via rapid axonal transport, which would move the virus along the microtubules to the neuronal soma (64). Supporting this hypothesis, it has been shown that some CoVs and other viruses such as rabies and hemagglutinating encephalomyelitis can enter and spread to the CNS via retrograde transsynaptic pathways (65–67), from peripheral nerve endings through membranous-coating-mediated endocytosis and exocytosis (66). Mechanisms have also been described by which viruses can enter and leave axons, both retrograde and anterograde, through coupled transport mediated by vesicles or separate transport which is not mediated by vesicles (68). For this reason, it would be interesting in the future to know if SARS-CoV2 uses transsynaptic transport and to trace neural circuits, using specific labeling techniques.
4) Finally, we suggest that SARS-CoV-2 might enter the CNS through CVOs. CVOs include the subfornical organ, the paraventricular nucleus, the nucleus tractus solitarius (NTS), and the rostral ventrolateral medulla, all of which express ACE2. Besides, these CVOs are highly vascularized and lack a BBB (69). Therefore, these areas would be more susceptible to the virus, triggering neurovascular damage, as we have discussed previously.

Figure 2. Schematic representation of the pathophysiological mechanisms of SARS-CoV-2. (A) Peripheral pathological events triggered by SARS-CoV-2 infection. (B) Possible CNS pathological mechanisms caused by the severe peripheral hyperinflammation associated with COVID-19. ACE2, angiotensin-converting enzyme 2 receptor; AJs, adherent junctions; Aβ; amyloid-beta; BBB, blood-brain barrier; C1q, the complement component 1q; CASP1, caspase1; CCL, chemokine (C-C motif) ligand; CNS, central nervous system; CXCL10, C–X–C motif chemokine 10; GSDMD, gasdermin-D; IL, interleukin; MMPs, metalloproteinases; NETs, neutrophil extracellular traps; NF-κB, Nuclear factor Kappa B; N-GSDMD, N-terminal gasdermin; NLRP3, nucleotide-binding domain-, leucine-rich repeat-containing receptor, pyrin domain-containing 3; TJs, tight junctions; TLR3, toll-like receptor 3; TNF-α, tumor necrosis factor-alpha; α-syn, alpha-synuclein.
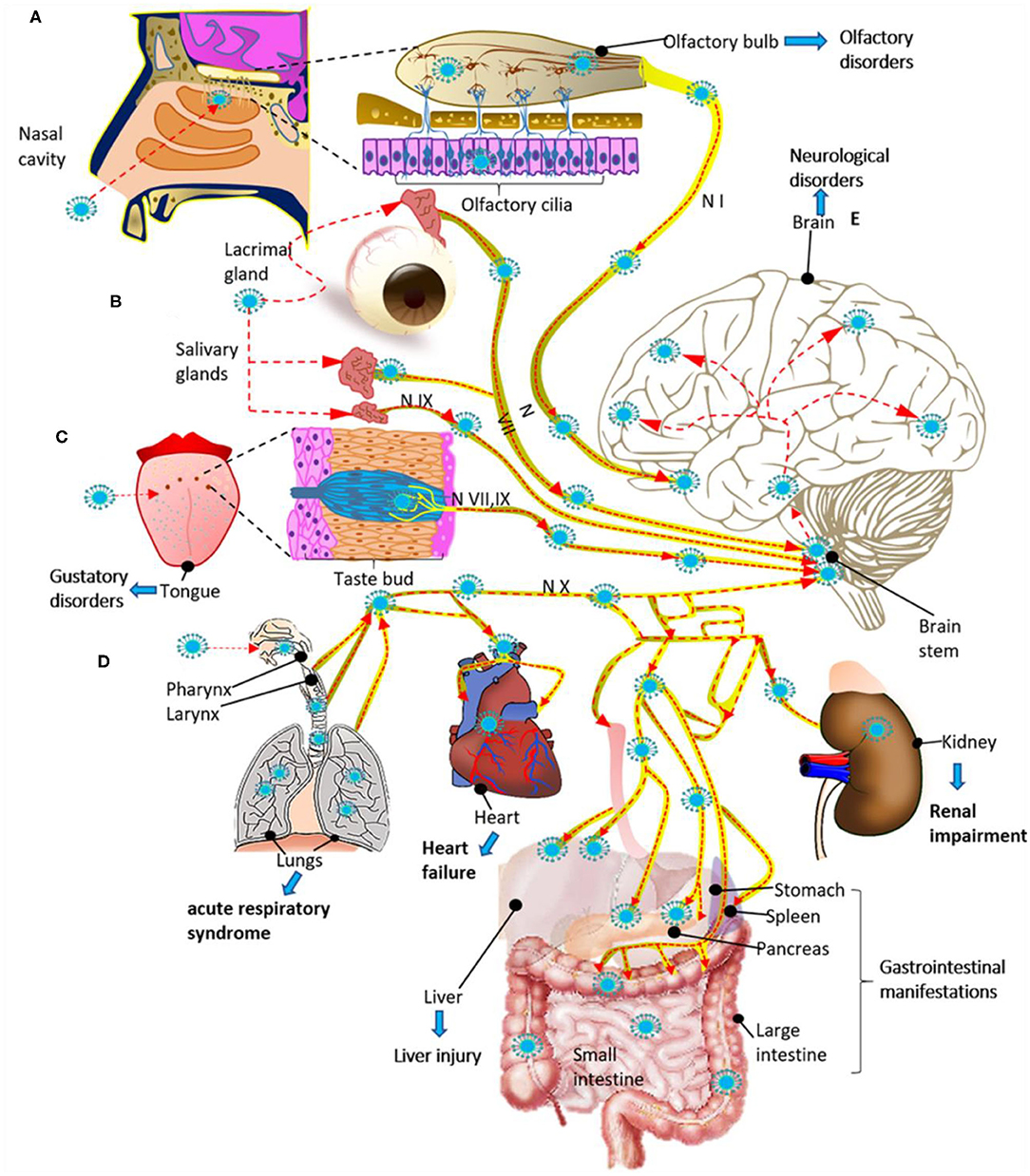
Figure 3. Potential routes for infection and spread of SARS-CoV-2 to systemic organs and the central nervous system through the cranial nerves (N). (A) SARS-CoV-2 could enter through the olfactory mucosa (causing anosmia), spread through the olfactory nerve (N I) and end in the olfactory cortex. (B) SARS-CoV-2 could also enter through the lacrimal and salivary glands, spread through the facial (N VII) and glossopharyngeal (N IX) nerves, and end in their respective brain stem nuclei. (C) The infection could spread from the taste buds (triggering ageusia) through the N VII and N IX nerves ending in the NTS located in the brain stem. (D) SARS-CoV-2 could also enter through the respiratory tract, reach the respiratory system and via the vagus nerve (N X), spread to other systemic organs innervated by this nerve, and end in the brain stem. (E) Finally, once the virus reaches the brain stem, it can spread to the brain through neuroanatomically interconnected pathways. The SARS-Cov2 infection can cause multiple organ dysfunction syndrome (A–E). The red dashed arrows indicate the possible dissemination route for SARS-CoV-2 through the cranial nerves.
Once SARS-CoV-2 enters the CNS, it could bind to CNS cells, such as neurons, astrocytes, oligodendrocytes, and microglia (70), due to the presence of ACE2 (28, 71) and TMPRSS2 (72) receptors and probably via binding to other receptors (Table 1). It is important to highlight that the expression of ACE2 is low in the human brain, with a higher expression in certain areas such as thalamus and choroid plexus. ACE2 also has access to peptides in the circulation in the cerebrospinal and interstitial fluid, and it is present in pericytes and smooth muscle cells of human brain vessels (95). ACE2 receptors have been reported in other organs, mainly enterocytes, renal tubules, gallbladder, cardiomyocytes, male reproductive cells, placental trophoblasts, ductal cells, eye, and vasculature. In the respiratory system, its expression is limited (96). ACE2 plays a role in attenuating microvascular pathology and protecting against atherogenesis, endothelial dysfunction, thrombus formation, oxidative stress, and inflammatory cascades responsible for monocyte-endothelial cell interaction (71, 97).
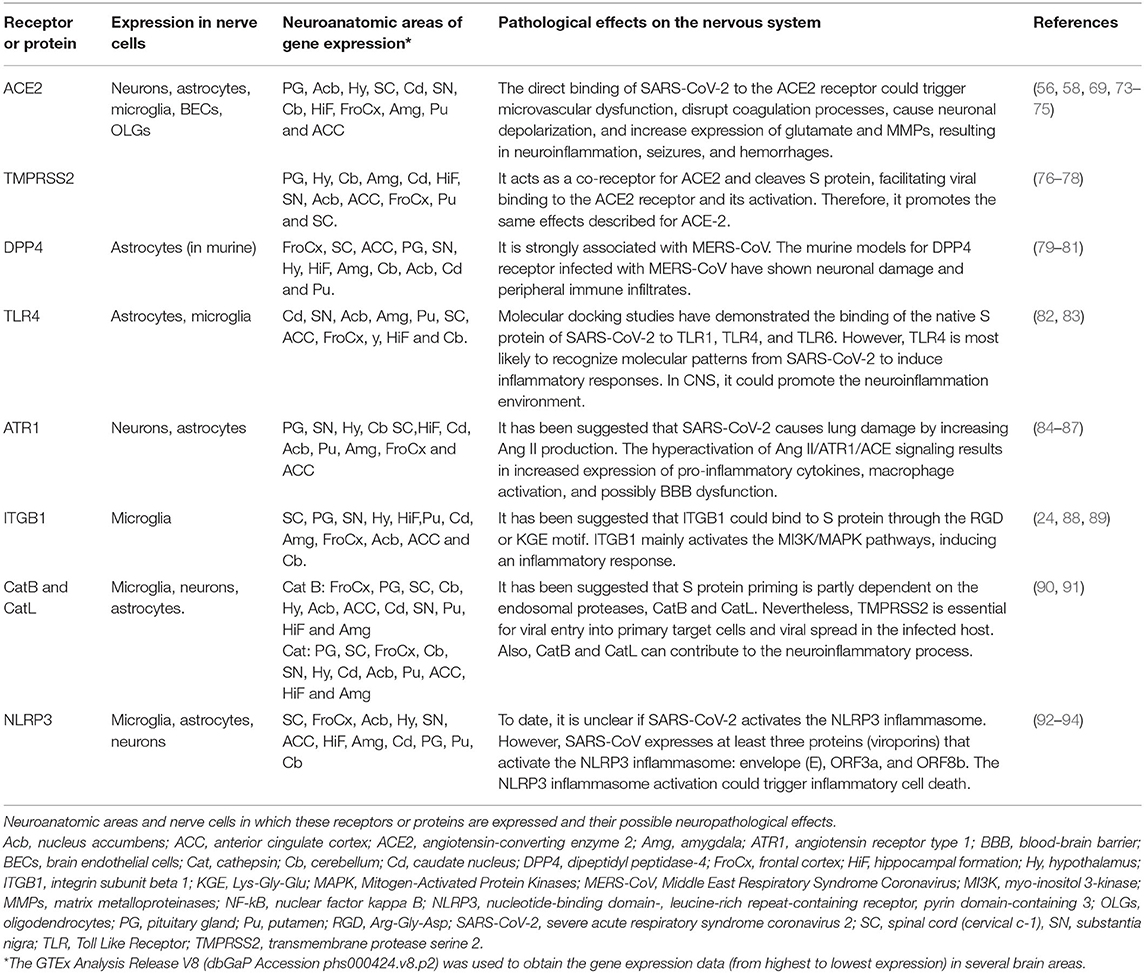
Table 1. Receptors or proteins related to SARS-CoV-2 infection in the nervous system.
SARS-CoV-2 interaction with ACE2 could cause astrogliosis and microgliosis, increase BBB permeability, allowing monocyte and leukocyte infiltration to the CNS in multiple brain regions (98, 99). These areas include the olfactory bulb, choroid plexus, cerebral cortex, caudate/putamen, ventral striatum, thalamus, hypothalamus (paraventricular nuclei), spinal cord, hippocampus, frontal cortex (52, 95, 100), substantia nigra, middle temporal gyrus (64, 101), and other brain areas (Table 1). Since many viruses have neurotropic properties (102), SARS-CoV-2 could spread through neuroanatomically interconnected pathways (103) and lead to nerve cell dysfunction and neurodegeneration in the CNS.
Can the Systemic Inflammation by COVID-19 Trigger Neurovascular Disturbance?
In this section, we highlight evidence at the systemic level and locally in the respiratory tract tissue of patients with COVID-19. Since this virus induces lung pathology, the detailed information of other organs and systems such as the CNS has yet to be fully investigated. Therefore, the nerve signaling pathways proposed here are based on the systemic evidence from similar viruses.
Channappanavar and Perlman focused on the systemic immune response against pathogenic human coronaviruses such as SARS-CoV and Middle East respiratory syndrome CoV (MERS-CoV). They proposed that a dysregulated immune response in the host is responsible for triggering the pulmonary pathology and fatal clinical manifestations (104).
On the other hand, high levels of viral replication in the host could contribute to tissue damage. Two mechanisms might be responsible: first, the delayed induction of interferon responses and second, the production of interferon inhibitory proteins by the human CoVs. Therefore, early unmitigated viral replication could be responsible for the high and exaggerated production of cytokines and chemokines by infected alveolar epithelial cells, macrophages, and leukocytes infiltrated into the lung tissue, which leads to severe damage (Figure 2A steps 1–3) (104). Other investigators have concluded that COVID-19 is characterized by an extreme hyper-inflammatory process followed by hyper-coagulation (Figure 2A step 4) (105).
CRS is a systemic inflammatory response that can be triggered by SARS CoV-2 infection, characterized by a drastic increase in the levels of the pro-inflammatory cytokines (Figure 2A) (106). The CRS may induce a MODS in COVID-19 patients, which is characterized by acute failure of different organs such as the liver, kidney, heart, and as well as hematological, gastrointestinal, and neurological disorders (Figure 3) (107). Besides, it has been proposed that patients who died from severe COVID-19 have a significant endothelial affectation or “endothelitis,” which may be associated with MODS. It has been suggested that endothelial dysfunction in several organs may be triggered by the interaction between SARS-CoV-2 with ACE2 receptors that express endothelial cells and the subsequent inflammatory response (108, 109). This inflammatory response can contribute to increased vascular permeability, edema, and the synthesis of coagulation factors (110). From a meta-analysis, it has been reported that levels of D-dimer, an indicator of fibrinolysis, have been reported following severe infection by COVID-19 (111). The formation of clots could result in the occlusion of blood vessels and cerebral arteries, which can lead to cerebral venous thrombosis. Therefore, we assume that some of the symptoms and even neurological complications may be caused by the systemic cytokine storm and subsequent endothelium and BBB dysfunction (Figure 2). In this way, systemic hyper-inflammation caused by maladaptive innate immunity may trigger neurovascular function damage, a BBB rupture, and activate the CNS innate immune signaling pathways (112). This BBB disruption could promote immune cell infiltration (113) (Figure 2B steps 1 and 2). The intracerebral cytokine storm also could contribute to the BBB rupture (114, 115), leading to a vicious cycle of increasing pathology. These events may also be responsible for developing other neuropathies such as necrotizing encephalopathy or Guillain-Barré syndrome (GBS) (116, 117). The coagulopathy observed in COVID-19 could make patients prone to thrombotic cerebrovascular or bleeding events (118).
On the other hand, microglia and astrocytes are the main cell lineages that mediate immunological processes within the CNS. Thus, microglia, the macrophage of the CNS par excellence, can also promote states of hyper-inflammation that exacerbate hypercoagulation by infiltration of professional immune cells and coagulation elements. Nevertheless, we hypothesize that SARS-CoV-2 could activate the microglia and, subsequently, induce the reactivation of A1 astrocytes via secreting IL-1α, TNF, and the complement component 1q (C1q), as occurs in other neurological diseases (119) (Figure 2B step 3). Besides, exposure to the viruses or their components promotes the expression and activation of Toll-like receptors (TLR) in astrocytes. This signaling promotes the production and release of pro-inflammatory mediators and induces inflammatory responses in the CNS (Figure 2B step 3), eliminating the pathogen as demonstrated for Flavivirus infections (82, 120). Therefore, this pathological signaling causes neuronal degeneration and dysfunction of the nerve cells short or long term.
Viroporins belong to a family of small transmembrane proteins that include CoV protein E (121, 122), which could generate neurotropism of SARS-CoV. According to previous studies, viroporins can promote the activation of the NLRP3 inflammasome (123, 124). The NLRP3 inflammasome is a subcellular multiprotein complex that is highly expressed in several nerve cells and CNS areas (Table 1). Activation of the NLRP3 occurs after infection by the influenza A virus and SARS-CoV (125) (Figure 2B step 4). After NLRP3 inflammasome activation, caspase-1 and other non-canonical inflammasome caspases (caspase-4, caspase-5, or caspase-11) activate gasdermin-D (GSDMD), which subsequently forms pores in the cell membrane. These pores facilitate the secretion of IL-1β and IL-18 and, importantly, they also enable the simultaneous influx of Na+ and water molecules, facilitating neuroinvasion by causing excessive cellular swelling, membrane rupture, and subsequent pyroptosis, an inflammatory form of cell death (126, 127). Therefore, it is possible that pyroptosis may occur in nerve cells (Figure 2B step 4).
Neurological and Neuropsychiatric Manifestations, Diagnostic, and Treatment in Patients With COVID-19
The respiratory symptoms caused by the SARS-CoV-2 virus are still the most readily identified and studied. However, neurological manifestations are beginning to take on unquestionable importance, mainly in the critically affected patient. Our understanding of the long-term neurological symptoms is limited and presents a real challenge (13, 14, 62, 128). A physiopathological explanation for the neurological and neuropsychiatric manifestations of COVID-19 has yet to be found. Although there are hypotheses about the direct effects of SARS-CoV-2 on the CNS and PNS, evidence suggests that these effects can be attributed to other causes such as: (1) the impact of the systemic inflammatory response caused by the virus and (2) the underlying comorbidities of the patients (62, 129, 130). Patients with mild COVID-19 have been reported to have non-specific neurological disorders such as headache and myalgias, dizziness, dysgeusia, and anosmia with variations in their prevalence (Table 2) (13, 14).

Table 2. SARS-CoV-2 infection in the central and peripheral nervous system: clinical manifestations, mechanism of pathogenicity, laboratory, and clinical findings and suggested treatment.
Some studies point to headache as the most common neurological symptom and often as the only symptom of COVID-19 (129, 147). However, other authors have defined the headache as a consequence of systemic disease. It has been suggested that the chronic release or exposure of vasoactive peptides such as Calcitonin Gene-Related Peptide (CGRP; pain and migraine-related peptide) (148) can activate trigeminal sensory fibers and thus modulate the transmission of impulses related to headache (149). Besides, in COVID-19, a close link between cytokine storm and headache has been proposed, due to the release of the vasoactive peptides (150).
In contrast, for hospitalized patients, encephalopathy with neuropsychiatric manifestations such as delirium and agitation have been observed. There has also been a considerable increase in the reports of patients with neuromuscular diseases, among which are GBS with some of its variants and several rhabdomyolysis cases. However, it has not been possible to find a clear relationship between these manifestations and COVID-19. An increasing incidence of neurological manifestations has been observed in COVID-19-infected patients that have been associated with severe health conditions and prolonged hospital stays (129, 134). However, there are also reports of neurological manifestations in outpatients with COVID-19 infection (129, 138). Therefore, it is difficult to determine the incidence of each of the neurological manifestations due to the different screening methods applied for each reported case.
Neurological disorders such as multiple sclerosis (MS), encephalopathy, and GBS, have been associated with SARS-CoV2. In some cases, CNS demyelination has occurred shortly after SARS-CoV-2 infection, suggesting a causal relationship between these two pathologies (151). Viruses, such as the Epstein-Barr virus (EBV), have been linked to MS, with high titers of EBV antibodies found in MS patients. Viral induced demyelination could be a direct result of viral infection of oligodendrocytes, which leads to cell death and myelin degeneration, or to the exacerbated inflammatory response caused by virus replication (152, 153). The cytokine storm caused by SARS-CoV-2 may cause the activation of glial cells and the start of the demyelination process (154). Conversely, other studies suggest that SARS-CoV-2 could act as an accelerating factor for MS but not the trigger for the disaese (155). Likewise, several case reports have reported the appearance of GBS after SARS-CoV-2 infection (156–158). Although hypoxic/metabolic changes caused by intense inflammatory response against the virus together with the presence of comorbidities, may result in encephalopathy (159) there is still insufficient evidence to prove that SARS-CoV-2 virus infection invades the CNS directly to provoke encephalopathy (151). In the same way, despite the complications associated with SARS-CoV-2 infection in patients with GBS, there is no clear evidence yet that COVID-19 initiates GBS (151).
Cerebral events have been associated with SARS-CoV-2 patients, with cerebral ischemic events being the most frequent. Cerebral hemorrhages and microhemorrhages are also noticed (62, 144) (Table 2).
As discussed previously, the cranial nerves might also be susceptible to a direct or indirect injury caused by SARS-CoV-2. According to a recent study, about 86 and 88% of patients with COVID-19 develop olfactory and gustatory alterations, respectively (138). These findings might be specific for SARS-CoV-2 infection and be useful to distinguish them from other causes. Interestingly, the presence of these dysfunctions can precede the onset of respiratory symptoms (160, 161) and may predict a mild clinical course of the disease (162) (Table 2). Besides, it has been proposed that pericytes of the olfactory bulb, which express high levels of the ACE2 receptor, may be responsible for triggering the cytokine storm and thus causing olfactory disorders in COVID-19 patients (163).
On the other hand, neuroimaging data could help us understand the pathological effects of SARS-CoV-2 in the CNS and PNS. Unfortunately, published brain imaging findings from confirmed COVID-19 patients are currently scarce and limited to small case series. However, it has been possible to correlate them with the potential pathophysiological mechanisms involved. For example, in a recent study, it was shown that patients with COVID-19 presented multifocal petechial hemorrhages associated with BBB rupture (164). In another study, microhemorrhages and macrohemorrhages were associated with posterior reversible encephalopathy syndrome (165) (Table 2). Although the underlying mechanism of brain abnormalities detected through neuroimaging remains to be understood, these findings provide further evidence that CNS damage can occur in COVID-19 patients. The correct understanding of pathophysiological mechanisms of neurological manifestations may reveal potential therapeutic targets (Table 3). Depending on the neurological complications associated with COVID-19, treatments would need to be adjusted accordingly (Table 2).

Table 3. Current drugs used against COVID-19.
Neurohistopathological Findings by COVID-19
The histopathological analysis of nervous tissue of patients who presented neurological complications and died due to COVID-19 is undoubtedly precious to our understanding of the pathophysiology and potential therapeutic strategies (Table 4).
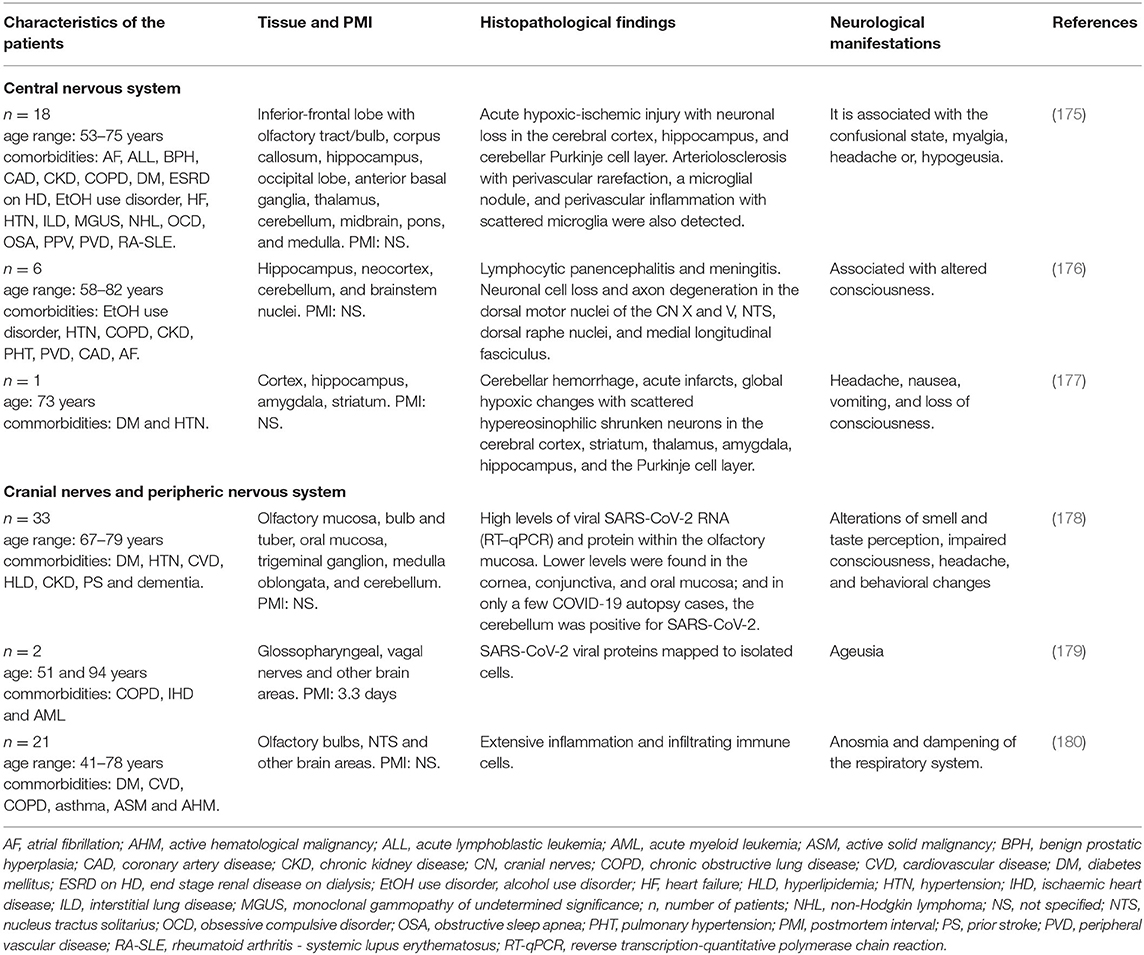
Table 4. Neurohistopathological findings in patients infected with SARS-CoV-2 and their association with neurological manifestations.
Solomon et al. analyzed nervous tissue from 18 patients infected with SARS-CoV-2 who had also presented with certain comorbidities such as DM, hypertension, cardiovascular disease, hyperlipidemia, chronic kidney disease, and dementia. The histological examination (Table 4) revealed a greater number of copies of SARS-CoV-2, acute hypoxic-ischemic injury, neuronal loss, and perivascular inflammation in several brain areas, and even pathological features of Alzheimer's disease (AD) were observed (175). A different case report described the brain from a 73-year-old man with unspecified neurological manifestations (Table 4), hypertension and DM and positive for SARS-CoV-2 with cranial computed tomography. The results showed right cerebellar intra-parenchymal hemorrhage, edema, medulla compression, and tonsillar herniation. After 18 h without improvement, the patient died of palliative extubation. Brain histopathology revealed severe global hypoxic changes with scattered hypereosinophilic shrunken neurons in several brain areas and mild perivascular inflammatory infiltrates (Table 4) (177). This study also suggested a preference of the virus to the cerebellar Purkinje cell layer. In addition, astrogliosis was noted in the superior frontal and orbital cortices, while microglial activation in the cortex was not evident (177).
A study focused on describing the SARS-CoV-2 tropism within the olfactory mucosa to the CNS examined autopsy material from 33 patients positive for the virus. The authors showed viral RNA for SARS-CoV-2 within the olfactory mucosa sampled directly beneath the cribiform plate. They also found viral RNA in anatomically distinct regions such as cornea, conjunctiva and oral mucosa. Using immunohistochemistry, in situ hybridization, and electron microscopy, they suggested that SARS-CoV-2 neuroinvasion to the CNS occurs via axonal transport, thus explaining the well-documented neurological symptoms (Table 4) (178). The authors also proposed that SARS-CoV-2 infection in the cerebellar region may occur by the migration of the virus-carrying leukocytes across the BBB, without directly connecting this area to the olfactory mucosa.
Other cranial nerves have been considered as the route for entrance of the virus to the CNS (Figure 3) (181, 182). Regarding ageusia, the pathogenesis may involve an alteration in the glossopharyngeal, facial, vagus nerve, or the nucleus tractus solitarii (NTS), at the brainstem level (183). In an immunohistochemistry analysis of the cranial nerves from two individuals, SARS-CoV-2 and viral proteins were found within the medulla oblongata and in both glossopharyngeal and vagal nerves from the lower brainstem (179), suggesting these areas as a potential route for virus entry into the CNS and peripheral tissue (Table 4).
Other authors found hypereosinophilia or nuclear and cytoplasmic condensation of neurons in the cerebrum and cerebellum of severe COVID-19 patients due to hypoxic brain changes (Table 4). The olfactory bulb histopathological analysis showed many activated microglia with enlarged bodies and T-cell extravasation into the parenchyma and elevated levels of reactive astrocytes (180). Interestingly, the NTS showed astrogliosis and a massive microglial activation with the formation of a microglia nodule and T cells in the leptomeninges of the medulla oblongata (180). This implies extensive inflammation in this area, which results in a dysregulation of the respiratory system (184). One of the main limitations of these studies is that it is not clear whether the histopathological findings are the result of patients' comorbidities/aging, or due to SARS-CoV2 neuro-infection. Further investigations are necessary to make the corresponding comparison with healthy subjects with appropriate age ranges and to understand the neuropathological mechanism of SARS-CoV-2 and its relationship with neurological manifestations and patient comorbidities.
The Potential Role of SARS-CoV-2 in the Pathogenesis of Neurodegenerative Diseases
The SARS-CoV-2 neurotropism has already been documented in several reports (Table 2) (185–187). However, it remains unknown whether SARS-CoV-2 contributes to neurodegenerative pathogenesis. It has been hypothesized that viruses can cause neurological problems by affecting neurotransmitter release, lysing the cells, inducing apoptosis, commanding neuronal transcriptional pathways or indirectly activating the immune response (188).
Neuroinvasive animal CoVs, such as the porcine hemagglutinating encephalitis virus (PHEV) or MHV have been shown to induce different types of neuropathology. Similarly, human CoVs, such as HCoV-229E and HCoV-OC43, have been implicated in establishing or exacerbating neurodegenerative diseases (189–192).
AD is the most common cause of dementia among the elderly. Neuropathological hallmarks of AD include the neurofibrillary tangles, consisting of intraneuronal and hyperphosphorylated tau, and the extracellular accumulation of amyloid β-peptide (Aβ) in the brain parenchyma in the form of neuritic plaques (193, 194). The neurovascular unit (NVU) and BBB dysregulation are also critical pathophysiological events in neurodegenerative diseases, including AD. Previous studies have suggested a relationship between AD and infectious agents. Chlamydia pneumoniae, Helicobacter pylori, Borrelia burgdorferi, and herpes simplex virus have been reported in post-mortem AD brain (195) and a relationship between virus infection and Aβ has been suggested. Soscia et al. noticed that Aβ exerted antimicrobial activity against relevant microorganisms and was modulated in response to some environmental stressors. Thus, transient viral infection could initiate or accelerate Aβ accumulation in the brain and neuronal damage (Figure 2B steps 5 and 6) (196). ACE2 can induce an increase of nitric oxide (NO) in the brain, which becomes neurotoxic. NO and other reactive species, which could be produced as a consequence of viral internalization and impairment of cell organelles (mitochondria, lysosomes), could, in turn, increase misfolding and aggregation of cellular proteins (197).
It has been proposed that SARS-CoV-2 could increase the hyperphosphorylation of tau in the axonal region (52, 198–200), promoting disassembly of microtubules and, subsequently, neuronal degeneration (Figure 2B steps 5 and 6) (201). Likewise, it has been suggested that persistent CoV infections can induce a neuroimmune response and a pro-inflammatory state, and activate glial cells (202). Microglial cells may be chronically activated by a single stimulus, such as pathogen infection, resulting in slow and progressive neuronal loss through multiple neurotoxic factors (203). Interferon (IFN), which has a role in mediating AD pathology, directly activates microglia and stimulates a pro-inflammatory response derived from SARS-CoV-2 infection (204). Therefore, it seems that the neurotropism of SARS-CoV-2 can lead to the activation of microglial cells, trigger chronic neuroinflammation, and finally, neurodegeneration (Figure 2B).
Finally, it has been found that ACE2 is upregulated in the cerebral vasculature of dementia cases. ACE2 increases the intracellular level of angiotensin 2, causing vasoconstriction and promoting brain degeneration (205). Buzhdygan et al. demonstrated that S1 could promote BBB alteration in an advanced 3D microfluidic model of the BBB, being able to induce different types of neuropathology (98).
Like AD, viral agents have been associated with parkinsonism disorders. These include the post-encephalitic parkinsonism linked to the 1918 influenza A H1N1 pandemic (206) and the parkinsonism associated with Epstein Barr, Coxsackie, West Nile, herpes and the human immunodeficiency (HIV) viruses (207–209). Parkinson's disease (PD) is the second most common and fastest-growing neurodegenerative disorder (210). It is characterized by dopaminergic neuronal loss in the substantia nigra pars compacta and the accumulation of misfolded α-synuclein (α-syn), which is found as intracytoplasmic inclusions called “Lewy bodies” (211). SARS-CoV-2 could play a role in the epidemiology of PD, since pro-inflammatory events triggered by viral infections could act as predisposing factors to the development of PD (Figure 2B steps 5 and 6) (Table 2) (212–214). This inflammatory environment can trigger the long-term neuronal loss, misfolding, aggregation, and spread of α-syn through the CNS (215, 216). Interestingly, it has been proposed that α-syn plays an essential role in response to infection, promoting a higher expression of α-syn, as occurs in the West Nile virus encephalitis (217, 218). Furthermore, α-syn aggregation can activate microglia, favoring the pro-inflammatory response and cellular damage signals, leading to slow and progressive neuronal death (219). However, it remains unknown whether SARS-CoV-2 could contribute to neurodegenerative pathogenesis, or whether it only uses the CNS as a reservoir, making it difficult for the virus to replicate, due to the low level of ACE2 receptors expressed in CNS (220).
Conclusion
COVID-19 pandemic has become a real challenge for the scientific community around the world. Although SARS-CoV-2 mainly affects the respiratory tract, more evidence suggests that this virus can also invade the CNS causing neurological manifestations. The possible routes of SARS-CoV-2 neuroinvasion include: (1) the hematopoietic pathway via the BBB, (2) via the B-CSF, (3) via retrograde axonal transport through the cranial nerves, and (4) via the circumventricular organs. Once the virus enters the CNS, it binds to cell receptors, including ACE2. This receptor is expressed in several brain areas and in both neuronal and non-neuronal cell types. The binding of SARS-CoV-2 with ACE2 can promote neuroinflammation, hypercoagulation, microhemorrhages, BBB dysfunction, generation of reactive species, phosphorylation of tau, protein misfolding and aggregation, and neuronal death, features that are closely related to the appearance or progression of neurodegenerative diseases. Further studies on the molecular changes in the brain triggered by SARS-CoV-2 infection would facilitate timely diagnosis and therapeutic approaches.
Author Contributions
MP-H, JL-M, and LS-R contributed to the idea formulation, reviewing of the literature, and writing and revision of the manuscript. YF-M, CH, MV-R, AL-A, PM-G, BC-C, VB-A, RA-P, CC-T, and JD-G contributed to reviewing of the literature and revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Fondo Nacional de Ciencia, Tecnologia, FONDOCyT, from the Ministry of Higher Education, Science and Technology, Dominican Republic (2015-3A2-127 to MP-H and 2018-2019-2A3-208 to JL-M and MP-H).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to express our gratitude to the Union Medical University Clinic, Dominican Republic, for their support and collaboration in the development of this research project. We also want to express our gratitude to the Mexican families who have donated the brain of their loved ones affected with Alzheimer's disease and made our research possible. This work is dedicated to the memory of Professor Dr. José Raúl Mena López†.
Footnotes
† ^Deceased.
References
1. Allam Z. “Chapter 1 - The First 50 days of COVID-19: A Detailed Chronological Timeline and Extensive Review of Literature Documenting the Pandemic” In: Surveying the Covid-19 Pandemic and its Implications. 1–7 doi: 10.1016/B978-0-12-824313-8.00001-2
2. World Health Organization. Coronavirus Disease (COVID-19). Situation Report. Available online at: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200730-covid-19-sitrep-192.pdf?sfvrsn=5e52901f_8
3. Zhang W, Du RH, Li B, Zheng XS, Yang XL, Hu B, et al. Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerg Microbes Infect. (2020) 9:386–9. doi: 10.1080/22221751.2020.1729071
4. Organization WH. COVID-19. (2021). Available online at: https://covid19.who.int/ (accessed 22 February, 2021).
5. Wu Y-C, Chen C-S, Chan Y-J. The outbreak of COVID-19: an overview. J Chinese Medical Assoc. (2020) 83:217–20. doi: 10.1097/JCMA.0000000000000270
6. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. (2020) 382:1708–20. doi: 10.1101/2020.02.06.20020974
7. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
8. Kanne JP, Little BP, Chung JH, Elicker BM, Ketai LH. Essentials for radiologists on COVID-19: an update-radiology scientific expert panel. Radiology. (2020) 296:E113–4. doi: 10.1148/radiol.2020200527
9. Castelli V, Cimini A, Ferri C. Cytokine storm in COVID-19: “when you come out of the storm, you won't be the same person who walked in.” Front Immunol. (2020) 11:2132. doi: 10.3389/fimmu.2020.02132
10. Jin JM, Bai P, He W, Wu F, Liu XF, Han DM, et al. Gender differences in patients with COVID-19: focus on severity and mortality. Front Public Health. (2020) 8:152. doi: 10.3389/fpubh.2020.00152
11. Guan WJ, Liang WH, Zhao Y, Liang HR, Chen ZS, Li YM, et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: a nationwide analysis. Eur Respir J. (2020) 55:2020. doi: 10.1183/13993003.00547-2020
12. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
13. Guadarrama-Ortiz P, Choreno-Parra JA, Sanchez-Martinez CM, Pacheco-Sanchez FJ, Rodriguez-Nava AI, Garcia-Quintero G. Neurological aspects of SARS-CoV-2 infection: mechanisms and manifestations. Front Neurol. (2020) 11:1039. doi: 10.3389/fneur.2020.01039
14. Puccioni-Sohler M, Poton AR, Franklin M, Silva SJD, Brindeiro R, Tanuri A. Current evidence of neurological features, diagnosis, and neuropathogenesis associated with COVID-19. Rev Soc Bras Med Trop. (2020) 53:e20200477. doi: 10.1590/0037-8682-0477-2020
15. Baig AM. Neurological manifestations in COVID-19 caused by SARS-CoV-2. CNS Neurosci Ther. (2020) 26:499–501. doi: 10.1111/cns.13372
16. Conti P, Ronconi G, Caraffa A, Gallenga CE, Ross R, Frydas I, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. (2020) 34:327–31. doi: 10.23812/CONTI-E
17. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. (2020) 11:827. doi: 10.3389/fimmu.2020.00827
18. Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, et al. Neurological associations of COVID-19. Lancet Neurol. (2020) 19:767–83. doi: 10.1016/S1474-4422(20)30221-0
19. Coronaviridae Study Group of the International Committee on Taxonomy of V. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. (2020) 5:536–44. doi: 10.1038/s41564-020-0695-z
20. Ludwig S, Zarbock A. Coronaviruses and SARS-CoV-2: a brief overview. Anesth Analg. (2020) 131:93–6. doi: 10.1213/ANE.0000000000004845
21. Woo PC, Lau SK, Huang Y, Yuen KY. Coronavirus diversity, phylogeny and interspecies jumping. Exp Biol Med. (2009) 234:1117–27. doi: 10.3181/0903-MR-94
22. Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. (2016) 3:237–61. doi: 10.1146/annurev-virology-110615-042301
23. Huang Y, Yang C, Xu XF, Xu W, Liu SW. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. (2020) 41:1141–9. doi: 10.1038/s41401-020-0485-4
24. Sigrist CJ, Bridge A, Le Mercier P. A potential role for integrins in host cell entry by SARS-CoV-2. Antiviral Res. (2020) 177:104759. doi: 10.1016/j.antiviral.2020.104759
25. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. (2003) 426:450–4. doi: 10.1038/nature02145
26. Lam SD, Bordin N, Waman VP, Scholes HM, Ashford P, Sen N, et al. SARS-CoV-2 spike protein predicted to form complexes with host receptor protein orthologues from a broad range of mammals. Sci Rep. (2020) 10:16471. doi: 10.1038/s41598-020-71936-5
27. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. (2014) 88:1293–307. doi: 10.1128/JVI.02202-13
28. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80 e278. doi: 10.1016/j.cell.2020.02.052
29. Lim YX, Ng YL, Tam JP, Liu DX. Human coronaviruses: a review of virus-host interactions. Diseases. (2016) 4:26. doi: 10.3390/diseases4030026
30. Satarker S, Nampoothiri M. Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch Med Res. (2020) 51:482–91. doi: 10.1016/j.arcmed.2020.05.012
31. Wong CK, Lam CW, Wu AK, Ip WK, Lee NL, Chan IH, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. (2004) 136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x
32. Fu Y, Cheng Y, Wu Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol Sin. (2020) 35:266–71. doi: 10.1007/s12250-020-00207-4
33. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. (2020) 76:14–20. doi: 10.1016/j.ejim.2020.04.037
34. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
35. Fu B, Xu X, Wei H. Why tocilizumab could be an effective treatment for severe COVID-19? J Transl Med. (2020) 18:164. doi: 10.1186/s12967-020-02339-3
36. Fung SY, Yuen KS, Ye ZW, Chan CP, Jin DY. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microbes Infect. (2020) 9:558–70. doi: 10.1080/22221751.2020.1736644
37. Nieto-Torres JL, Dediego ML, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. (2014) 10:e1004077. doi: 10.1371/journal.ppat.1004077
38. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. (2020) 395:507–13. doi: 10.1016/S0140-6736(20)30211-7
39. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. (2020) 63:364–74. doi: 10.1007/s11427-020-1643-8
40. Tay MZ, Poh CM, Renia L, Macary PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. (2020) 20:363–74. doi: 10.1038/s41577-020-0311-8
41. Iwasaki A, Yang Y. The potential danger of suboptimal antibody responses in COVID-19. Nat Rev Immunol. (2020) 20:339–41. doi: 10.1038/s41577-020-0321-6
42. Wang SF, Tseng SP, Yen CH, Yang JY, Tsao CH, Shen CW, et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem Biophys Res Commun. (2014) 451:208–14. doi: 10.1016/j.bbrc.2014.07.090
43. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med. (2017) 377:562–72. doi: 10.1056/NEJMra1608077
44. Mahmudpour M, Roozbeh J, Keshavarz M, Farrokhi S, Nabipour I. COVID-19 cytokine storm: the anger of inflammation. Cytokine. (2020) 133:155151. doi: 10.1016/j.cyto.2020.155151
45. Sanclemente-Alaman I, Moreno-Jimenez L, Benito-Martin MS, Canales-Aguirre A, Matias-Guiu JA, Matias-Guiu J, et al. Experimental models for the study of central nervous system infection by SARS-CoV-2. Front Immunol. (2020) 11:2163. doi: 10.3389/fimmu.2020.02163
46. Gruslin E, Moisan S, St-Pierre Y, Desforges M, Talbot PJ. Transcriptome profile within the mouse central nervous system and activation of myelin-reactive T cells following murine coronavirus infection. J Neuroimmunol. (2005) 162:60–70. doi: 10.1016/j.jneuroim.2005.01.007
47. Dandekar AA, Perlman S. Virus-induced demyelination in nude mice is mediated by gamma delta T cells. Am J Pathol. (2002) 161:1255–63. doi: 10.1016/S0002-9440(10)64402-1
48. Choi KS, Aizaki H, Lai MM. Murine coronavirus requires lipid rafts for virus entry and cell-cell fusion but not for virus release. J Virol. (2005) 79:9862–71. doi: 10.1128/JVI.79.15.9862-9871.2005
49. Singh M, Khan RS, Dine K, Das Sarma J, Shindler KS. Intracranial inoculation is more potent than intranasal inoculation for inducing optic neuritis in the mouse hepatitis virus-induced model of multiple sclerosis. Front Cell Infect Microbiol. (2018) 8:311. doi: 10.3389/fcimb.2018.00311
50. Iadecola C, Anrather J, Kamel H. Effects of COVID-19 on the nervous system. Cell. (2020) 183:16–27 e11. doi: 10.1016/j.cell.2020.08.028
51. Buzhdygan TP, Deore BJ, Baldwin-Leclair A, Mcgary H, Razmpour R, Galie PA, et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in vitro models of the human blood-brain barrier. bioRxiv. (2020). doi: 10.1101/2020.06.15.150912
52. Liu JM, Tan BH, Wu S, Gui Y, Suo JL, Li YC. Evidence of central nervous system infection and neuroinvasive routes, as well as neurological involvement, in the lethality of SARS-CoV-2 infection. J Med Virol. (2020) 93:1304–13. doi: 10.1002/jmv.26570
53. Perrin P, Collongues N, Baloglu S, Bedo D, Bassand X, Lavaux T, et al. Cytokine release syndrome-associated encephalopathy in patients with COVID-19. Eur J Neurol. (2020) 28:248–58. doi: 10.1111/ene.14491
54. Ur A, Verma K. Cytokine storm in COVID19: a neural hypothesis. ACS Chem Neurosci. (2020) 11:1868–70. doi: 10.1021/acschemneuro.0c00346
55. Pena-Silva RA, Faraci FM, Heistad DD. Response to letter regarding article, “impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging.” Stroke. (2013) 44:e35. doi: 10.1161/STROKEAHA.111.000481
56. Maclean MA, Kamintsky L, Leck ED, Friedman A. The potential role of microvascular pathology in the neurological manifestations of coronavirus infection. Fluids Barriers CNS. (2020) 17:55. doi: 10.1186/s12987-020-00216-1
57. Brinker T, Stopa E, Morrison J, Klinge P. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS. (2014) 11:10. doi: 10.1186/2045-8118-11-10
58. Desforges M, Le Coupanec A, Stodola JK, Meessen-Pinard M, Talbot PJ. Human coronaviruses: viral and cellular factors involved in neuroinvasiveness and neuropathogenesis. Virus Res. (2014) 194:145–58. doi: 10.1016/j.virusres.2014.09.011
59. Fenrich M, Mrdenovic S, Balog M, Tomic S, Zjalic M, Roncevic A, et al. SARS-CoV-2 dissemination through peripheral nerves explains multiple organ injury. Front Cell Neurosci. (2020) 14:229. doi: 10.3389/fncel.2020.00229
60. Baig AM, Sanders EC. Potential neuroinvasive pathways of SARS-CoV-2: deciphering the spectrum of neurological deficit seen in coronavirus disease-2019 (COVID-19). J Med Virol. (2020) 92:1845–57. doi: 10.1002/jmv.26105
61. Li CW, Syue LS, Tsai YS, Li MC, Lo CL, Tsai CS, et al. Anosmia and olfactory tract neuropathy in a case of COVID-19. J Microbiol Immunol Infect. (2020) 54:93–6. doi: 10.1016/j.jmii.2020.05.017
62. Lin E, Lantos JE, Strauss SB, Phillips CD, Campion TR Jr, Navi BB, et al. Brain imaging of patients with COVID-19: findings at an academic institution during the height of the outbreak in New York City. AJNR Am J Neuroradiol. (2020) 41:2001–8. doi: 10.3174/ajnr.A6793
63. Lu S, Wei N, Jiang J, Wu L, Sheng J, Zhou J, et al. First report of manic-like symptoms in a COVID-19 patient with no previous history of a psychiatric disorder. J Affect Disord. (2020) 277:337–40. doi: 10.1016/j.jad.2020.08.031
64. Zubair AS, Mcalpine LS, Gardin T, Farhadian S, Kuruvilla DE, Spudich S. Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: a review. JAMA Neurol. (2020) 77:1018–27. doi: 10.1001/jamaneurol.2020.2065
65. Gosztonyi G. Propagation of viruses along neuron networks by transsynaptic passage–a contribution to the pathogenesis of rabies. Tierarztl Prax. (1986) 14:199–204.
66. Li YC, Bai WZ, Hirano N, Hayashida T, Taniguchi T, Sugita Y, et al. Neurotropic virus tracing suggests a membranous-coating-mediated mechanism for transsynaptic communication. J Comp Neurol. (2013) 521:203–12. doi: 10.1002/cne.23171
67. Li YC, Bai WZ, Hashikawa T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol. (2020) 92:552–5. doi: 10.1002/jmv.25728
68. Taylor MP, Enquist LW. Axonal spread of neuroinvasive viral infections. Trends Microbiol. (2015) 23:283–8. doi: 10.1016/j.tim.2015.01.002
69. Tremblay M-E, Madore C, Bordeleau M, Tian L, Verkhratsky A. Neuropathobiology of COVID-19: the role for Glia. Front Cell Neurosci. (2020) 14:592214. doi: 10.3389/fncel.2020.592214
70. Sun SH, Chen Q, Gu HJ, Yang G, Wang YX, Huang XY, et al. A mouse model of SARS-CoV-2 infection and pathogenesis. Cell Host Microbe. (2020) 28:124–33 e124. doi: 10.1016/j.chom.2020.05.020
71. Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. (2007) 292:R373–381. doi: 10.1152/ajpregu.00292.2006
72. Kerslake R, Hall M, Randeva HS, Spandidos DA, Chatha K, Kyrou I, et al. Coexpression of peripheral olfactory receptors with SARSCoV2 infection mediators: potential implications beyond loss of smell as a COVID19 symptom. Int J Mol Med. (2020) 46:949–56. doi: 10.3892/ijmm.2020.4646
73. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, Van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. (2004) 203:631–7. doi: 10.1002/path.1570
74. Vazana U, Veksler R, Pell GS, Prager O, Fassler M, Chassidim Y, et al. Glutamate-mediated blood-brain barrier opening: implications for neuroprotection and drug delivery. J Neurosci. (2016) 36:7727–39. doi: 10.1523/JNEUROSCI.0587-16.2016
75. Merkler AE, Parikh NS, Mir S, Gupta A, Kamel H, Lin E, et al. Risk of ischemic stroke in patients with covid-19 versus patients with influenza. MedRxiv. (2020). doi: 10.1101/2020.05.18.20105494
76. Bilinska K, Jakubowska P, Von Bartheld CS, Butowt R. Expression of the SARS-CoV-2 entry proteins, ACE2 and TMPRSS2, in cells of the olfactory epithelium: identification of cell types and trends with age. ACS Chem Neurosci. (2020) 11:1555–62. doi: 10.1021/acschemneuro.0c00210
77. Mollica V, Rizzo A, Massari F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol. (2020) 16:2029–33. doi: 10.2217/fon-2020-0571
78. Qiao J, Li W, Bao J, Peng Q, Wen D, Wang J, et al. The expression of SARS-CoV-2 receptor ACE2 and CD147, and protease TMPRSS2 in human and mouse brain cells and mouse brain tissues. Biochem Biophys Res Commun. (2020) 533:867–71. doi: 10.1016/j.bbrc.2020.09.042
79. Mentzel S, Dijkman HB, Van Son JP, Koene RA, Assmann KJ. Organ distribution of aminopeptidase A and dipeptidyl peptidase IV in normal mice. J Histochem Cytochem. (1996) 44:445–61. doi: 10.1177/44.5.8627002
80. Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev. (2014) 35:992–1019. doi: 10.1210/er.2014-1035
81. Meyerholz DK, Lambertz AM, Mccray PB Jr. Dipeptidyl peptidase 4 distribution in the human respiratory tract: implications for the middle east respiratory syndrome. Am J Pathol. (2016) 186:78–86. doi: 10.1016/j.ajpath.2015.09.014
82. Li L, Acioglu C, Heary RF, Elkabes S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav Immun. (2020) 91:740–55. doi: 10.1016/j.bbi.2020.10.007
83. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. (2020) 92:2105–13. doi: 10.1002/jmv.25987
84. Singh KD, Karnik SS. Angiotensin receptors: structure, function, signaling and clinical applications. J Cell Signal. 1:111. doi: 10.4172/jcs.1000111
85. Krasniqi S, Daci A. Role of the angiotensin pathway and its target therapy in epilepsy management. Int J Mol Sci. (2019) 20:30726. doi: 10.3390/ijms20030726
86. Gurwitz D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev Res. (2020) 81:537–40. doi: 10.1002/ddr.21656
87. D'ardes D, Boccatonda A, Rossi I, Guagnano MT, Santilli F, Cipollone F, et al. COVID-19 and RAS: unravelling an unclear relationship. Int J Mol Sci. (2020) 21:83003. doi: 10.3390/ijms21083003
88. Maginnis MS. Virus-Receptor interactions: the key to cellular invasion. J Mol Biol. (2018) 430:2590–611. doi: 10.1016/j.jmb.2018.06.024
89. Yan S, Sun H, Bu X, Wan G. New strategy for COVID-19: an evolutionary role for RGD motif in SARS-CoV-2 and potential inhibitors for virus infection. Front Pharmacol. (2020) 11:912. doi: 10.3389/fphar.2020.00912
90. Stoka V, Turk V, Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev. (2016) 32:22–37. doi: 10.1016/j.arr.2016.04.010
91. Pezzini A, Padovani A. Lifting the mask on neurological manifestations of COVID-19. Nat Rev Neurol. (2020) 16:636–44. doi: 10.1038/s41582-020-0398-3
92. Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, et al. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia. (2015) 63:2260–73. doi: 10.1002/glia.22891
93. Voet S, Srinivasan S, Lamkanfi M, Van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. (2019) 11:10248. doi: 10.15252/emmm.201810248
94. Deora V, Lee JD, Albornoz EA, Mcalary L, Jagaraj CJ, Robertson A, et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia. (2020) 68:407–21. doi: 10.1002/glia.23728
95. Chen R, Wang K, Yu J, Howard D, French L, Chen Z, et al. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front Neurol. (2020) 11:573095. doi: 10.3389/fneur.2020.573095
96. Hikmet F, Mear L, Edvinsson A, Micke P, Uhlen M, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol Syst Biol. (2020) 16:e9610. doi: 10.15252/msb.20209610
97. Lovren F, Pan Y, Quan A, Teoh H, Wang G, Shukla PC, et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am J Physiol Heart Circ Physiol. (2008) 295:H1377–84. doi: 10.1152/ajpheart.00331.2008
98. Buzhdygan TP, Deore BJ, Baldwin-Leclair A, Bullock TA, Mcgary HM, Khan JA, et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol Dis. (2020) 146:105131. doi: 10.1016/j.nbd.2020.105131
99. Vargas G, Medeiros Geraldo LH, Gedeao Salomao N, Viana Paes M, Regina Souza Lima F, Carvalho Alcantara Gomes F. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and glial cells: insights and perspectives. Brain Behav Immun Health. (2020) 7:100127. doi: 10.1016/j.bbih.2020.100127
100. Jha NK, Ojha S, Jha SK, Dureja H, Singh SK, Shukla SD, et al. Evidence of coronavirus (CoV) pathogenesis and emerging pathogen SARS-CoV-2 in the nervous system: a review on neurological impairments and manifestations. J Mol Neurosci. (2021) 19:1–8. doi: 10.1007/s12031-020-01767-6
101. Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. (2020) 116:1097–100. doi: 10.1093/cvr/cvaa078
102. Ludlow M, Kortekaas J, Herden C, Hoffmann B, Tappe D, Trebst C, et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. (2016) 131:159–84. doi: 10.1007/s00401-015-1511-3
103. Sporns O. Structure and function of complex brain networks. Dialogues Clin Neurosci. (2013) 15:247–62. doi: 10.31887/DCNS.2013.15.3/osporns
104. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. (2017) 39:529–39. doi: 10.1007/s00281-017-0629-x
105. Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. (2020) 20:355–62. doi: 10.1038/s41577-020-0331-4
106. Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents. (2020) 55:105954. doi: 10.1016/j.ijantimicag.2020.105954
107. Mokhtari T, Hassani F, Ghaffari N, Ebrahimi B, Yarahmadi A, Hassanzadeh G. COVID-19 and multiorgan failure: a narrative review on potential mechanisms. J Mol Histol. (2020) 51:613–28. doi: 10.1007/s10735-020-09915-3
108. Bhaskar S, Sinha A, Banach M, Mittoo S, Weissert R, Kass JS, et al. Cytokine storm in COVID-19-immunopathological mechanisms, clinical considerations, and therapeutic approaches: the REPROGRAM consortium position paper. Front Immunol. (2020) 11:1648. doi: 10.3389/fimmu.2020.01648
109. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
110. Fotuhi M, Mian A, Meysami S, Raji CA. Neurobiology of COVID-19. J Alzheimers Dis. (2020) 76:3–19. doi: 10.3233/JAD-200581
111. Xiong M, Liang X, Wei YD. Changes in blood coagulation in patients with severe coronavirus disease 2019 (COVID-19): a meta-analysis. Br J Haematol. (2020) 189:1050–2. doi: 10.1111/bjh.16725
112. Najjar S, Najjar A, Chong DJ, Pramanik BK, Kirsch C, Kuzniecky RI, et al. Central nervous system complications associated with SARS-CoV-2 infection: integrative concepts of pathophysiology and case reports. J Neuroinflammation. (2020) 17:231. doi: 10.1186/s12974-020-01896-0
113. Larochelle C, Alvarez JI, Prat A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. (2011) 585:3770–80. doi: 10.1016/j.febslet.2011.04.066
114. Najjar S, Pahlajani S, De Sanctis V, Stern JNH, Najjar A, Chong D. Neurovascular unit dysfunction and blood-brain barrier hyperpermeability contribute to schizophrenia neurobiology: a theoretical integration of clinical and experimental evidence. Front Psychiatry. (2017) 8:83. doi: 10.3389/fpsyt.2017.00083
115. Dantzer R. Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol Rev. (2018) 98:477–504. doi: 10.1152/physrev.00039.2016
116. Ottaviani D, Boso F, Tranquillini E, Gapeni I, Pedrotti G, Cozzio S, et al. Early Guillain-Barre syndrome in coronavirus disease 2019 (COVID-19): a case report from an Italian COVID-hospital. Neurol Sci. (2020) 41:1351–4. doi: 10.1007/s10072-020-04449-8
117. Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B. COVID-19-associated acute hemorrhagic necrotizing encephalopathy: imaging features. Radiology. (2020) 296:E119–20. doi: 10.1148/radiol.2020201187
118. Al Saiegh F, Ghosh R, Leibold A, Avery MB, Schmidt RF, Theofanis T, et al. Status of SARS-CoV-2 in cerebrospinal fluid of patients with COVID-19 and stroke. J Neurol Neurosurg Psychiatry. (2020) 91:846–8. doi: 10.1136/jnnp-2020-323522
119. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
120. Potokar M, Jorgacevski J, Zorec R. Astrocytes in flavivirus infections. Int J Mol Sci. (2019) 20:30691. doi: 10.3390/ijms20030691
121. Madan V, Garcia Mde J, Sanz MA, Carrasco L. Viroporin activity of murine hepatitis virus E protein. FEBS Lett. (2005) 579:3607–12. doi: 10.1016/j.febslet.2005.05.046
122. Achar A, Ghosh C. COVID-19-associated neurological disorders: the potential route of CNS invasion and blood-brain relevance. Cells. (2020) 9:112360. doi: 10.3390/cells9112360
123. Guo HC, Jin Y, Zhi XY, Yan D, Sun SQ. NLRP3 inflammasome activation by viroporins of animal viruses. Viruses. (2015) 7:3380–91. doi: 10.3390/v7072777
124. Farag NS, Breitinger U, Breitinger HG, El Azizi MA. Viroporins and inflammasomes: a key to understand virus-induced inflammation. Int J Biochem Cell Biol. (2020) 122:105738. doi: 10.1016/j.biocel.2020.105738
125. Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. (2010) 11:404–10. doi: 10.1038/ni.1861
126. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. (2017) 27:673–84. doi: 10.1016/j.tcb.2017.05.005
127. Lieberman J, Wu H, Kagan JC. Gasdermin D activity in inflammation and host defense. Sci Immunol. (2019) 4:aav1447. doi: 10.1126/sciimmunol.aav1447
128. Munhoz RP, Pedroso JL, Nascimento FA, Almeida SM, Barsottini OGP, Cardoso FEC, et al. Neurological complications in patients with SARS-CoV-2 infection: a systematic review. Arq Neuropsiquiatr. (2020) 78:290–300. doi: 10.1590/0004-282x20200051
129. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. (2020) 77:683–90. doi: 10.1001/jamaneurol.2020.1127
130. Pugin D, Vargas MI, Thieffry C, Schibler M, Grosgurin O, Pugin J, et al. COVID-19-related encephalopathy responsive to high-dose glucocorticoids. Neurology. (2020) 95:543–6. doi: 10.1212/WNL.0000000000010354
131. Arca KN, Starling AJ. Treatment-refractory headache in the setting of COVID-19 pneumonia: migraine or meningoencephalitis? case report. SN Compr Clin Med. (2020) 369:1–4. doi: 10.1007/s42399-020-00369-y
132. Toptan T, Aktan C, Basari A, Bolay H. Case series of headache characteristics in COVID-19: headache can be an isolated symptom. Headache. (2020) 60:1788–92. doi: 10.1111/head.13940
133. Uygun O, Ertas M, Ekizoglu E, Bolay H, Ozge A, Kocasoy Orhan E, et al. Headache characteristics in COVID-19 pandemic-a survey study. J Headache Pain. (2020) 21:121. doi: 10.1186/s10194-020-01188-1
134. Liotta EM, Batra A, Clark JR, Shlobin NA, Hoffman SC, Orban ZS, et al. Frequent neurologic manifestations and encephalopathy-associated morbidity in Covid-19 patients. Ann Clin Transl Neurol. (2020) 7:2221–30. doi: 10.1002/acn3.51210
135. Beach SR, Praschan NC, Hogan C, Dotson S, Merideth F, Kontos N, et al. Delirium in COVID-19: a case series and exploration of potential mechanisms for central nervous system involvement. Gen Hosp Psychiatry. (2020) 65:47–53. doi: 10.1016/j.genhosppsych.2020.05.008
136. Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. (2020) 382:2268–70. doi: 10.1056/NEJMc2008597
137. Tan YK, Goh C, Leow AST, Tambyah PA, Ang A, Yap ES, et al. COVID-19 and ischemic stroke: a systematic review and meta-summary of the literature. J Thromb Thrombolysis. (2020) 50:587–95. doi: 10.1007/s11239-020-02228-y
138. Lechien JR, Chiesa-Estomba CM, De Siati DR, Horoi M, Le Bon SD, Rodriguez A, et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study. Eur Arch Otorhinolaryngol. (2020) 277:2251–61. doi: 10.1007/s00405-020-05965-1
139. Aragao M, Leal MC, Cartaxo Filho OQ, Fonseca TM, Valenca MM. Anosmia in COVID-19 associated with injury to the olfactory bulbs evident on MRI. AJNR Am J Neuroradiol. (2020) 41:1703–6. doi: 10.3174/ajnr.A6675
140. Gori A, Leone F, Loffredo L, Cinicola BL, Brindisi G, De Castro G, et al. COVID-19-related anosmia: the olfactory pathway hypothesis and early intervention. Front Neurol. (2020) 11:956. doi: 10.3389/fneur.2020.00956
141. Meng X, Deng Y, Dai Z, Meng Z. COVID-19 and anosmia: a review based on up-to-date knowledge. Am J Otolaryngol. (2020) 41:102581. doi: 10.1016/j.amjoto.2020.102581
142. Vaira LA, Salzano G, Fois AG, Piombino P, De Riu G. Potential pathogenesis of ageusia and anosmia in COVID-19 patients. Int Forum Allergy Rhinol. (2020) 10:1103–4. doi: 10.1002/alr.22593
143. Guidon AC, Amato AA. COVID-19 and neuromuscular disorders. Neurology. (2020) 94:959–69. doi: 10.1212/WNL.0000000000009566
144. Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, et al. Guillain-Barre syndrome associated with SARS-CoV-2. N Engl J Med. (2020) 382:2574–6. doi: 10.1056/NEJMc2009191
145. Abu-Rumeileh S, Abdelhak A, Foschi M, Tumani H, Otto M. Guillain-Barre syndrome spectrum associated with COVID-19: an up-to-date systematic review of 73 cases. J Neurol. (2020) 25:1–38. doi: 10.1007/s00415-020-10124-x
146. Zhao H, Shen D, Zhou H, Liu J, Chen S. Guillain-Barre syndrome associated with SARS-CoV-2 infection: causality or coincidence? Lancet Neurol. (2020) 19:383–4. doi: 10.1016/S1474-4422(20)30109-5
147. Ozdag Acarli AN, Samanci B, Ekizoglu E, Cakar A, Sirin NG, Gunduz T, et al. Coronavirus disease 2019 (COVID-19) from the point of view of neurologists: observation of neurological findings and symptoms during the combat against a pandemic. Noro Psikiyatr Ars. (2020) 57:154–9. doi: 10.29399/npa.26148
148. Benemei S, Nicoletti P, Capone JG, Geppetti P. CGRP receptors in the control of pain and inflammation. Curr Opin Pharmacol. (2009) 9:9–14. doi: 10.1016/j.coph.2008.12.007
149. Durham PL. Calcitonin gene-related peptide (CGRP) and migraine. Headache. (2006) 46(Suppl.1):S3–8. doi: 10.1111/j.1526-4610.2006.00483.x
150. Porta-Etessam J, Matias-Guiu JA, Gonzalez-Garcia N, Gomez Iglesias P, Santos-Bueso E, Arriola-Villalobos P, et al. Spectrum of headaches associated with SARS-CoV-2 infection: study of healthcare professionals. Headache. (2020) 60:1697–704. doi: 10.1111/head.13902
151. Zhao F, Han Z, Wang R, Luo Y. Neurological manifestations of COVID-19: causality or coincidence? Aging Dis. (2021) 12:27–35. doi: 10.14336/AD.2020.0917
152. Stohlman SA, Hinton DR. Viral induced demyelination. Brain Pathol. (2001) 11:92–106. doi: 10.1111/j.1750-3639.2001.tb00384.x
153. Savarin C, Bergmann CC. Viral-induced suppression of self-reactive T cells: lessons from neurotropic coronavirus-induced demyelination. J Neuroimmunol. (2017) 308:12–6. doi: 10.1016/j.jneuroim.2017.01.003
154. Dziedzic A, Saluk-Bijak J, Miller E, Niemcewicz M, Bijak M. The impact of SARS-CoV-2 infection on the development of neurodegeneration in multiple sclerosis. Int J Mol Sci. (2021) 22:1804. doi: 10.3390/ijms22041804
155. Palao M, Fernandez-Diaz E, Gracia-Gil J, Romero-Sanchez CM, Diaz-Maroto I, Segura T. Multiple sclerosis following SARS-CoV-2 infection. Mult Scler Relat Disord. (2020) 45:102377. doi: 10.1016/j.msard.2020.102377
156. Diez-Porras L, Verges E, Gil F, Vidal MJ, Massons J, Arboix A. Guillain-Barre-Strohl syndrome and COVID-19: case report and literature review. Neuromuscul Disord. (2020) 30:859–61. doi: 10.1016/j.nmd.2020.08.354
157. Paybast S, Gorji R, Mavandadi S. Guillain-Barre syndrome as a neurological complication of novel COVID-19 infection: a case report and review of the literature. Neurologist. (2020) 25:101–3. doi: 10.1097/NRL.0000000000000291
158. Zito A, Alfonsi E, Franciotta D, Todisco M, Gastaldi M, Cotta Ramusino M, et al. COVID-19 and Guillain-Barre syndrome: a case report and review of literature. Front Neurol. (2020) 11:909. doi: 10.3389/fneur.2020.00909
159. Garg RK, Paliwal VK, Gupta A. Encephalopathy in patients with COVID-19: a review. J Med Virol. (2021) 93:206–22. doi: 10.1002/jmv.26207
160. Lorenzo Villalba N, Maouche Y, Alonso Ortiz MB, Cordoba Sosa Z, Chahbazian JB, Syrovatkova A, et al. Anosmia and dysgeusia in the absence of other respiratory diseases: should COVID-19 infection be considered? Eur J Case Rep Intern Med. (2020) 7:001641. doi: 10.12890/2020_001641
161. Spinato G, Fabbris C, Polesel J, Cazzador D, Borsetto D, Hopkins C, et al. Alterations in smell or taste in mildly symptomatic outpatients with SARS-CoV-2 infection. JAMA. (2020) 323:2089–90. doi: 10.1001/jama.2020.6771
162. Yan CH, Faraji F, Prajapati DP, Ostrander BT, Deconde AS. Self-reported olfactory loss associates with outpatient clinical course in COVID-19. Int Forum Allergy Rhinol. (2020) 10:821–31. doi: 10.1002/alr.22592
163. Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van Den Berge K, Gong B, et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv. (2020) 6:abc5801. doi: 10.1126/sciadv.abc5801
164. Nicholson P, Alshafai L, Krings T. Reply. AJNR Am J Neuroradiol. (2020) 41:E91. doi: 10.3174/ajnr.A6827
165. Coolen T, Lolli V, Sadeghi N, Rovai A, Trotta N, Taccone FS, et al. Early postmortem brain MRI findings in COVID-19 non-survivors. Neurology. (2020) 95:e2016–27. doi: 10.1212/WNL.0000000000010116
166. Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of covid-19 - final report. N Engl J Med. (2020) 383:1813–26. doi: 10.1056/NEJMoa2007764
167. Breining P, Frolund AL, Hojen JF, Gunst JD, Staerke NB, Saedder E, et al. Camostat mesylate against SARS-CoV-2 and COVID-19-Rationale, dosing and safety. Basic Clin Pharmacol Toxicol. (2021) 128:204–12. doi: 10.1111/bcpt.13533
168. Doi K, Ikeda M, Hayase N, Moriya K, Morimura N, Group C-US. Nafamostat mesylate treatment in combination with favipiravir for patients critically ill with Covid-19: a case series. Crit Care. (2020) 24:392. doi: 10.1186/s13054-020-03078-z
169. Hoffmann M, Schroeder S, Kleine-Weber H, Muller MA, Drosten C, Pohlmann S. Nafamostat mesylate blocks activation of SARS-CoV-2: new treatment option for COVID-19. Antimicrob Agents Chemother. (2020) 64:20. doi: 10.1128/AAC.00754-20
170. Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD, et al. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N Engl J Med. (2021) 384:20–30. doi: 10.1056/NEJMoa2030340
171. Huet T, Beaussier H, Voisin O, Jouveshomme S, Dauriat G, Lazareth I, et al. Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol. (2020) 2:e393–400. doi: 10.1016/S2665-9913(20)30164-8
172. Kooistra EJ, Waalders NJB, Grondman I, Janssen NaF, De Nooijer AH, Netea MG, et al. Anakinra treatment in critically ill COVID-19 patients: a prospective cohort study. Crit Care. (2020) 24:688. doi: 10.1186/s13054-020-03364-w
173. De Luca G, Cavalli G, Campochiaro C, Della-Torre E, Angelillo P, Tomelleri A, et al. GM-CSF blockade with mavrilimumab in severe COVID-19 pneumonia and systemic hyperinflammation: a single-centre, prospective cohort study. Lancet Rheumatol. (2020) 2:e465–e473. doi: 10.1016/S2665-9913(20)30170-3
174. Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in hospitalized patients with covid-19. N Engl J Med. (2021) 384:693–704. doi: 10.1056/NEJMoa2021436
175. Solomon IH, Normandin E, Bhattacharyya S, Mukerji SS, Keller K, Ali AS, et al. Neuropathological Features of Covid-19. N Engl J Med. (2020) 383:989–92. doi: 10.1056/NEJMc2019373
176. Von Weyhern CH, Kaufmann I, Neff F, Kremer M. Early evidence of pronounced brain involvement in fatal COVID-19 outcomes. Lancet. (2020) 395:e109. doi: 10.1016/S0140-6736(20)31282-4
177. Al-Dalahmah O, Thakur KT, Nordvig AS, Prust ML, Roth W, Lignelli A, et al. Neuronophagia and microglial nodules in a SARS-CoV-2 patient with cerebellar hemorrhage. Acta Neuropathol Commun. (2020) 8:147. doi: 10.1186/s40478-020-01024-2
178. Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. (2020) 24:168–75. doi: 10.1038/s41593-020-00758-5
179. Matschke J, Lutgehetmann M, Hagel C, Sperhake JP, Schroder AS, Edler C, et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. (2020) 19:919–29. doi: 10.1016/S1474-4422(20)30308-2
180. Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, De Boer HH, et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe. (2020) 1:e290–9. doi: 10.1016/S2666-5247(20)30144-0
181. Dinkin M, Gao V, Kahan J, Bobker S, Simonetto M, Wechsler P, et al. COVID-19 presenting with ophthalmoparesis from cranial nerve palsy. Neurology. (2020) 95:221–3. doi: 10.1212/WNL.0000000000009700
182. Jakhmola S, Indari O, Chatterjee S, Jha HC. SARS-CoV-2, an underestimated pathogen of the nervous system. SN Compr Clin Med. (2020) 7:1–10. doi: 10.1007/s42399-020-00522-7
183. Luers JC, Rokohl AC, Loreck N, Wawer Matos PA, Augustin M, Dewald F, et al. Olfactory and gustatory dysfunction in coronavirus disease 2019 (COVID-19). Clin Infect Dis. (2020) 71:2262–4. doi: 10.1093/cid/ciaa525
184. Chamberlin NL. Functional organization of the parabrachial complex and intertrigeminal region in the control of breathing. Respir Physiol Neurobiol. (2004) 143:115–25. doi: 10.1016/j.resp.2004.03.015
185. Chao CC, Tsai LK, Chiou YH, Tseng MT, Hsieh ST, Chang SC, et al. Peripheral nerve disease in SARS: report of a case. Neurology. (2003) 61:1820–1. doi: 10.1212/01.WNL.0000099171.26943.D0
186. Tsai LK, Hsieh ST, Chao CC, Chen YC, Lin YH, Chang SC, et al. Neuromuscular disorders in severe acute respiratory syndrome. Arch Neurol. (2004) 61:1669–73. doi: 10.1001/archneur.61.11.1669
187. Saad M, Omrani AS, Baig K, Bahloul A, Elzein F, Matin MA, et al. Clinical aspects and outcomes of 70 patients with Middle East respiratory syndrome coronavirus infection: a single-center experience in Saudi Arabia. Int J Infect Dis. (2014) 29:301–6. doi: 10.1016/j.ijid.2014.09.003
188. Van Den Pol AN. Viral infection leading to brain dysfunction: more prevalent than appreciated? Neuron. (2009) 64:17–20. doi: 10.1016/j.neuron.2009.09.023
189. Fazzini E, Fleming J, Fahn S. Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson's disease. Mov Disord. (1992) 7:153–8. doi: 10.1002/mds.870070210
190. Murray RS, Brown B, Brian D, Cabirac GF. Detection of coronavirus RNA and antigen in multiple sclerosis brain. Ann Neurol. (1992) 31:525–33. doi: 10.1002/ana.410310511
191. Arbour N, Cote G, Lachance C, Tardieu M, Cashman NR, Talbot PJ. Acute and persistent infection of human neural cell lines by human coronavirus OC43. J Virol. (1999) 73:3338–50. doi: 10.1128/JVI.73.4.3338-3350.1999
192. Desforges M, Le Coupanec A, Brison E, Meessen-Pinard M, Talbot PJ. Neuroinvasive and neurotropic human respiratory coronaviruses: potential neurovirulent agents in humans. Adv Exp Med Biol. (2014) 807:75–96. doi: 10.1007/978-81-322-1777-0_6
193. Sagare AP, Bell RD, Zlokovic BV. Neurovascular defects and faulty amyloid-beta vascular clearance in Alzheimer's disease. J Alzheimers Dis. (2013) 33(Suppl.1):S87–100. doi: 10.3233/JAD-2012-129037
194. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. (2016) 8:595–608. doi: 10.15252/emmm.201606210
195. Harris SA, Harris EA. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer's disease. J Alzheimers Dis. (2015) 48:319–53. doi: 10.3233/JAD-142853
196. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE. (2010) 5:e9505. doi: 10.1371/journal.pone.0009505
197. Singh K, Chen YC, Judy JT, Seifuddin F, Tunc I, Pirooznia M. Network analysis and transcriptome profiling identify autophagic and mitochondrial dysfunctions in SARS-CoV-2 infection. bioRxiv. (2020) 2020:092536. doi: 10.1101/2020.05.13.092536
198. Bullen CK, Hogberg HT, Bahadirli-Talbott A, Bishai WR, Hartung T, Keuthan C, et al. Infectability of human BrainSphere neurons suggests neurotropism of SARS-CoV-2. ALTEX. (2020) 37:665–71. doi: 10.14573/altex.2006111s
199. Ramani A, Muller L, Ostermann PN, Gabriel E, Abida-Islam P, Muller-Schiffmann A, et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. (2020) 39:e106230. doi: 10.15252/embj.2020106230
200. Song E, Zhang C, Israelow B, Lu-Culligan A, Prado AV, Skriabine S, et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. bioRxiv. (2020). doi: 10.1101/2020.06.25.169946
201. Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. (2009) 4:13. doi: 10.1186/1750-1326-4-13
202. Abate G, Memo M, Uberti D. Impact of COVID-19 on Alzheimer's disease risk: viewpoint for research action. Healthcare. (2020) 8:30286. doi: 10.3390/healthcare8030286
203. Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. (2010) 7:354–65. doi: 10.1016/j.nurt.2010.05.014
204. Mcnab F, Mayer-Barber K, Sher A, Wack A, O'garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
205. Kai H, Kai M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors—lessons from available evidence and insights into COVID-19. Hypertension Res. (2020) 43:648–54. doi: 10.1038/s41440-020-0455-8
206. Jang H, Boltz DA, Webster RG, Smeyne RJ. Viral parkinsonism. Biochim Biophys Acta. (2009) 1792:714–21. doi: 10.1016/j.bbadis.2008.08.001
207. Yamada T. Viral etiology of Parkinson's disease: focus on influenza A virus. Parkinsonism Relat Disord. (1996) 2:113–21. doi: 10.1016/1353-8020(96)00006-5
208. Limphaibool N, Iwanowski P, Holstad MJV, Kobylarek D, Kozubski W. Infectious etiologies of parkinsonism: pathomechanisms and clinical implications. Front Neurol. (2019) 10:652. doi: 10.3389/fneur.2019.00652
209. Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. Lancet. (2020) 395:470–3. doi: 10.1016/S0140-6736(20)30185-9
210. Dorsey ER, Sherer T, Okun MS, Bloem BR. The emerging evidence of the parkinson pandemic. J Parkinsons Dis. (2018) 8:S3–8. doi: 10.3233/JPD-181474
211. Balestrino R, Schapira AHV. Parkinson disease. Eur J Neurol. (2020) 27:27–42. doi: 10.1111/ene.14108
212. Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat Disord. (2005) 11:493–8. doi: 10.1016/j.parkreldis.2005.07.005
213. Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. (2009) 27:119–45. doi: 10.1146/annurev.immunol.021908.132528
214. Ringheim GE, Conant K. Neurodegenerative disease and the neuroimmune axis (Alzheimer's and Parkinson's disease, and viral infections). J Neuroimmunol. (2004) 147:43–9. doi: 10.1016/j.jneuroim.2003.10.013
215. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. (2018) 14:576–90. doi: 10.1038/s41574-018-0059-4
216. Caggiu E, Arru G, Hosseini S, Niegowska M, Sechi G, Zarbo IR, et al. Inflammation, infectious triggers, and Parkinson's disease. Front Neurol. (2019) 10:122. doi: 10.3389/fneur.2019.00122
217. Lesteberg KE, Beckham JD. Immunology of west nile virus infection and the role of alpha-synuclein as a viral restriction factor. Viral Immunol. (2019) 32:38–47. doi: 10.1089/vim.2018.0075
218. Massey AR, Beckham JD. Alpha-synuclein, a novel viral restriction factor hiding in plain sight. DNA Cell Biol. (2016) 35:643–5. doi: 10.1089/dna.2016.3488
219. Gelders G, Baekelandt V, Van Der Perren A. Linking neuroinflammation and neurodegeneration in Parkinson's disease. J Immunol Res. (2018) 2018:4784268. doi: 10.1155/2018/4784268
Keywords: SARS-CoV-2, storm cytokine syndrome, neuroinflammation, blood-brain barrier, neurological alterations, neurodegenerative diseases, Alzheimer's disease
Citation: Pacheco-Herrero M, Soto-Rojas LO, Harrington CR, Flores-Martinez YM, Villegas-Rojas MM, León-Aguilar AM, Martínez-Gómez PA, Campa-Córdoba BB, Apátiga-Pérez R, Corniel-Taveras CN, Dominguez-García JdJ, Blanco-Alvarez VM and Luna-Muñoz J (2021) Elucidating the Neuropathologic Mechanisms of SARS-CoV-2 Infection. Front. Neurol. 12:660087. doi: 10.3389/fneur.2021.660087
Received: 28 January 2021; Accepted: 09 March 2021;
Published: 12 April 2021.
Edited by:
Jorge Matias-Guiu, Complutense University of Madrid, SpainReviewed by:
Ulises Gomez-Pinedo, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos, SpainNiraj Kumar Jha, Sharda University, India
Ghadha Ibrahim Fouad, National Research Centre, Egypt
Copyright © 2021 Pacheco-Herrero, Soto-Rojas, Harrington, Flores-Martinez, Villegas-Rojas, León-Aguilar, Martínez-Gómez, Campa-Córdoba, Apátiga-Pérez, Corniel-Taveras, Dominguez-García, Blanco-Alvarez and Luna-Muñoz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José Luna-Muñoz, jluna_tau67@comunidad.unam.mx; Mar Pacheco-Herrero, mpacheco@pucmm.edu.do
†These authors have contributed equally to this work