 Maria Pia Giannoccaro
Maria Pia Giannoccaro Sukhvir K. Wright3,4
Sukhvir K. Wright3,4 Angela Vincent
Angela Vincent- 1Department of Biomedical and Neuromotor Sciences, University of Bologna and IRCCS Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy
- 2Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom
- 3School of Life and Health Sciences & Aston Neuroscience Institute, Aston University, Birmingham, United Kingdom
- 4Department of Neurology, Birmingham Children's Hospital, Birmingham, United Kingdom
Over the last two decades, the discovery of antibodies directed against neuronal surface antigens (NSA-Abs) in patients with different forms of encephalitis has provided a basis for immunotherapies in previously undefined disorders. Nevertheless, despite the circumstantial clinical evidence of the pathogenic role of these antibodies in classical autoimmune encephalitis, specific criteria need to be applied in order to establish the autoimmune nature of a disease. A growing number of studies have begun to provide proof of the pathogenicity of NSA-Abs and insights into their pathogenic mechanisms through passive transfer or, more rarely, through active immunization animal models. Moreover, the increasing evidence that NSA-Abs in the maternal circulation can reach the fetal brain parenchyma during gestation, causing long-term effects, has led to models of antibody-induced neurodevelopmental disorders. This review summarizes different methodological approaches and the results of the animal models of N-methyl-d-aspartate receptor (NMDAR), leucine-rich glioma-inactivated 1 (LGI1), contactin-associated protein 2 (CASPR2), and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) antibody-mediated disorders and discuss the results and the limitations. We also summarize recent experiments that demonstrate that maternal antibodies to NMDAR and CASPR2 can alter development in the offspring with potential lifelong susceptibility to neurological or psychiatric disorders.
Introduction
Over the last two decades, it has become clear that antibodies against neuronal surface antigens, particularly receptor-gated ion channels of ion-channel-associated proteins, can reach the brain to cause a group of disorders referred to as antibody-mediated or autoimmune encephalitis (AE) (1). These are immune disorders of the central nervous system (CNS) characterized by a wide range of neurological and psychiatric clinical features and associated with antibodies against different proteins expressed on the neuronal surface, mainly at excitatory, and inhibitory synapses (Figure 1). Distinct from classical paraneoplastic syndromes that are associated with onconeural antibodies (3), in AE, the neuronal surface antibodies (NSAbs) are considered to be pathogenic, and patients respond substantially to immunotherapies that reduce antibody levels (4).
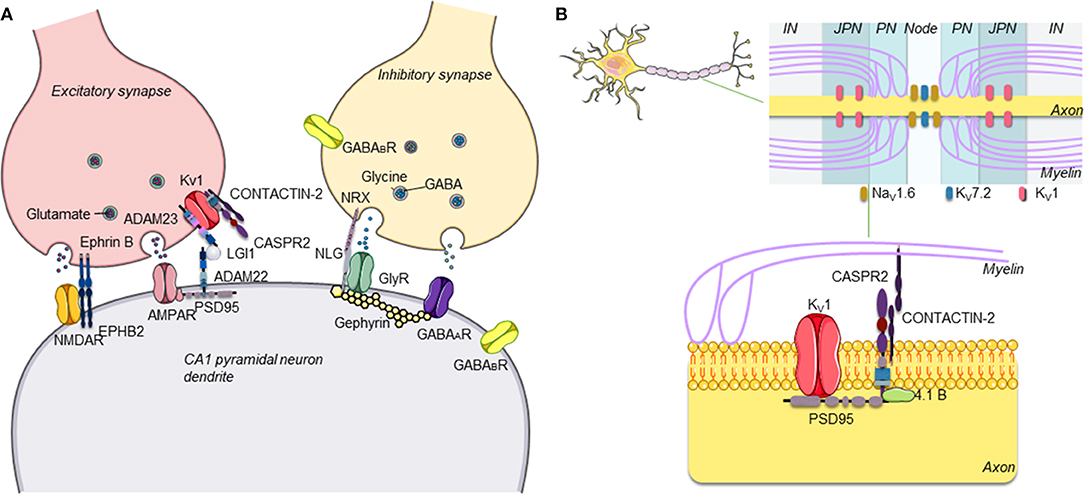
Figure 1. (A) Schematic representation of central excitatory and inhibitory synapses and main antibody targets. The proteins targeted by antibodies associated with autoimmune encephalitis are proteins expressed on the neuronal surface, often at both presynaptic and postsynaptic levels on inhibitory (GABAergic) and/or excitatory (glutamatergic) neurons in the central nervous system (CNS). (B) Schematic representation of CASPR2. CASPR2 localizes at the juxtaparanode of myelinated axons. CASPR2 binds to contactin-2/TAG-1 via its extracellular domain and links to PDZ-binding proteins and to the cytoskeleton via protein 4.1B, stabilizing Kv1 channels [adapted with permission from Giannoccaro et al. (2)].
Interestingly, these pathogenic antibodies can be either predominantly immunoglobulin G1 (IgG1) or IgG4, depending on the target antigen. In vitro studies have helped to decipher the mechanisms by which they lead to neuronal dysfunction: in many cases, divalent antibodies (IgG1 > IgG3, IgG2) cause internalization of adjacent surface proteins, leading to their loss from the membrane; complement activation by these antibodies can be demonstrated in vitro but may not always occur in vivo. By contrast, in some disorders, IgG4 antibodies predominate and act principally or exclusively by direct inhibition of the function of the target antigen [see (5) and Figure 2].
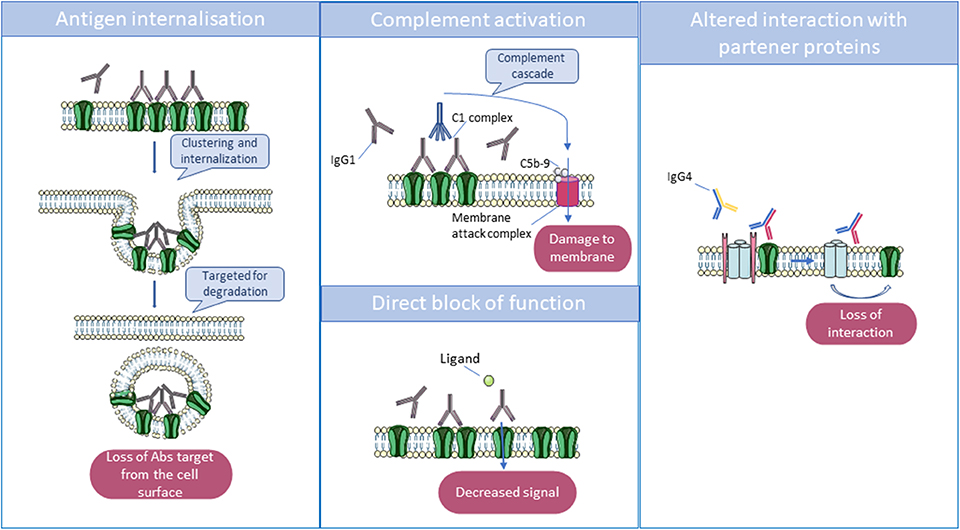
Figure 2. Main mechanisms by which antibodies act to reduce the function of their targets. Immunoglobulin G1 (IgG1) and IgG3 can cross-link antigenic targets, leading to internalization, and degradation of the antigen in lysosomes. Also, IgG1 and IgG3 can activate the complement cascade via their Fc domains, which interact with complement proteins C1 and C1q. The complement cascade culminates in the formation of the membrane attack complex which disrupts the phospholipid bilayer, resulting in cell damage. Finally, some autoantibodies can directly block receptors by binding to an essential transmitter or regulatory binding site, but monovalent IgG4 can only act by disrupting the function of the target or the interaction between their target and partner proteins.
However, an effect of the antibodies in vitro does not necessarily reflect a pathogenic role in vivo. For instance, IgG, IgA, and IgM N-methyl-D-aspartate receptor (NMDAR) antibodies (NMDAR-Abs) have been identified in a small proportion of healthy humans and mammals (6–9) and cause internalization of the NMDAR in cultured neurons (9, 10), similar to the antibodies found in patients with the IgG NMDAR-Ab encephalitis (NMDARE) (11). This suggests that other factors are likely required to induce the clinical syndrome, factors that may be difficult to model in vitro alone.
Indeed, according to the modified Witebsky criteria (12), direct and indirect evidence of pathogenicity requires the reproduction of the disease in a recipient through direct transfer of the antibodies (passive transfer) or through active immunization, respectively. Animal models not only provide evidence of pathogenicity but can also offer insight into sites of action, pathogenic mechanisms, and therapeutic approaches.
Accordingly, over the last few years, animal models, usually in mice, have been established for the most commonly encountered NSAbs in clinical practice. Below, we describe the approaches used and the results of these models and discuss their advantages and limitations. We also summarize recent experiments that demonstrate that maternal antibodies to these or other NSAbs can alter development in the offspring with potential lifelong susceptibility to neurological or psychiatric diseases.
Different Models of Antibody-Mediated Disorders
Animal models of autoimmune disorders can be divided into two main categories: (1) spontaneous models where, comparably to humans, animals develop an autoimmune disease spontaneously and (2) induced models where an autoimmune disease is artificially provoked. Spontaneous forms of AE have been reported in different species, but they are uncommon (13, 14). Most of the models of AE have been obtained through induction by passive or active immunization. Passive immunization is based on the reproduction of the disease in a healthy recipient by transfer of serum, purified immunoglobulins, monoclonal antibodies, or, more rarely, antibody-producing cells isolated from an affected human or animal donor. Active immunization is based on the exposure to an antigen, often in association with adjuvants, to generate an adaptive immune response. The antigen can be in the form of purified proteins, recombinant or synthesized peptides (15).
Work on myasthenia gravis (MG) provides examples of both active and passive immunization and has helped to shape our understanding of antibody-mediated diseases (Table 1). Passive transfer is the best way to assess the acute effects of human autoantibody-mediated diseases and has been used extensively to study patients' derived antibodies in MG [(16); see a brief review by Phillips and Vincent (17). By contrast, active immunization (see (18)] has been particularly useful to investigate more broadly the immunological factors underlying the disease, though with the limitations of possible differences between the function of the human and rodent immune systems and between different strains of mice. For instance, C57B1/6 mice were very susceptible to active immunization with acetylcholine receptor (AChR), whereas AKR/J mice were resistant (19, 20). Moreover, the use of the target antigen as a whole protein often induces high titers of antibodies, but if the protein is from a different species, not all of the antibodies will necessarily cross-react with the mouse antigen or be directed against the disease-causing epitope(s). Therefore, active immunization models are not always relevant to the human pathology but, when successful in producing an appropriate clinical and physiological phenotype, provide a long-term model of the disease that is suitable for testing experimental therapies.

Table 1. Example of antibody-mediated diseases: clinical and experimental evidence for MG.
In contrast to conditions such as MG, where the target antigens of the antibodies are peripheral and thereby easily accessible from the systemic circulation, the blood–brain barrier (BBB) limits the access of immune molecules to the brain. One way to overcome this limitation, in models of CNS antibody-mediated diseases, is to infuse the antibodies directly into the cerebrospinal fluid (CSF) within the cerebral ventricle(s) (intracerebroventricular, icv) or to inject them into the brain parenchyma. However, in the majority of autoimmune forms of encephalitis, the antibody levels are higher in the serum than in the CSF, suggesting that the antibodies could initiate the disease by diffusion through an incomplete or temporarily disrupted BBB (32) or at sites of limited BBB protection such as the choroid plexus. Therefore, another approach is to administer the antibodies in the periphery, using the intravenous (iv) or intraperitoneal (ip) route and if necessary to induce artificially a breach in the BBB to allow the antibodies to reach their targets. Classically, the latter is obtained by one or two ip injections of lipopolysaccharide (LPS), which induces a transient disruption of the BBB, particularly in the frontal cortex, thalamus, pons–medulla, and cerebellum (33). It is not yet clear whether the icv or ip route of administration is most appropriate and whether they could lead to different CNS changes.
Finally, there is a possibility of transfer from a mouse dam to developing embryos. Although the BBB interfaces are formed early in development (34), maternal IgG antibodies can cross into the fetal brain parenchyma during gestation (32). It is long established that a neonatal form of MG can result from the transfer of IgG antibodies from an affected mother to her fetus in utero (27, 35). Human MG AChR antibodies injected intraperitoneally into pregnant mice were shown to cross efficiently from the mouse dam to her fetuses and to cause neuromuscular changes in utero (36); this model has since been used to study the effects of human serum antibodies on brain development (as described below).
Models of Neuronal Antibody-Mediated Disorders
The clinical and investigative features of the patients with antibodies to neuronal surface proteins, and the results of the existing models, are summarized in Table 2.

Table 2. Summary of main features of NSAb diseases and the models.
NMDAR-Ab Encephalitis
Clinical Disease and in vitro Mechanisms
NMDARE, the classical syndrome associated with IgG1 NMDAR-Abs, is the most commonly recognized AE in clinical practice. It is characterized by psychiatric symptoms, such as confusion, abnormal behavior, paranoia, and hallucinations, in addition to memory problems, seizures, dyskinesia, autonomic instability, catatonia, hypoventilation, lethargy, and language deficits (56). In vitro, pathogenic NSAbs bind and cause clustering (57), cross-linking, and internalization of NMDAR, leading to a loss of functional receptors on the cell surface (NMDAR hypofunction), which is reversible on removal of the NMDAR-Abs (11). Moreover, NMDAR-Abs induce dispersal of GluN2A-NMDAR, through the blockade of the interaction between the extracellular domains of GluN1/GluN2 subunits and ephrin-B2 receptors (EPHB2R) (58).
In a high proportion of younger women, the disease is caused by the presence in an ovarian teratoma of neuronal tissue expressing NMDARs and inducing an immune response (59, 60). In others, particularly young children, the disease can follow herpes simplex virus encephalitis (HSVE), probably as a secondary response to the neuronal damage caused by the virus (61).
Spontaneous or Genetic Disease
NMDAR-Abs have been described in other mammals (9) and are present at a low percentage (around 1%) in healthy individuals. In 2014, a retrospective study showed that Knut, the polar bear of the Berlin Zoological Garden who drowned in 2011 following seizures, had high levels of NMDAR-Abs in his serum and CSF, making him the first non-human case of NMDARE and reaffirming the epileptogenicity of these antibodies in mammals. Pathological examination showed a patchy distribution of infiltrating immune cells, with numerous plasma cells around vessels and within the parenchymal infiltrates, in the absence of marked neuronal abnormalities (14).
Mutations in GRIN1 [which encodes the GluN1 (NR1) subunit of NMDAR] have been associated with a phenotype consisting of severe intellectual disability, seizures, hyperkinetic and stereotyped movement disorders, and dysmorphic features (62–64). In mice, selective deletion of GluN1 in CA1 and CA3 pyramidal neurons abolished long-term potentiation (LTP) and induced memory impairment (65, 66).
Passive Transfer Models
Animal models of NMDARE have been published recently with results that recapitulate some of the specific features of the human disease. In rats, stereotactic parenchymal injection of CSF or purified IgGs from patients with NMDARE produced different outcomes. Infusion in the CA1 and premotor cortex increased the levels of extracellular glutamate and, consequently, neuronal excitability (46). On the other hand, several studies showed that a single injection of CSF from patients with NMDARE into the hippocampus produced a reduction of LTP in the CA1, CA3, and dentate gyrus (47–49). Behaviorally, effects ranging from impaired Morris water maze memory performance (47) to a lack of novel object recognition (49) were reported, in the absence of significant changes in locomotor activity or anxiety-like behavior (49).
Continuous icv infusions of CSFs pooled from individuals with NMDARE into mice over 14 days reproduced some of the neuropsychiatric features observed in patients such as memory deficits, anhedonia, and depressive-like behaviors. Seizures or movement disorders were not observed. IgG deposition and a decrease in NMDAR clusters on hippocampal neurons was observed in NMDAR-Ab-injected mice, which resolved within days after discontinuing the infusion (38). Further studies have also shown disruption of the normal interaction with other synaptic proteins, in particular EphrinB2R. Administration of ephrin-B2 (the ligand of the EphrinB2 receptor) in the 14-day infusion animal model prevented the pathogenic effects of NMDAR-Abs on memory and behavior, levels of cell-surface NMDAR, and synaptic plasticity (41). Recently developed human-derived monoclonal antibodies to the NMDAR have produced similar pathogenic effects in vivo and in vitro and offer a promising less-limited resource (compared to human CSF and IgG) for future experimental studies (43).
In another mouse model, icv injection of purified plasmapheresis IgG from individuals with NMDARE induced, in association with a subthreshold dose of the chemo-convulsant pentylenetetrazol (PTZ), more frequent and severe seizures than a single injection of IgG from control individuals [(39); see Figure 3]; cognitive and other features were not examined in these mice. Continuous wireless electroencephalogram (EEG) recording did not identify any spontaneous seizure activity. However, there was IgG bound to the hippocampus at 48 h post icv infusion, particularly to the CA3 region, and it correlated with the number and severity of seizures seen in the mice, but there was no apparent loss of NMDARs (Figure 3). In a more recent study, EEG recordings of mice infused intraventricularly for 14 days with CSF NMDAR-Abs showed a higher frequency of seizures compared with control mice, associated with variable behavior ranging from sleeping or normal exploratory activity to freezing and myoclonic jerks (42). Two main seizure patterns were observed, one, more frequent, characterized by high-amplitude rhythmic spikes that occurred at relatively constant rates or at irregular intervals and another, less common, characterized by high-amplitude fast rhythmic activity that fluctuated in amplitude in a spindle-like fashion (42). Continuous EEG recordings may be necessary to detect reliably spontaneous non-motor seizures in models of antibody-mediated encephalitis. Neuropathology showed absence of neuronal death and only mild astrocytic activation (42).
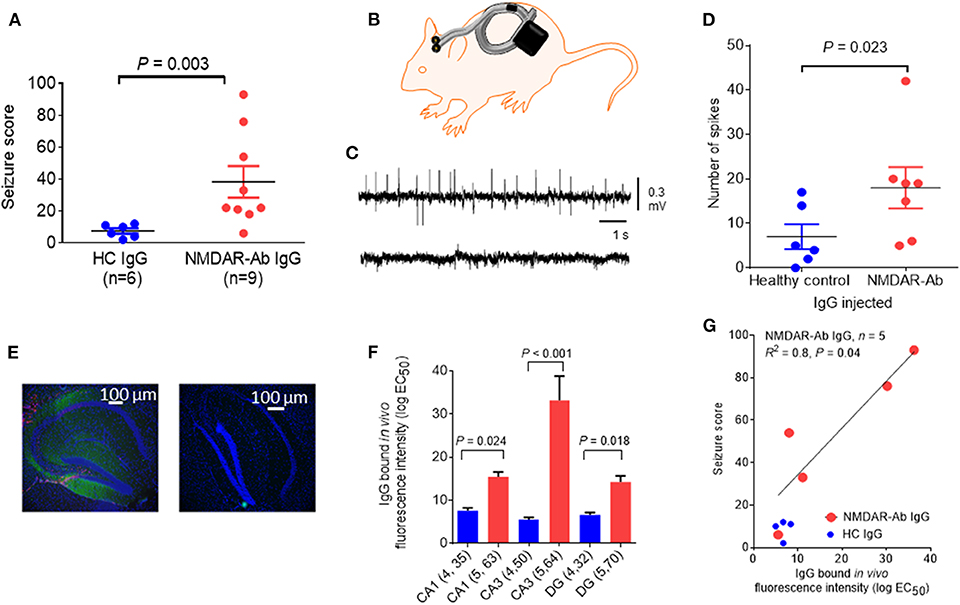
Figure 3. Epileptogenic effects of a single intracerebroventricular (icv) injection of N-methyl-D-aspartate receptor antibody (NMDAR-Ab)-positive immunoglobulin G (IgG). (A) The seizure score of mice injected with NMDAR-Ab IgG was higher than that of those injected with control IgG following exposure to a subthreshold dose of PTZ. (B) Diagram showing placement of subcutaneous wireless electroencephalogram (EEG) transmitter that allows continuous EEG recording in injected mice with no need for tethering (Open Source, Hashemi Instruments, USA). (C) A representative EEG of an NMDAR-Ab-injected mouse post-PTZ shows a number of “spikes” corresponding to convulsive seizures (upper trace), compared with the EEG of a healthy control IgG-injected mouse, which has minimal spike activity (lower trace). (D) When analyzed using the computer-based event detection program and blinded observer verification, the number of spikes seen in the hour following PTZ injection was greater in the NMDAR-Ab (n = 7) compared with the healthy control IgG (n = 6) injected mice (P = 0.023, Mann–Whitney). Results are mean ± SEM. (E) Human IgG injected in vivo was detected postmortem in NMDAR-Ab IgG-injected mice with antihuman IgG (green) merged with the nuclear stain 4′,6-diamidino-2-phenylindole (DAPI) (blue). The typical pattern of NMDAR-Ab in the molecular cell layer with sparing of the granule cell layer was found (left image). Control IgG-injected mice had no detectable IgG (right image). (F) Bound human IgG in the hippocampi, as determined by the mean fluorescence intensity analysis of brain sections, was higher in the NMDAR-Ab IgG-injected mice than in healthy control IgG-injected mice in the CA1, CA3, and dentate gyrus (DG). (G) For the NMDAR-Ab animals (n = 5), there was a linear correlation between IgG binding and seizure score (R2 = 0.8; P = 0.04). The contents of this figure are taken from Wright et al. (39) with permission from Oxford University Press.
In another study using continuous icv infusion, mice receiving patients' CSF showed memory impairment in the Morris water maze, but not in the novel object recognition test, and a tendency to a reduced expression of NMDAR in the mouse brains. No overt inflammatory changes were observed, but an increase of the chemokine CXCL10 was detected (40), a finding that has been observed also in patients with NMDARE (67). Intravenous infusion of monoclonal NMDAR-Abs followed by LPS increased mouse voluntary locomotor activity at the mouse wheel-running test, similarly to that observed in mice treated with low doses of the NMDAR inhibitor MK-801 (44).
Overall, the passive-transfer animal models support the proposed mechanisms of cross-linking and internalization as well as the relevant role of altered NMDAR trafficking in the pathogenesis. However, these models have not demonstrated all the clinical features; for example, none have reproduced the (often-striking) movement disorders or shown long-term cognitive deficits and structural hippocampal damage as seen in some patients (68). A possibility is that some inflammatory changes are not reproduced by passive transfer. The discrepancies observed between different models might also relate to different protocols, to the use of different species and strains, and to different effects of the antibodies in relation to acute or chronic exposure.
Active Immunization
In a recent mice active immunization model, Pan et al. (9) showed that mice immunized against NMDAR1 peptides did not show behavioral changes at the open-field test. Even in the presence of high titers of NMDAR-Ab, an increase of locomotor activity, a psychosis-like behavior, was obtained only upon MK-801 challenge in ApoE−/− mice, which present a disrupted BBB. No lymphocyte (CD3) infiltrates nor microglial activation was detected on immunopathology. On the contrary, immunization with purified GluN1/GluN2B fully assembled tetrameric NMDARs (holoreceptors) embedded in liposomes induced a phenotype characterized by hyperactivity, stereotyped motor features (tight curling), and seizures in association with neuroinflammation and immune cell infiltrates (37). Distinct from the passive-transfer models, these immunized mice produced GluN1 and GluN2 antibodies that reacted with the linear epitopes of the NMDAR protein, and not the amino-terminal domain of GluN1 as seen in the human-derived antibodies (69). Nevertheless, this model may prove useful for testing novel treatments acting on the cellular inflammatory component of the disease.
Finally, a recent small study investigated the mechanisms involved in the pathogenesis of post-HSV-1 NMDARE (45). Following intranasal inoculation of HSV-1, 67% (four out of six) of mice developed serum NMDAR-Abs. The same mice showed reduced hippocampal NMDAR compared with mice without antibodies, inferring IgG-mediated loss, but the authors did not demonstrate IgG antibodies bound to the hippocampus. This model could be a useful platform to further explore the mechanisms of post-HSV encephalitis with secondary NMDARE.
CASPR2-Ab Encephalitis
Clinical Disease and in vitro Mechanisms
CASPR2 is a neurexin-related cell adhesion molecule expressed in the CNS and peripheral nervous system, and CASPR2 antibodies (CASPR2-Abs) react with both the brain and peripheral nerve tissues [(70); see Figure 1]. This expression pattern well-explains why CASPR2-Abs have been associated not only with peripheral nerve hyperexcitability (often called neuromyotonia) but also with CNS symptoms including cognitive impairment, memory loss, hallucinations, delusions, cerebellar symptoms, and epilepsy. Some patients present with Morvan syndrome (MoS), characterized by the combination of neuromyotonia, neuropathic pain, encephalopathy with hallucinations, and a sleep disorder, described as agrypnia excitata (71, 72); the latter is characterized by severe insomnia, dream-like stupor (hallucinations and enacted dreams), sympathetic hyperactivity (hyperthermia, perspiration, tachypnea, tachycardia, and hypertension), and motor agitation. CASPR2-Abs are mainly IgG4, but most patients have IgG1 antibodies as well.
CASPR2 is essential for clustering Kv1.1 and Kv1.2 channels at the juxtaparanodes of myelinated axons, where the channels are important for repolarization of the nerve axon, avoiding repetitive firing and helping to maintain the internodal resting potential. Their functions at CNS synapses are not well-defined.
The in vitro effects of CASPR2-Abs are complex. In one study, the antibodies inhibited CASPR2 interaction with contactin-2 but did not lead to CASPR2 internalization (73). However, in two others, in vitro exposure induced CASPR2 internalization in vitro (51, 52) with variable effects on CASPR2 expression, ranging from absent (51) to significant (52) loss of surface expression.
Spontaneous or Genetic Disorders
Interestingly, mutations in the CNTNAP2 gene, encoding CASPR2, are associated with focal epilepsy, schizophrenia, and autism spectrum disorder (ASD) (74). CNTNAP2-knockout (KO) mice were shown to have social deficits, abnormal motor activity, cognitive deficits, and seizures (75).
Passive Transfer Models
Intraperitoneal injection of purified IgG from two CASPR2-Ab-positive patients to mice over 14–18 days, without attempt to breach the BBB, reduced the thresholds for mechanical stimuli, a signature of pain (50). The effects induced by the antibodies on pain sensitivity were also observed in KO mice lacking CASPR2 (CNTNAP2−/−). These mice demonstrated enhanced pain-related hypersensitivity to noxious mechanical stimuli, although more severe than that obtained with the antibodies, and also to heat and algogens. Nevertheless, either immune or genetic-mediated ablation of CASPR2 enhanced the excitability of dorsal root ganglia (DRG) neurons through regulation of Kv1 channel expression at the soma membrane (50). CASPR2-IgG did not cause neuronal loss nor overt inflammation, although a modest increase in microglial cell count was observed in the spinal cord (50).
To explore the effects of CASPR2-Ab in the CNS, a similar protocol was used with eight daily injections of IgG purified from one patient with AE and from one healthy control (Figure 4). A single dose of LPS was added at day 3 to disrupt the BBB (51). Mice injected with CASPR2-IgG showed less alternation in the continuous spontaneous alternation tests, suggestive of memory impairment, and longer latency to interact and increased immobility during the social interaction test (Figure 4). These changes had not been seen during isolated open-field or other tests, suggesting that the effects could be indicative of anxiety in the context of a novel mouse, rather than an effect on normal exploratory activity. At neuropathology, CASPR2-IgG injected mice showed human IgG deposition, particularly in the cortex, hippocampus, and thalamus; mild loss of Purkinje cells and c-Fos activation as well as microglial and astrocyte activation without B- or T-cell infiltration (Figure 4). Microglial activation has been reported in neuropathological cases of patients with CASPR2-Ab encephalitis (76, 77).
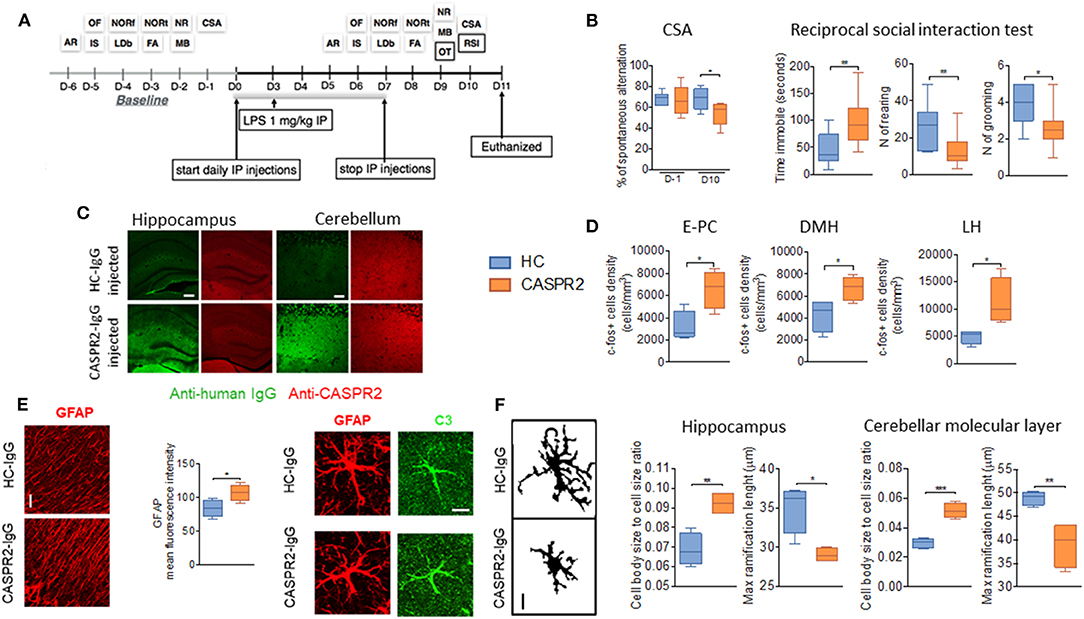
Figure 4. Intraperitoneal (ip) injection of CASPR2 immunoglobulin G (IgG) with lipopolysaccharide (LPS) causes behavioral and neuropathological changes in mice. (A) Experimental design and selected behavioral tests. The behavioral tasks assessed locomotion (open field, OF), strength (inverted screen, IS), coordination (accelerating rotarod, AR; and narrow beam, NB), working memory (continuous spontaneous alternation, CSA), short- (forced alternation, FA) and long-term memory (novel object recognition, NOR—NORf, familiarization phase; NORt, test phase), anxiety (light-dark box, LDb), compulsive-like behavior (marble burying test, MB), social behavior (reciprocal social interaction tests, RSI), and olfaction (olfaction test, OT). (B) Continuous spontaneous alternations were reduced in CASPR2-IgG-injected mice compared with HC-IgG-injected mice (P = 0.044). In the RSI test, there was reduced latency to interact (P = 0.04; Mann–Whitney test) but no differences in the interaction time or number of interactions. However, in the non-social aspects of the test, there was increased time spent immobile (U = 0.008), reduced rearing (U = 0.02), and reduced grooming (U = 0.018). (C) Bound human IgG in the hippocampi and cerebellum of CASPR2- and HC-IgG-injected mice. CASPR2-IgG-injected animals had higher levels of IgG in the cortex (Cx) (P = 0.03), hippocampus (Hip) (P = 0.023), and thalamus (Th) (P = 0.0004) compared with HC-IgG-injected mice. No differences were observed in the levels of CASPR2 expression in the same areas (n = 4 per group). (D) c-Fos expression in the entorhinal–piriform cortex (P = 0.020), dorsomedial hypothalamus (DMH) (P = 0.037), and lateral hypothalamus (LH) (P = 0.031) was higher in the CASPR2-IgG-injected mice than in the HC-IgG-injected mice (n = 4 per group). (E) Representative images of glial fibrillary acidic protein (GFAP) staining in the molecular layer of the cerebellum and quantification of the mean fluorescence intensity in the same area showing higher GFAP expression in the CASPR2-IgG-injected mice (P = 0.043) (n = 4 per group; 40X, 10 μm). On the right, representative images of complement C3 expression on GFAP-positive cells. Percentage of C3/GFAP area ratio per cell showed increased C3 expression of astrocytes in CASPR2-IgG-injected mice. (F) Representative images of the z-stack projected IBA1 staining used for morphological analysis (40X, 10 μm). Quantification of morphological data in the hippocampus and molecular layer of the cerebellum showed that microglia from CASPR2-IgG-injected mice had a higher cell soma/cell total body size ratio [t(6) = 4.74, P = 0.0032] and shorter [t(6) = 3.68] ramifications than HC-IgG-injected mice, compatible with an activated phenotype in both the hippocampus (P = 0.017 and P = 0.010, respectively) and the cerebellum (P = 0.0003 and P = 0.008, respectively). ***The contents of this figure are taken from Giannoccaro et al. (51) with permission from Oxford University Press. * < 0.05, ** ≤ 0.01, *** ≤ 0.001.
Although this model showed evidence of pathogenicity of CASPR2-IgG, it failed to recapitulate the wide range of defects found in the patients (e.g., autonomic, sleep disturbance, and hormonal/neuropeptide abnormalities) who would require substantial additional tests. Moreover, it does not explain how CASPR2-Abs cause their effects. Indeed, IgG deposition was not associated with a reduction of CASPR2 expression on immunohistology. On the contrary, a trend toward higher levels of mouse CASPR2 was seen in the brain extracts of CASPR2-IgG-injected mice, suggesting some compensatory upregulation.
Injection of a mixture of CASPR2-Abs in mouse visual cortex produced impaired localization of mouse Caspr2 to excitatory synapses and significantly decreased AMPAR-mediated currents in layer 2/3 pyramidal neurons; this implied a dysfunction of glutamatergic transmission in the pathogenesis of CASPR2-Ab encephalitis (52). Future studies should evaluate in parallel the effects of CASPR2-Abs on its partner protein network and on neuronal activity.
LGI1-Ab Encephalitis
Clinical Disease and in vitro Mechanisms
Autoantibodies to LGI1 (LGI1-Abs) are the most common autoantibody in patients with limbic encephalitis (LE), a clinical syndrome characterized by the acute development of mood changes, anxiety, short-term memory deficit, and seizures due to an inflammatory process involving the limbic system that includes the medial temporal lobes, hippocampus, amygdala, and frontobasal and cingulate cortices (1). In patients with LGI1-Abs, the onset of an overt limbic dysfunction can be preceded by episodes of faciobrachial or crural seizures that last a few seconds and occur many times during the day; these episodes have been described as faciobrachial dystonic seizures (FBDS) (78).
LGI1 is a protein secreted by the presynaptic terminals of neurons that bind to ADAM22 and ADAM23, two proteins involved in cell–cell adhesion and located presynaptically and postsynaptically, respectively (Figure 1). Binding to ADAM22, LGI1 regulates AMPAR-mediated synaptic currents in the hippocampus (79). Binding to ADAM23, LGI1 selectively prevents inactivation of the presynaptic voltage-gated potassium channel Kv1.1 (80) mediated by a cytoplasmic regulatory protein, Kvβ (81).
In cultured hippocampal neurons, LGI1-Abs disrupt the ligand–receptor interaction of LGI1 with ADAM22, resulting in reversible reduction in synaptic AMPARs [(82); see Figure 1]; these antibodies could be IgG4. However, in the few postmortem studies on patients who have died unexpectedly, there appears to be IgG deposition, some complement deposition, and loss of neurons. These findings would be compatible with the presence of IgG1 antibodies; although they are in the minority compared with IgG4, they tend to be much higher in patients with cognitive impairment (70, 83). IgG1-induced neuronal loss would explain why, despite a good response to immunotherapy, many patients are left with hippocampal atrophy (84), and only 35% of patients return to their baseline cognitive function (85).
Spontaneous or Genetic Disorders
LGI1 mutations have been associated with an autosomal dominant lateral temporal lobe epilepsy (ADLTLE) manifesting with focal seizures often with auditory features (86). The majority of mutations prevent LGI1 secretion, whereas others alter its interactions with ADAM22/ADAM23 (87). Animal models of LGI1 depletion all present spontaneous seizures (88–92), although the mechanisms behind this increased epileptic susceptibility have not been fully elucidated and both enhanced excitatory transmission (90, 91, 93) and reduced AMPAR function (79, 80, 89) have been reported.
Intriguingly, a spontaneous model of LGI1-Ab encephalitis has been observed in cats with feline complex partial seizures with orofacial involvement (FEPSO) (13, 94–96). Clinically, they presented with acute onset of complex partial seizures with orofacial involvement (salivation, facial twitching, lip smacking, chewing, licking, or swallowing), motor arrest (motionless starring), and behavioral changes associated with bilateral hyperintensities at brain MRI (13, 94, 95). Postmortem analysis of three cases showed IgG and complement deposition associated with neuronal loss, consistent with the findings in the few available postmortem examinations from patients with LGI1-related encephalitis (3, 95). Subsequent neuropathological studies in cats showed also that, whereas T-cell infiltrates were present brainwide, BBB leakage was more restricted to limbic areas (96). This observation suggests that a local BBB vulnerability might be responsible for the selective involvement of the limbic system, even though LGI1 is expressed throughout the brain.
Passive Transfer Model
More recently, the pathogenicity of LGI1-Abs has been confirmed by a passive transfer mouse model based on cerebroventricular transfer of patient- or control-derived IgG (53). LGI1-Ab-injected mice showed memory impairment which slowly reversed after stopping the infusion. However, in contrast to the spontaneous feline model and LGI1-KO animals, no epileptic seizures were observed. Nevertheless, LGI1-Ab caused a significant decrease of the density of total and synaptic Kv1.1 and AMPAR clusters due to the disruption of LGI1 interactions with presynaptic ADAM23 and postsynaptic ADAM22. Consistent with decreased Kv1.1 expression and previous in vitro studies (97), increased presynaptic excitability and glutamatergic transmission were observed in acute brain slice preparations, resulting in increased evoked excitatory postsynaptic currents (eEPSCs) and reduced failure rate of synaptic transmission after minimal-stimulation excitatory postsynaptic currents (msEPSCs). Exposure to LGI1-Ab was also associated with impaired LTP, which was however independent of Kv1.1 blockade and possibly related to reduced availability of AMPAR during LTP. However, these changes were not sufficient to cause seizures in this model. It is likely that the changes induced by the antibodies are not as severe as those induced by genetic mutation or ablation of the LGI1 gene. On the other hand, complement activation and neuronal loss may play a major role in the human and feline diseases and mouse serum has a low intrinsic complement activity (98). Further studies are needed to investigate this aspect and its relevance to the clinical phenotype.
AMPAR-Ab Encephalitis
Clinical Disease and in vitro Mechanisms
AMPAR antibodies (AMPAR-Abs) are usually associated with a typical LE, sometimes associated with extra limbic manifestations, although they can rarely present with rapidly progressive dementia or psychosis (99, 100).
AMPAR is a heterotetrameric ionotropic glutamate receptor that mediates most of the fast-excitatory transmission in the brain (101). AMPAR-Abs can be directed against the GluA1 or GluA2 subunits or both (100). Incubation of cultured rodent neurons with patients' IgG to GluA2 led to a decrease of synaptic AMPAR clusters, resulting in reduced frequency and peak amplitude of AMPAR-mediated miniature excitatory postsynaptic currents (mEPSCs) (100, 102).
Spontaneous or Genetic Disorders
Mutations in the GluA1 or GluA2 subunits have been associated with neurodevelopmental disorders (NDs) including intellectual disability and autism (103, 104). GluA1-KO mice present impaired hippocampal synaptic plasticity (105, 106) and working memory (107–109), whereas GluA2-KO mice are hypomorphs with poor motor coordination and low explorative activity (110, 111). Conditional ablation of GluA1 or GluA2 in mice causes memory deficits and remodeling of AMPAR subunit distribution (112–115).
Passive Transfer Models
In accordance with these findings, in vitro studies and in vivo hippocampal injection of human antibodies against the GluA2 subunit in mice was associated with synaptic downregulation of GluA2 and increased single-channel conductance in recordings of the GluA2 IgG-injected mouse, suggestive of GluA2 endocytosis and compensatory synaptic incorporation of GluA1-containing AMPARs, which have higher channel permeability (54), as observed in conditional KO models (113–115). Consistently, this compensatory increase in single-channel conductance was abrogated in KO mice deficient for GluA1 stereotactically injected with GluA2 antibodies (GluA2-Abs). Despite these compensatory mechanisms, injection of GluA2-Abs was associated with impaired LTP in the region of GluA2-IgG deposition. Both continuous icv infusion of GluA2-Abs over a 2-week period and stereotactic bilateral injections of patient IgG directly into the DG, CA1 and CA3 regions of the hippocampus, were associated with impaired memory and increased anxiety-like behavior in mice (54). Despite the observed AMPAR subunit rearrangement, mice did not show seizures. Therefore, future studies have to evaluate if these changes are associated with neuronal hyperactivity and how they are related to seizures in patients. Moreover, the pathogenicity and mechanisms associated with antibodies directed against the GluA1 subunit of AMPAR remain to be established.
Animal Models of NDs Induced by in utero Exposure TO NSAbs
There has been growing interest in the possibility that maternal antibodies to neuronal antigens could cause neurodevelopmental diseases, presenting neonatally or later in life. This sprung initially from studies in mothers with MG whose babies developed arthrogryposis. The maternal antibodies were found to inhibit the function of the fetal AChR and, when crossing the placenta in the second trimester, paralyzed the babies in utero; consecutive pregnancies were affected (28, 116). A mouse model of maternal antibody transfer to the mouse fetus was developed to show that the maternal serum antibodies were pathogenic (36), and the model was then used to study a mother who had two consecutive children with NDs (one healthy, one with autism, and one with language disorder). The serum contained antibodies that bound to fetal cerebellar neurons in rat tissue sections and impaired motor behavior in the adult mouse offspring of injected dams (117). Since then, many studies looking for maternal antibodies in autism and testing their effects in mouse or non-human primates have been performed [see (118)], but until recently, none had defined a specific neuronal antigen that was likely to be the target of fetopathogenic antibodies.
As mentioned above, mutations in the gene encoding CASPR2 are not common but can be associated with a variety of neurological and psychiatric disorders, ranging from ASD or mental retardation and epilepsy to learning disability, schizophrenia, and Tourette syndrome (119). Mutations in the GluN genes that encode the N-methyl-D-aspartate (NMDA) subunits are found in children with a variety of NDs and epileptic syndromes (120). Both these proteins could be targets for antibodies that, during development, altered neurodevelopment. Table 3 summarizes the most recent work in this field.

Table 3. Neurodevelopmental antigens and models.
Evidence For Antibodies to NSAbs in Pregnancy
CASPR2-Abs
Only one study to our knowledge has looked for antibodies to these proteins in gestational samples from women whose children have subsequently been diagnosed with specific or non-specific neurodevelopmental conditions, comparing with mothers with no such history in their children. Coutinho et al. (121) measured a range of neuronal antibodies in Danish cohorts of early or mid-gestational sera. LGI1-Abs, AMPAR-Abs, and GABAB receptor antibodies were not found. NMDAR-Abs were not uncommon (overall 5.8%) and more common in mothers who developed psychosis at some time after the pregnancy. By contrast, CASPR2-Abs were present (4.9%) in mothers of children with a diagnosis of mental retardation or other disorders of psychological development in their children compared with only 0.9% of coded age-matched mothers with no such history. This supported the possibility that CASPR2-Abs could be a cause or contributor to neurodevelopmental diseases in the offspring. Surprisingly, CASPR2-Abs were low in mothers of autistic children and not different from the appropriate controls.
A maternal-to-fetal transfer of disease was performed in mice. The offspring of CASPR2-injected dams were normal postnatally but as adults showed changes in social interaction tests, and after termination, there was clear evidence of microglial activation and reduced glutamatergic synapses, suggesting that microglia activated by CASPR2-Abs induced changes that resulted in persistent synaptic loss (122).
A similar model was undertaken using a monoclonal CASPR2-Ab cloned from a mother of an autistic child (123). In this study, male mice exposed in utero to CASPR2-Abs showed an ASD-like phenotype, abnormal cortical development, and altered hippocampal neurons. Postnatal samples from selected mothers of autistic children were more often positive for CASPR2-Abs than from mothers of children with typical development or women of childbearing age. These sera were not gestational and in many cases obtained from mothers years after the affected birth.
NMDAR-Abs
In Coutinho et al. (121), NMDAR-Abs were relatively frequent (5.8%) during pregnancy. Although NMDAR-Abs were more frequent in mothers with NDs in their children (ND mothers) than coded age- and gestation-matched mothers with no such histories (HC mothers), this difference was not significant (7.7 vs. 4.6%). Indeed, among the few reported cases of NMDARE during pregnancy, the majority of newborns were healthy, except for three cases with neurological sequelae, including neurodevelopmental delay, movement disorders, and seizures, and three cases of miscarriages and abortion (125–127). Whether these complications are due to the antibodies or to the mothers' condition severity and related pharmacological treatments during gestation is not yet clear.
Jurek et al. (124) showed a marginal increase in NMDAR-Ab titers in postnatal sera from mothers of a mixed population of neuropsychiatric disorders in a recent study, compared with mothers of unaffected children. These authors preformed a similar model of in utero exposure to human NMDAR-Abs, but in this case using recombinant human monoclonal NR1-reactive IgG antibodies (124). The placentally transferred antibodies bound to synaptic structures in the fetal brain, and the pups demonstrated increased mortality and transiently reduced NMDAR brain density with impaired excitatory neurotransmission. The animals displayed hyperactivity, lower anxiety, and impaired sensorimotor gaiting during adolescence and adulthood. In aged mice (10 months), the volumes of the cerebellum, midbrain, and brain stem were all reduced (124). This study suggests that prenatal exposure to NMDAR-Abs may result in children's lifelong neurodevelopmental changes that are potentially treatable and preventable, if identified in the mothers during pregnancy, although there is no evidence of that so far. Such changes might predispose to specific NDs such as autism or schizophrenia.
Discussion and Conclusions
Animal models have helped to elucidate pathogenic mechanisms of several NSAbs. However, they often fail to recapitulate the entire phenotypic spectrum associated with human diseases. In particular, no movement disorders have been found in the models of NMDARE, and no seizures were detected in mice injected with LGI1-Abs. This could be related to several factors. Firstly, the choice of the species and strains is relevant. Nowadays, mice are the preferred animals for the majority of immune models; however, certain strains used can be resistant to development of diseases, as shown by MG models of active immunization. The gender is another potentially relevant factor, as hormones can significantly impact several immunological and neuronal aspects.
Different immunization models have different advantages and disadvantages. Intraventricular or intraparenchymal administration routes are useful in exposing the antibodies to their targets, but they may be misleading when peripheral antibodies play a major role as appears to be the case for CASPR2-Abs and LGI1-Abs. On the other hand, peripheral injection of the antibodies often requires “opening” the BBB by some method, and these methods may bias the results, allowing the antibodies to access certain brain areas and not others that are more relevant to the human disease (128, 129).
Passive transfer of antibodies is ideal to investigate the downstream mechanisms by which the patient antibodies affect their targets with possible secondary effects, but by itself, it does not appear to enlist cellular mechanisms that might be important in the human condition. Thus, it does not provide insight into the immunological mechanisms behind the generation of the antibodies nor the immunological effectors. For instance, the poor ability of human IgG to fix mouse complement is a limitation if complement activation plays a relevant part in the disease. Overall, the immune cells and the Fc receptors relevant for the human immune response might be different in animal models due to the use of alternative pathways, different effectors, and different cellular receptor affinities (130–132). Future passive transfer studies of patient-derived immune cells into humanized models or studies in non-human primates might help define the involvement of specific immune cells in the pathogenesis of these disorders.
Active immunization models could be helpful in overcoming some of these limitations and could also be more helpful in studying the effector immune mechanisms, but few studies have used this approach to date. Moreover, using peptide sequences for immunization is unlikely to generate the most appropriate pathogenic antibodies if the natural disease recognizes the native membrane protein rather than peptide or polypeptide sequences.
It is also important to note that the failure to reproduce some clinical features observed in patients might be related to the experimental approach or timing of protocols. For example, as shown for NMDAR-Ab, the presence of spontaneous seizures could be overlooked in the absence of continuous EEG monitoring (42). Similarly, antibodies may manifest their maximum effects up to 18 days after CSF infusion (38). Behavioral testing has to be carefully tailored and should take into account the effects of habituation and test repetition.
Future research and refinement of these animal models require a collaborative approach and sharing of optimal methods. Effective and reliable preclinical testing of novel treatments demands rigorous and reproducible protocols that not only allow study of the underlying neurobiology but also facilitate therapeutic studies with rapid translation to the clinic.
Author Contributions
MG: conception and drafting of the manuscript. SW: drafting, editing, and review of the manuscript. AV: conception, drafting, editing, and review of the manuscript.
Funding
SW was funded by an Epilepsy Research UK Postdoctoral Fellowship (F1601) and a Wellcome Trust Clinical Research Career Development Fellowship (216613/Z/19/Z) during the course of this work.
Conflict of Interest
The University of Oxford and AV hold patents for LGI1 and CASPR2 antibody tests, licensed to Euroimmun AG. AV receives a proportion of royalties.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SB declared a past co-authorship with one of the authors AV to the handling Editor.
Acknowledgments
We are very grateful to the Department of Biomedical and Neuromotor Sciences (MG) for its support and to Dr. Ester Coutinho for her helpful comments on the manuscript.
References
1. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. (2016) 15:391–404. doi: 10.1016/S1474-4422(15)00401-9
2. Giannoccaro MP, Crisp SJ, Vincent A. Antibody-mediated central nervous system diseases. Brain Neurosci Adv. (2018). doi: 10.1177/2398212818817497. [Epub ahead of print].
3. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain. (2012) 135(Pt 5):1622–38. doi: 10.1093/brain/aws082
4. Titulaer M. J., McCracken L, Gabilondo I, Armangué T., Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. (2013) 12:157–65. doi: 10.1016/S1474-4422(12)70310-1
5. Crisp SJ, Kullmann DM, Vincent A. Autoimmune synaptopathies. Nat Rev Neurosci. (2016) 17:103–17. doi: 10.1038/nrn.2015.27
6. Steiner J, Teegen B, Schiltz K, Bernstein HG, Stoecker W, Bogerts B. Prevalence of N-methyl-D-aspartate receptor autoantibodies in the peripheral blood: healthy control samples revisited. JAMA Psychiatr. (2014) 71:838–9. doi: 10.1001/jamapsychiatry.2014.469
7. Zerche M, Weissenborn K, Ott C, Dere E, Asif AR, Worthmann H, et al. Preexisting serum autoantibodies against the NMDAR subunit NR1 modulate evolution of lesion size in acute ischemic stroke. Stroke. (2015) 46:1180–6. doi: 10.1161/STROKEAHA.114.008323
8. Castillo-Gomez E, Kastner A, Steiner J, Schneider A, Hettling B, Poggi G, et al. The brain as immunoprecipitator of serum autoantibodies against N-Methyl-D-aspartate receptor subunit NR1. Ann Neurol. (2016) 79:144–51. doi: 10.1002/ana.24545
9. Pan H, Oliveira B, Saher G, Dere E, Tapken D, Mitjans M, et al. Uncoupling the widespread occurrence of anti-NMDAR1 autoantibodies from neuropsychiatric disease in a novel autoimmune model. Mol Psychiatr. (2019) 24:1489–501. doi: 10.1038/s41380-017-0011-3
10. Castillo-Gomez E, Oliveira B, Tapken D, Bertrand S, Klein-Schmidt C, Pan H, et al. All naturally occurring autoantibodies against the NMDA receptor subunit NR1 have pathogenic potential irrespective of epitope and immunoglobulin class. Mol Psychiatr. (2016) 22:1776–8. doi: 10.1038/mp.2016.125
11. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. (2010) 30:5866–75. doi: 10.1523/JNEUROSCI.0167-10.2010
12. Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky's postulates revisited). Immunol Today. (1993) 14:426–30. doi: 10.1016/0167-5699(93)90244-F
13. Pakozdy A, Halasz P, Klang A, Bauer J, Leschnik M, Tichy A, et al. Suspected limbic encephalitis and seizure in cats associated with voltage-gated potassium channel (VGKC) complex antibody. J Vet Intern Med. (2013) 27:212–4. doi: 10.1111/jvim.12026
14. Pruss H, Leubner J, Wenke NK, Czirjak GA, Szentiks CA, Greenwood AD. Anti-NMDA receptor encephalitis in the polar bear (Ursus maritimus) knut. Sci Rep. (2015) 5:12805. doi: 10.1038/srep12805
15. Yu X, Petersen F. A methodological review of induced animal models of autoimmune diseases. Autoimmun Rev. (2018) 17:473–9. doi: 10.1016/j.autrev.2018.03.001
16. Toyka KV, Drachman DB, Griffin DE, Pestronk A, Winkelstein JA, Fishbeck KH, et al. Myasthenia gravis study of humoral immune mechanisms by passive transfer to mice. N Engl J Med. (1977) 296:125–31. doi: 10.1056/NEJM197701202960301
17. Phillips WD, Vincent A. Pathogenesis of myasthenia gravis: update on disease types, models, and mechanisms. F1000Res. (2016) 5:F1000. doi: 10.12688/f1000research.8206.1
18. Kusner LL, Losen M, Vincent A, Lindstrom J, Tzartos S, Lazaridis K, et al. Guidelines for pre-clinical assessment of the acetylcholine receptor–specific passive transfer myasthenia gravis model-Recommendations for methods and experimental designs. Exp Neurol. (2015) 270:3–10. doi: 10.1016/j.expneurol.2015.02.025
19. Berman PW, Patrick J. Linkage between the frequency of muscular weakness and loci that regulate immune responsiveness in murine experimental Myasthenia gravis. J Exp Med. (1980) 152:507–20. doi: 10.1084/jem.152.3.507
20. Berman PW, Patrick J. Experimental myasthenia gravis. A murine system. J Exp Med. (1980) 151:204–23. doi: 10.1084/jem.151.1.204
21. Newsom-Davis J, Wilson SG, Vincent A, Ward CD. Long-term effects of repeated plasma exchange in myasthenia gravis. Lancet. (1979) 1:464–8.
22. Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology. (1976) 26:1054–9. doi: 10.1212/WNL.26.11.1054
23. Vincent A, Newsom-Davis J. Acetylcholine receptor antibody characteristics in myasthenia gravis. I Patients with generalized myasthenia or disease restricted to ocular muscles. Clin Exp Immunol. (1982) 49:257–65.
24. Engel AG, Lambert EH, Howard FM. Immune complexes (IgG and C3) at the motor end-plate in myasthenia gravis: ultrastructural and light microscopic localization and electrophysiologic correlations. Mayo Clin Proc. (1977) 52:267–80.
25. Scadding GK, Vincent A, Newsom-Davis J, Henry K. Acetylcholine receptor antibody synthesis by thymic lymphocytes: correlation with thymic histology. Neurology. (1981) 31:935–43. doi: 10.1212/WNL.31.8.935
26. Wolfe GI, Kaminski HJ, Sonnett JR, Aban IB, Kuo HC, Cutter GR. Randomized trial of thymectomy in myasthenia gravis. J Thorac Dis. (2016) 8:E1782–3. doi: 10.21037/jtd.2016.12.80
27. Simpson JA, Myasthenia Gravis A. New hypothesis. Scottish Med J. (1960) 5:419–36. doi: 10.1177/003693306000501001
28. Vincent A, Newland C, Brueton L, Beeson D, Riemersma S, Huson SM, et al. Arthrogryposis multiplex congenita with maternal autoantibodies specific for a fetal antigen. Lancet. (1995) 346:24–5. doi: 10.1016/S0140-6736(95)92652-6
29. Rodríguez Cruz PM, Palace J, Beeson D. The neuromuscular junction and wide heterogeneity of congenital myasthenic syndromes. Int J Mol Sci. (2018) 19:1677. doi: 10.3390/ijms19061677
30. Engel AG. The immunopathological basis of acetylcholine receptor deficiency in myasthenia gravis. Prog Brain Res. (1979) 49:423–34.
31. Vincent A. Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol. (2002) 2:797–804. doi: 10.1038/nri916
32. Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. (2014) 6:263ra158. doi: 10.1126/scitranslmed.3009759
33. Banks WA, Gray AM, Erickson MA, Salameh TS, Damodarasamy M, Sheibani N, et al. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J Neuroinflamm. (2015) 12:223. doi: 10.1186/s12974–015-0434–1
34. Saunders NR, Liddelow SA, Dziegielewska KM. Barrier mechanisms in the developing brain. Front Pharmacol. (2012) 3:46. doi: 10.3389/fphar.2012.00046
35. Morel E, Eymard B, Vernet-der Garabedian B, Pannier C, Dulac O, Bach JF. Neonatal Myasthenia gravis: a new clinical and immunologic appraisal on 30 cases. Neurology. (1988) 38:138–42. doi: 10.1212/WNL.38.1.138
36. Jacobson L, Polizzi A, Morriss-Kay G, Vincent A. Plasma from human mothers of fetuses with severe arthrogryposis multiplex congenita causes deformities in mice. J Clin Invest. (1999) 103:1031–8. doi: 10.1172/JCI5943
37. Jones BE, Tovar KR, Goehring A, Jalali-Yazdi F, Okada NJ, Gouaux E, et al. Autoimmune receptor encephalitis in mice induced by active immunization with conformationally stabilized holoreceptors. Sci Transl Med. (2019) 11:eaaw0044. doi: 10.1126/scitranslmed.aaw0044
38. Planagum à J., Leypoldt F, Mannara F, Gutiérrez-Cuesta J, Martín-García E, Aguilar E, et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain. (2015) 138(Pt 1):94–109. doi: 10.1093/brain/awu310
39. Wright S, Hashemi K, Stasiak L, Bartram J, Lang B, Vincent A, et al. Epileptogenic effects of NMDAR antibodies in a passive transfer mouse model. Brain. (2015) 138(Pt 11):3159–67. doi: 10.1093/brain/awv257
40. Li Y, Tanaka K, Wang L, Ishigaki Y, Kato N. Induction of memory deficit in mice with chronic exposure to cerebrospinal fluid from patients with anti-N-methyl-D-aspartate receptor encephalitis. Tohoku J Exp Med. (2015) 237:329–38. doi: 10.1620/tjem.237.329
41. Planagumà J., Haselmann H, Mannara F, Petit-Pedrol M, Grünewald B, Aguilar E, et al. Ephrin-B2 prevents N-methyl-D-aspartate receptor antibody effects on memory and neuroplasticity. Ann Neurol. (2016) 80:388–400. doi: 10.1002/ana.24721
42. Taraschenko O, Fox HS, Pittock SJ, Zekeridou A, Gafurova M, Eldridge E, et al. A mouse model of seizures in anti-N-methyl-d-aspartate receptor encephalitis. Epilepsia. (2019) 60:452–63. doi: 10.1111/epi.14662
43. Malviya M, Barman S, Golombeck KS, Planagumà J, Mannara F, Strutz-Seebohm N, et al. NMDAR encephalitis: passive transfer from man to mouse by a recombinant antibody. Ann Clin Transl Neurol. (2017) 4:768–83. doi: 10.1002/acn3.444
44. Sharma R, Al-Saleem FH, Panzer J, Lee J, Puligedda RD, Felicori LF, et al. Monoclonal antibodies from a patient with anti-NMDA receptor encephalitis. Ann Clin Transl Neurol. (2018) 5:935–51. doi: 10.1002/acn3.592
45. Linnoila J, Pulli B, Armangué T., Planagumà J., Narsimhan R, Schob S, et al. Mouse model of anti-NMDA receptor post-herpes simplex encephalitis. Neurol Neuroimmunol Neuroinflamm. (2018) 6:e529. doi: 10.1212/NXI.0000000000000529
46. Manto M, Dalmau J, Didelot A, Rogemond V, Honnorat J. In vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: further evidence of synaptic glutamatergic dysfunction. Orphanet J Rare Dis. (2010) 5:31. doi: 10.1186/1750-1172-5-31
47. Würdemann T, Kersten M, Tokay T, Guli X, Kober M, Rohde M, et al. Stereotactic injection of cerebrospinal fluid from anti-NMDA receptor encephalitis into rat dentate gyrus impairs NMDA receptor function. Brain Res. (2016) 1633:10–8. doi: 10.1016/j.brainres.2015.12.027
48. Blome R, Bach W, Guli X, Blome R, Bach W, Guli X, et al. Differentially altered NMDAR dependent and independent long-term potentiation in the CA3 subfield in a model of anti-NMDAR encephalitis. Front Synaptic Neurosci. (2018) 10:26. doi: 10.3389/fnsyn.2018.00026
49. Kersten M, Rabbe T, Blome R, Porath K, Sellmann T, Bien CG, et al. Novel object recognition in rats with NMDAR dysfunction in CA1 after stereotactic injection of anti-NMDAR encephalitis cerebrospinal fluid. Front Neurol. (2019) 10:586. doi: 10.3389/fneur.2019.00586
50. Dawes JM, Weir GA, Middleton SJ, Patel R, Chisholm KI, Pettingill P, et al. Immune or genetic-mediated disruption of CASPR2 causes pain hypersensitivity due to enhanced primary afferent excitability. Neuron. (2018) 97:806–822.e10. doi: 10.1016/j.neuron.2018.01.033
51. Giannoccaro MP, Menassa DA, Jacobson L, Coutinho E, Prota G, Lang B, et al. Behaviour and neuropathology in mice injected with human contactin-associated protein 2 antibodies. Brain. (2019) 142:2000–12. doi: 10.1093/brain/awz119
52. Fernandes D, Santos SD, Coutinho E, Whitt JL, Beltrão N, Rondão T, et al. Disrupted AMPA receptor function upon genetic- or antibody-mediated loss of autism-associated CASPR2. Cereb Cortex. (2019) 29:bhz032. doi: 10.1093/cercor/bhz032
53. Petit-Pedrol M, Sell J, Planagumà J., Mannara F, Radosevic M, Haselmann H, et al. LGI1antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain. (2018) 141:3144–59. doi: 10.1093/brain/awy253
54. Haselmann H, Mannara F, Werner C, Planagumà J., Miguez-Cabello F, Schmidl L, et al. Human autoantibodies against the AMPA receptor subunit GluA2 induce receptor reorganization and memory dysfunction. Neuron. (2018) 100:91–105.e9. doi: 10.1016/j.neuron.2018.07.048
55. Crisp SJ, Dixon CL, Jacobson L, Chabrol E, Irani SR, Leite MI, et al. Glycine receptor autoantibodies disrupt inhibitory neurotransmission. Brain. (2019) 142:3398–410. doi: 10.1093/brain/awz297
56. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. (2011) 10:63–74. doi: 10.1016/S1474-4422(10)70253-2
57. Ladépêche L, Planagumà J., Thakur S, Suárez I, Hara M, Borbely JS, et al. NMDA receptor autoantibodies in autoimmune encephalitis cause a subunit-specific nanoscale redistribution of NMDA receptors. Cell Rep. (2018) 23:3759–68. doi: 10.1016/j.celrep.2018.05.096
58. Mikasova L, De Rossi P, Bouchet D, Georges F, Rogemond V, Didelot A, et al. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain. (2012). 135(Pt 5):1606–21. doi: 10.1093/brain/aws092
59. Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med. (2018) 378:840–51. doi: 10.1056/NEJMra1708712
60. Makuch M, Wilson R, Al-Diwani A, Varley J, Kienzler AK, Taylor J, et al. N-methyl-D-aspartate receptor antibody production from germinal center reactions: therapeutic implications. Ann Neurol. (2018) 83:553–61. doi: 10.1002/ana.25173
61. Armangue T, Spatola M, Vlagea A, Mattozzi S, Cárceles-Cordon M, Martinez-Heras E, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol. (2018) 17:760–72. doi: 10.1016/S1474-4422(18)30244-8
62. Ohba C, Shiina M, Tohyama J, Haginoya K, Lerman-Sagie T, Okamoto N, et al. GRIN1 mutations cause encephalopathy with infantile-onset epilepsy, and hyperkinetic and stereotyped movement disorders. Epilepsia. (2015) 56:841–8. doi: 10.1111/epi.12987
63. Redin C, Gérard B, Lauer J, Herenger Y, Muller J, Quartier A, et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet. (2014) 51:724–36. doi: 10.1136/jmedgenet-2014-102554
64. Lemke JR, Geider K, Helbig KL, Heyne HO, Schütz H, Hentschel J, et al. Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology. (2016) 86:2171–8. doi: 10.1212/WNL.0000000000002740
65. Nakazawa K, Quirk MC, Chitwood RA, Watanabe M, Yeckel MF, Sun LD, et al. Requirement for hippocampal CA3 NMDA receptors in associative memory recall. Science. (2002) 297:211–8. doi: 10.1126/science.1071795
66. Shimizu E, Tang YP, Rampon C, Tsien JZ. NMDA receptor-dependent synaptic reinforcement as a crucial process for memory consolidation. Science. 290:1170–4. doi: 10.1126/science.290.5494.1170
67. Liba Z, Kayserova J, Elisak M, Marusic P, Nohejlova H, Hanzalova J, et al. Anti-N-methyl-D-aspartate receptor encephalitis: the clinical course in light of the chemokine and cytokine levels in cerebrospinal fluid. J Neuroinflammation. (2016) 13:55. doi: 10.1186/s12974-016-0507-9
68. Finke C, Kopp UA, Pajkert A, Behrens JR, Leypoldt F, Wuerfel JT, et al. Structural hippocampal damage following anti-N-Methyl-D-aspartate receptor encephalitis. Biol Psychiatr. (2016) 79:727–34. doi: 10.1016/j.biopsych.2015.02.024
69. Gleichman AJ, Spruce LA, Dalmau J, Seeholzer SH, Lynch DR. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci. (2012) 32:11082–94. doi: 10.1523/JNEUROSCI.0064-12.2012
70. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain. (2010) 133:2734–48. doi: 10.1093/brain/awq213
71. Liguori R, Vincent A, Clover L, Avoni P, Plazzi G, Cortelli P, et al. Morvan's syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain. (2001) 124(Pt 12):2417–26. doi: 10.1093/brain/124.12.2417
72. Provini F, Marconi S, Amadori M, Guaraldi P, Pierangeli G, Cortelli P, et al. Morvan chorea and agrypnia excitata: when video-polysomnographic recording guides the diagnosis. Sleep Med. (2011) 12:1041–3. doi: 10.1016/j.sleep.2011.05.005
73. Patterson KR, Dalmau J, Lancaster E. Mechanisms of Caspr2 antibodies in autoimmune encephalitis and neuromyotonia. Ann Neurol. (2018) 83:40–51. doi: 10.1002/ana.25120
74. Friedman JI, Vrijenhoek T, Markx S, Janssen IM, van der Vliet WA, Faas BH, et al. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatr. (2008) 13:261–6. doi: 10.1038/sj.mp.4002049
75. Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. (2011) 147:235–46. doi: 10.1016/j.cell.2011.08.040
76. Körtvelyessy P, Bauer J, Stoppel CM, Brück W, Gerth I, Vielhaber S, et al. Complement-associated neuronal loss in a patient with CASPR2 antibody-associated encephalitis. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e75. doi: 10.1212/NXI.0000000000000075
77. Sundal C, Vedeler C, Miletic H, Andersen O. Morvan syndrome with Caspr2 antibodies. Clin Autopsy Rep J Neurol Sci. (2017) 372:453–5. doi: 10.1016/j.jns.2016.10.046
78. Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. (2011) 69:892–900. doi: 10.1002/ana.22307
79. Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. (2006) 313:1792–5. doi: 10.1126/science.1129947
80. Owuor K, Harel NY, Englot DJ, Hisama F, Blumenfeld H, Strittmatter SM. LGI1-associated epilepsy through altered ADAM23-dependent neuronal morphology. Mol Cell Neurosci. (2009) 42:448–57. doi: 10.1016/j.mcn.2009.09.008
81. Schulte U, Thumfart JO, Klöcker N, Sailer CA, Bildl W, Biniossek M, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. (2006) 49:697–706. doi: 10.1016/j.neuron.2006.01.033
82. Ohkawa T, Fukata Y, Yamasaki M, Miyazaki T, Yokoi N, Takashima H, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci. (2013) 33:18161–74. doi: 10.1523/JNEUROSCI.3506-13.2013
83. Thompson J, Bi M, Murchison AG, Makuch M, Bien CG, Chu K, et al. Faciobrachial dystonic seizures study, group, the importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain. (2018) 141:348–56. doi: 10.1093/brain/awx323
84. Malter MP, Frisch C, Schoene-Bake JC, Helmstaedter C, Wandinger KP, Stoecker W, et al. Outcome of limbic encephalitis with VGKC-complex antibodies: relation to antigenic specificity. J Neurol. (2014) 261:1695–705. doi: 10.1007/s00415-014-7408-6
85. Ariño H, Armangué T., Petit-Pedrol M, Sabater L, Martinez-Hernandez E, Hara M, et al. Anti-LGI1-associated cognitive impairment: Presentation and long-term outcome. Neurology. (2016) 87:759–65. doi: 10.1212/WNL.0000000000003009
86. Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Martinelli Boneschi F, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. (2002) 30:335–41. doi: 10.1038/ng832
87. Dazzo E, Leonardi E, Belluzzi E, Malacrida S, Vitiello L, Greggio E, et al. Secretion-positive LGI1 mutations linked to lateral temporal epilepsy impair binding to ADAM22 and ADAM23 receptors. PLoS Genet. (2016) 12:e1006376. doi: 10.1371/journal.pgen.1006376
88. Chabrol E, Navarro V, Provenzano G, Cohen I, Dinocourt C, Rivaud-Péchoux S, et al. Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain. (2010) 133:2749–62. doi: 10.1093/brain/awq171
89. Fukata Y, Lovero KL, Iwanaga T, Watanabe A, Yokoi N, Tabuchi K, et al. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci USA. (2010) 107:3799–804. doi: 10.1073/pnas.0914537107
90. Yu YE, Wen L, Silva J, Li Z, Head K, Sossey-Alaoui K, et al. Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum Mol Genet. (2010) 19:1702–11. doi: 10.1093/hmg/ddq047
91. Boillot M, Huneau C, Marsan E, Lehongre K, Navarro V, Ishida S, et al. Glutamatergic neuron-targeted loss of LGI1 epilepsy gene results in seizures. Brain. (2014) 137(Pt 11):2984–96. doi: 10.1093/brain/awu259
92. Baulac S, Ishida S, Mashimo T, Boillot M, Fumoto N, Kuwamura M, et al. A rat model for LGI1-related epilepsies. Hum Mol Genet. (2012) 21:3546–57. doi: 10.1093/hmg/dds184
93. Seagar M, Russier M, Caillard O, Maulet Y, Fronzaroli-Molinieres L, De San Feliciano M, et al. LGI1 tunes intrinsic excitability by regulating the density of axonal Kv1 channels. Proc Natl Acad Sci USA. (2017) 114:7719–24. doi: 10.1073/pnas.1618656114
94. Pakozdy A, Glantschnigg U, Leschnik M, Hechinger H, Moloney T, Lang B, et al. EEG-confirmed epileptic activity in a cat with VGKC-complex/LGI1 antibody-associated limbic encephalitis. Epileptic Disord. (2014) 16:116–20. doi: 10.1684/epd.2014.0635
95. Klang A, Schmidt P, Kneissl S, Bag ó Z., Vincent A, Lang B, et al. IgG and complement deposition and neuronal loss in cats and humans with epilepsy and voltage-gated potassium channel complex antibodies. J Neuropathol Exp Neurol. (2014) 73:403–13. doi: 10.1097/NEN.0000000000000063
96. Tröscher AR, Klang A, French M, Quemada-Garrido L, Kneissl SM, Bien CG, et al. Selective limbic blood-brain barrier breakdown in a feline model of limbic encephalitis with LGI1 antibodies. Front Immunol. (2017) 8:1364. doi: 10.3389/fimmu.2017.01364
97. Lalic T, Pettingill P, Vincent A, Capogna M. Human limbic encephalitis serum enhances hippocampal mossy fiber-CA3 pyramidal cell synaptic transmission. Epilepsia. (2011) 52:121–31. doi: 10.1111/j.1528-1167.2010.02756.x
98. Ratelade J, Verkman AS. Inhibitor(s) of the classical complement pathway in mouse serum limit the utility of mice as experimental models of neuromyelitis optica. Mol Immunol. (2014) 62:104–13. doi: 10.1016/j.molimm.2014.06.003
99. Höftberger R, van Sonderen A, Leypoldt F, Houghton D, Geschwind M, Gelfand J, et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology. (2015) 84:2403–12. doi: 10.1212/WNL.0000000000001682
100. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. (2009) 65:424–34. doi: 10.1002/ana.21589
101. Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. (2007) 23:613–43. doi: 10.1146/annurev.cellbio.23.090506.123516
102. Peng X, Hughes EG, Moscato EH, Parsons TD, Dalmau J, Balice-Gordon RJ. Cellular plasticity induced by anti-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann Neurol. (2015) 77:381–98. doi: 10.1002/ana.24293
103. Geisheker MR, Heymann G, Wang T, Coe BP, Turner TN, Stessman HAF, et al. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nat Neurosci. (2017) 20:1043–51. doi: 10.1038/nn.4589
104. Salpietro V, Dixon CL, Guo H, Bello OD, Vandrovcova J, Efthymiou S, et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat Commun. (2019) 10:3094. doi: 10.1038/s41467-019-10910-w
105. Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. (1999) 284:1805–11. doi: 10.1126/science.284.5421.1805
106. Romberg C, Raffel J, Martin L, Sprengel R, Seeburg PH, Rawlins JN, et al. Induction and expression of GluA1 (GluR-A)-independent LTP in the hippocampus. Eur J Neurosci. (2009) 29:1141–52. doi: 10.1111/j.1460-9568.2009.06677.x
107. Mack V, Burnashev N, Kaiser KM, Rozov A, Jensen V, Hvalby O, et al. Conditional restoration of hippocampal synaptic potentiation in Glur,-A-,deficient mice. Science. (2001) 292:2501–4. doi: 10.1126/science.1059365
108. Barkus C, Sanderson DJ, Rawlins JN, Walton ME, Harrison PJ, Bannerman DM. What causes aberrant salience in schizophrenia? A role for impaired short-term habituation and the GRIA1 (GluA1) AMPA receptor subunit. Mol Psychiatr. (2014) 19:1060–70. doi: 10.1038/mp.2014.91
109. Sanderson DJ, Good MA, Skelton K, Sprengel R, Seeburg PH, Rawlins JN, et al. Enhanced long-term and impaired short-term spatial memory in GluA1 AMPA receptor subunit knockout mice: evidence for a dual-process memory model. Learn Mem. (2009) 16:379–86. doi: 10.1101/lm.1339109
110. Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, et al. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. (1996) 17:945–56. doi: 10.1016/S0896-6273(00)80225-1
111. Jia Z, Lu YM, Agopyan N, Roder J. Gene targeting reveals a role for the glutamate receptors mGluR5 and GluR2 in learning and memory. Physiol Behav. (2001) 73:793–802. doi: 10.1016/S0031-9384(01)00516-9
112. Inta D, Vogt MA, Elkin H, Weber T, Lima-Ojeda JM, Schneider M, et al. Phenotype of mice with inducible ablation of GluA1 AMPA receptors during late adolescence: relevance for mental disorders. Hippocampus. (2014) 24:424–35. doi: 10.1002/hipo.22236
113. Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, et al. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. (2009) 62:254–68. doi: 10.1016/j.neuron.2009.02.027
114. Shimshek DR, Bus T, Kim J, Mihaljevic A, Mack V, Seeburg PH, et al. Enhanced odor discrimination and impaired olfactory memory by spatially controlled switch of AMPA receptors. PLoS Biol. (2005) 3:e354. doi: 10.1371/journal.pbio.0030354
115. Shimshek DR, Jensen V, Celikel T, Geng Y, Schupp B, Bus T, et al. Forebrain-specific glutamate receptor B deletion impairs spatial memory but not hippocampal field long-term potentiation. J Neurosci. (2006) 26:8428–40. doi: 10.1523/JNEUROSCI.5410-05.2006
116. Riemersma S, Vincent A, Beeson D, Newland C, Hawke S, Vernet-der Garabedian B, et al. Association of arthrogryposis multiplex congenita with maternal antibodies inhibiting fetal acetylcholine receptor function. J Clin Invest. (1996) 98:2358–63. doi: 10.1172/JCI119048
117. Dalton P, Deacon R, Blamire A, Pike M, McKinlay I, Stein J, et al. Maternal neuronal antibodies associated with autism and a language disorder. Ann Neurol. (2003) 53:533–7. doi: 10.1002/ana.10557
118. Edmiston E, Ashwood P, Van de Water J. Autoimmunity, autoantibodies, and autism spectrum disorder. Biol Psychiatr. (2017) 81:383–90. doi: 10.1016/j.biopsych.2016.08.031
119. Rodenas-Cuadrado P, Ho J, Vernes SC. Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet. (2014) 22:171–8. doi: 10.1038/ejhg.2013.100
120. Burnashev N, Szepetowski P. NMDA receptor subunit mutations in neurodevelopmental disorders. Curr Opin Pharmacol. (2015) 20:73–82. doi: 10.1016/j.coph.2014.11.008
121. Coutinho E, Jacobson L, Pedersen MG, Benros ME, Nørgaard-Pedersen B, Mortensen PB, et al. CASPR2 autoantibodies are raised during pregnancy in mothers of children with mental retardation and disorders of psychological development but not autism. J Neurol Neurosurg Psychiatr. (2017) 88:718–21. doi: 10.1136/jnnp-2016-315251
122. Coutinho E, Menassa DA, Jacobson L, West SJ, Domingos J, Moloney TC, et al. Persistent microglial activation and synaptic loss with behavioral abnormalities in mouse offspring exposed to CASPR2-antibodies in utero. Acta Neuropathol. (2017) 134:567–83. doi: 10.1007/s00401-017-1751-5
123. Brimberg L, Mader S, Jeganathan V, Berlin R, Coleman TR, Gregersen PK, et al. Caspr2-reactive antibody cloned from a mother of an ASD child mediates an ASD-like phenotype in mice. Mol Psychiatr. (2016) 21:1663–71. doi: 10.1038/mp.2016.165
124. Jurek B, Chayka M, Kreye J, Lang K, Kraus L, Fidzinski P, et al. Human gestational N-methyl-d-aspartate receptor autoantibodies impair neonatal murine brain function. Ann Neurol. (2019) 86:656–70. doi: 10.1002/ana.25552
125. Shi YC, Chen XJ, Zhang HM, Wang Z, Du DY. Anti-N-Methyl-d-Aspartate receptor (NMDAR) encephalitis during pregnancy: Clinical analysis of reported cases. Taiwan J Obstet Gynecol. (2017) 56:315–9. doi: 10.1016/j.tjog.2017.04.009
126. Chourasia N, Watkins MW, Lankford JE, Kass JS, Kamdar A. An infant born to a mother with anti-N-methyl-d-aspartate receptor encephalitis. Pediatr Neurol. (2018) 79:65–8. doi: 10.1016/j.pediatrneurol.2017.11.010
127. Kalam S, Baheerathan A, McNamara C, Singh-Curry V. Anti-NMDAR encephalitis complicating pregnancy. Pract Neurol. (2019) 19:131–5. doi: 10.1136/practneurol-2018-002042
128. Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B. Immunity and behavior: antibodies alter emotion. Proc Natl Acad Sci USA. (2006) 103:678–83. doi: 10.1073/pnas.0510055103
129. Kowal C, DeGiorgio LA, Nakaoka T, Hetherington H, Huerta PT, Diamond B, et al. Cognition and immunity; antibody impairs memory. Immunity. (2004) 21:179–88. doi: 10.1016/j.immuni.2004.07.011
130. Stavnezer J, Amemiya CT. Evolution of isotype switching. Semin Immunol. (2004) 16:257–75. doi: 10.1016/j.smim.2004.08.005
131. Kacskovics I. Fc receptors in livestock species. Vet Immunol Immunopathol. (2004) 102:351–62. doi: 10.1016/j.vetimm.2004.06.008
Keywords: animal models, neuronal surface antibodies, passive transfer, maternal transfer, active immunization
Citation: Giannoccaro MP, Wright SK and Vincent A (2020) In vivo Mechanisms of Antibody-Mediated Neurological Disorders: Animal Models and Potential Implications. Front. Neurol. 10:1394. doi: 10.3389/fneur.2019.01394
Received: 11 October 2019; Accepted: 19 December 2019;
Published: 05 February 2020.
Edited by:
Marc J. Ruitenberg, University of Queensland, AustraliaReviewed by:
Enrico Castroflorio, Medical Research Council Harwell (MRC), United KingdomMichael Levy, Massachusetts General Hospital, Harvard Medical School, United States
Stefan Blum, University of Queensland, Australia
Copyright © 2020 Giannoccaro, Wright and Vincent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Pia Giannoccaro, mpgiannoccaro@gmail.com