 Jia Wei
Jia Wei Zihao Xu
Zihao Xu- Department of Urology, Children’s Hospital, Zhejiang University School of Medicine, Hangzhou, China
Renal fibrosis causes structural and functional impairment of the kidney, which is a dominant component of chronic kidney disease. Recently, a novel mechanism, macrophage-to-myofibroblast transition (MMT), has been identified as a crucial component in renal fibrosis as a response to chronic inflammation. It is a process by which bone marrow-derived macrophages differentiate into myofibroblasts during renal injury and promote renal fibrosis. Here, we summarized recent evidence and mechanisms of MMT in renal fibrosis. Understanding this phenomenon and its underlying signal pathway would be beneficial to find therapeutic targets for renal fibrosis in chronic kidney disease.
Introduction
Chronic kidney disease (CKD) affects around 11.7%–18% of the global population (1). While therapies can significantly delay the progression of CKD, the long-term prognosis remains worrisome (1, 2). Renal fibrosis is a dominant component of CKD, which is also the typical final stage of majority of the chronic and progressive nephropathies. It will eventually result in kidney failure (3). The renal fibrosis process consists of five steps, as follows (4): firstly, activation of renal tubular epithelia due to inflammation and infiltration of plentiful monocytes/macrophages into the kidney; secondly, excessive production of fibrogenic associated cytokines, growth factors, and other profibrogenic factors; thirdly, imbalance in the synthesis and degradation of the extracellular matrix (ECM) and the overdeposition and accumulation of ECM in the renal interstitium, which is a major stage of renal injury in structure and function; fourthly, mesenchymal changes in renal intrinsic cells and the decrease in the number of renal intrinsic cells; and finally, renal microangiopathy leading to ischemia and anoxia of the renal interstitium.
In renal fibrosis, activated myofibroblasts are a critical matrix-secreting cell type that plays a key role in ECM accumulation (5–7). Myofibroblasts are a heterogeneous population that may be derived from a variety of origins, including epithelia through epithelial-to-mesenchymal transition (EMT) (8, 9), endothelia through endothelial-to-mesenchymal transition (EndoMT) (10), and local fibroblast or pericyte proliferation (11). So far, literature indicates that monocytes/macrophages play an important role in the process of renal fibrosis, and the macrophage-to-myofibroblast transition (MMT) is recently discovered as another extrarenal genesis for myofibroblasts (12).
MMT is the transformation of macrophages into myofibroblasts in response to an inflammatory stimulation. The MMT cells are capable of producing collagen and are distinguished by the co-expression of a macrophage marker (CD68) and a myofibroblast marker [alpha smooth muscle actin (α-SMA)] (12). A study found that approximately 35% of the myofibroblasts in the unilateral ureteral obstruction (UUO) model of renal fibrosis originated from bone marrow cells (13). Another research discovered that 37% of myofibroblasts in the renal allograft were derived from the bone marrow (14). Thus, MMT acts a crucial role in the development of renal fibrosis. This review provides an update on current advancements in macrophages, with a particular emphasis on MMT-related mechanisms in renal fibrosis.
Macrophages and macrophage-to-myofibroblast transition inrenal fibrosis
Previously, it is believed that macrophages contribute to renal fibrosis indirectly by secreting cytokines and chemokines that attract and proliferate fibroblasts. It has been well-documented that macrophages are phenotypic heterogeneous. According to various microenvironments, macrophages can polarize into a pro-inflammatory M1 phenotype or anti-inflammatory M2 phenotype. M2 macrophages could secrete interleukin (IL)-10 and transforming growth factor-β (TGF-β) to stimulate myofibroblast proliferation. Furthermore, M2 macrophages could also express procollagen I that contributes to fibrosis (15). Infiltrating macrophages are the major source of TGF-β in glomerulonephritis, and TGF-β plays a crucial role in the development of renal fibrosis (16). Increased Ccl2 mRNA expression in infiltrating macrophages is associated with progressive renal fibrosis. The Ccl2 gene encodes monocyte chemoattractant protein 1 (MCP-1), which is an emerging biomarker in acute kidney injury and a crucial chemoattractant regulator of macrophages in the renal inflammatory response. A prospective multicenter cohort clinical study reported that a higher level of MCP-1 was associated with significant estimated glomerular filtration rate decline and an increased incidence of CKD and CKD progression (17).
So far, literature has revealed that bone marrow-derived macrophages (BMDMs) can also directly contribute to renal fibrosis through MMT. Both in vivo and in vitro, infiltrating macrophages at the renal glomerular level could transition into myofibroblasts through the MMT process in diabetic nephropathy, which finally leads to renal fibrosis (18). Biopsy tissues from renal fibrosis patients showed that only in acute and active renal fibrosis does the number of CD68+α-SMA+ MMT cells correlate with the total α-SMA+ myofibroblast population. In acute inflammatory lesions, CD68+ macrophages were infiltrated, but few CD68+α-SMA+ MMT cells were found. In kidneys with advanced sclerosis, the number of α-SMA+ myofibroblasts was increased, but both CD68+ macrophages and CD68+α-SMA+ MMT cells were reduced. However, MMT cells were not found in normal human kidneys or those affected by minimal change disease (12). Depletion of myeloid lineage cells prevented the appearance of MMT cells and substantially reduced myofibroblast accumulation and collagen deposition (12).
The origins of macrophage-to-myofibroblast transition in renal fibrosis
Urinary obstruction is a common cause of kidney fibrosis. Based on cell tracing in a murine UUO model, MMT cells were derived from bone marrow and were located in fibrosing mouse kidneys (19). The results found that more than 90% of monocytes/macrophages in the injured kidney came from bone marrow, while α-SMA was expressed in 22% of BMDMs, accounting for almost 80% of all α-SMA+ myofibroblasts in the injured kidney. Collagen I can be produced by active myofibroblasts, and the UUO mouse model revealed that about 70% of collagen I+ cells were transitioned from bone marrow monocytes/macrophages. Consequently, the infiltration of monocytes/macrophages into the injured kidney and the formation of a myofibroblastic phenotype are the principal contributions of cells to the development of renal fibrosis. In contrast, MMT cells were not detected in the kidneys of the sham-operated group. A study reported that BMDMs can develop into collagen-producing myofibroblasts during renal fibrosis in chimeric mice, supporting the idea that the myofibroblasts may originate from sources other than the renal system (13).
Renal chronic allograft rejection is also one of the major causes of renal fibrosis. BMDMs are a source of myofibroblasts in chronic allograft rejection and the development of renal interstitial fibrosis through the MMT process. Approximately half of the total myofibroblast population originated from BMDMs in human active chronic renal allograft injury. In patients with chronic renal allograft rejection, the number of CD68+ α-SMA+ MMT cells, not all myofibroblasts, was related to allograft functions and interstitial fibrosis severity (14). Lineage tracing in mice revealed that approximately 90% of the MMT cells originated in recipient bone marrow and contributed 37% of total myofibroblasts in the renal allograft. The results indicated that collagen-producing MMT cells accounted for an important component of the myofibroblast population in renal allograft injury and contribute to the development of fibrosis in renal allografts. It was established that the myeloid lineage of MMT cells comprised a significant proportion of the myofibroblast population and that the MMT process is a prevalent mechanism of renal interstitial fibrosis.
Regulation of macrophage-to-myofibroblast transition and inflammatory signal pathways
The classical signaling pathways in renal fibrosis include TGF-β1/Smad, nuclear factor-κB (NF-κB), Notch, Wnt, Hedgehog, phosphatidylinositol-3 kinase (PI3K/AKT), Janus kinase/signal transducers and activators of transcription (JAK-STAT), RHO/Rho coil kinase (ROCK), and tumor necrosis factor α (TNF-α) (20, 21). Some of these signal pathways are involved in renal fibrosis development by regulating the MMT process (Figure 1).
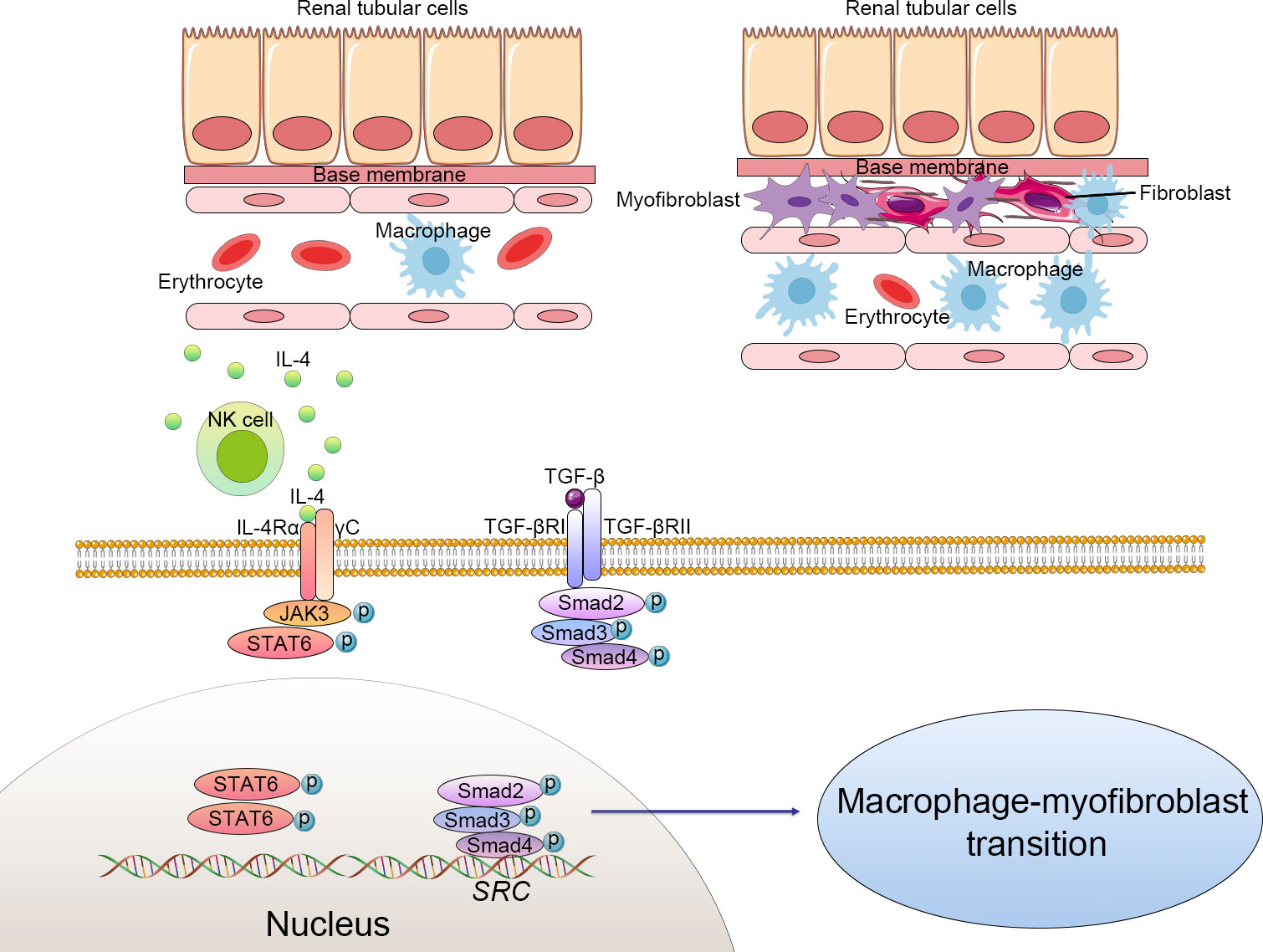
Figure 1 Cellular alteration and signaling pathway of macrophage-to-myofibroblast transition (MMT) in renal fibrosis. The macrophages derived from bone marrow differentiate to myofibroblasts, which produce excessive extracellular matrix and progressively cause structural and functional impairment of the kidney. Macrophage-to-myofibroblast transition plays a crucial role in renal fibrosis through multiple mechanisms, including triggering the transforming growth factor-β1 (TGF-β1)/Smad3 signaling pathways and natural killer T cells (NKT)/IL-4 signaling pathways. Activated NKT cells produce excessive IL-4, which subsequently combines with IL-4 receptor α and triggers the JAK3/STAT6 signaling to enhance the transformation of myofibroblasts. TGF-β binds to the TGF-β receptor complex and then phosphorylates the Smad family complex and finally activates the Src-centric gene network in bone marrow-derived macrophages to promote the MMT process. .
The TGF-β1-Smad3 signal pathway has been confirmed as the main mediator of the MMT process in BMDMs (14) (22). In brief, TGFR1 was activated by the interaction of TGF-β1 to its receptor TGF-β receptor 2 (TGFR2) (23). Activated TGFR1 induced the phosphorylation of Smad3 and Smad2, which then form complexes with Smad4. These complexes then enter the nucleus and activate the Src-centric gene network in BMDMs via transcriptional regulation, thereby promoting the MMT process in the fibrosing kidney. In this signaling pathway, Smad7 functions as a negative inhibitor, combining with activated TGFR1 and reducing the phosphorylation of Smad3 and Smad2. Smad7-deficient mice are more susceptible to renal fibrosis (24). The rerouting of TGF-β signaling from β-catenin/T-cell factor (TCF) to β-catenin/Forkhead Box O1(Foxo1) may affect the destiny of BMDMs. TGF-β signaling can be profibrotic or antifibrotic and anti-inflammatory, depending on which transfection factors are bound to β-catenin, TCF, or Foxo1 (25). An inhibitor of TCF (ICG-001) effectively suppressed the MMT process and reduced renal fibrosis. In vitro studies also showed that macrophage colony-stimulating factor (M-CSF) did not increase α-SMA and collagen I levels in Smad3+/+F4/80+ BMDMs. However, the expression of α-SMA and collagen Iα 1 could be detected in Smad3+/+ bone marrow macrophages incubated with TGF-β (26).
Smad3 is capable of directly binding specific Smad DNA-binding domains and promoting transcription, whereas Smad2 and Smad4 are transcriptional regulators but do not bind genetic elements. In the fibrotic kidney, the recruited Smad3−/− macrophages failed to differentiate into myofibroblasts. This had a functional effect, as mice transplanted with Smad3−/− bone marrow showed a remarkable reduction of collagen I deposition and α-SMA, indicating reduced renal fibrosis in the UUO kidney (26). Smad3 was required for the effective transition of recruited macrophages into collagen I+ α-SMA+ myofibroblasts in the injured kidney. Furthermore, the antifibrotic effect shown in Smad3−/− chimeric mice suggests that BMDMs contribute to the development of renal fibrosis via MMT, while the process is mediated by TGF-β/Smad3 signaling. Using Smad3 inhibitors, such as a combination of asiatic acid and naringenin, could reduce the activation of TGF-β/Smad3 signaling and inhibit renal fibrosis (27).
The TGF-β1-Smad3 signal pathway plays an important role in regulating the MMT process in renal fibrosis. Researchers determined that POU Class 4 Homeobox 1 (Pou4f1), a direct Smad3 target gene in the TGF-β1-induced MMT process in BMDMs, was a potential therapeutic target to prevent renal fibrosis. It was found that Smad3 could bind to the promoter of Pou4f1 gene and enhance gene transcription. Then, Pou4f1 promotes MMT-mediated tissue fibrosis through a fibrogenic gene network. Pou4f1 silencing effectively inhibited MMT-induced tissue fibrosis in vitro and in vivo (28). Those processes in Smad3 mediating renal fibrosis were via miR-29 and miR-200 downregulation and miR-21, miR-129, and miR-210 upregulation (20).
Src is a tyrosine kinase that could be activated by numerous cytokines and growth factors [e.g., TGF-β1 and epidermal growth factor (EGF)], which has been identified to be associated with fibrogenesis (29). Inhibition of Src suppressed the activation TGF-β receptor and epidermal growth factor receptor (EGFR) and protected against renal fibrosis (30). Src is upstream of Smad3 signaling and acts as a direct Smad3 target gene, which is specifically upregulated in macrophages during MMT. Bioinformatic analysis indicated that Src is a key gene in the network of differentially expressed genes in TGF-β1-induced MMT (29). Smad3 can directly activate the tyrosine kinase Src and trigger collagen production, which inhibits the degradation of ECM, renal interstitial fibroblast activation, and renal fibrosis (30). Furthermore, activated Src has been reported to promote endocytosis and aggravate renal interstitial fibrosis (31). However, the Src inhibitor lacks specificity, which severely limits its application in the treatment of renal fibrosis.
JAK3/STAT6 signaling was involved in MMT. STAT6-deficient mice or mice treated with a JAK3 inhibitor (CP690,550) had fewer accumulations of bone marrow-derived fibroblasts in the experimental obstructed kidney model and developed less renal fibrosis. Treatment with a JAK3 inhibitor significantly affected the transformation of myofibroblasts, the production of matrix proteins, and the development of fibrosis in the obstructed kidneys (32). Recent studies have shown that STAT6 could suppress M2 macrophage polarization and be involved in the monocyte-to-fibroblast transition in folic acid nephropathy (33). Furthermore, STAT6-specific inhibitor (AS 1517499) has been proven to prevent M2 macrophage polarization and transformation of myofibroblasts, which lead to a reduction of collagen deposition and ECM production in the injured kidney (34). This evidence supports that JAK3/STAT6 signaling takes a crucial part in renal fibrosis via regulating the MMT.
In human kidney fibrosis, the majority of CD68+α-SMA+ MMT cells expressed CD206, indicating a predominant M2 phenotype (26). MMT and the bone marrow-derived fibroblasts are activated by natural killer T cells (NKT)/IL-4 signaling. As has been well-documented, IL-4 promotes macrophages toward M2 polarization, which plays a crucial role in bone marrow-derived fibroblast activation and renal fibrosis (35). According to research, IL-4Rα disruption decreased the number of CD45+α-SMA+ and CD206+PDGFRβ+ cells and renal fibrosis induced by renal obstructive injury. In addition, activation of NKT cells exacerbated the accumulation of MMT in the process of renal fibrosis. Furthermore, administration of IL-4 to CD1d-deficient mice increased bone marrow-derived myofibroblasts, promoted the MMT process, and developed a fibrotic process in the injured kidney (5).
Chemokines induced in cells of the injured kidney are responsible for the recruitment of circulating monocytes/macrophages. To suppress excessive BMDM-to-myofibroblast transition, therapy options could target chemokines and their receptors, such as chemokine ligand 2 (CCL2), CCL5, CCL21, C-X-C motif chemokine ligand 6 (CXCL6), CXCL16, and phosphatase and tensin homolog (PTEN) (36–39).
Discussion and perspectives
In this review, we present the latest literature to identify that BMDMs promote renal fibrosis by both direct and indirect mechanisms. In a direct way, a part of BMDMs transition into myofibroblasts, which coexpress CD68 and α-SMA through MMT and produce collagen I to contribute to renal fibrosis. MMT plays a crucial part in the progression of renal fibrosis. The main signaling pathway of MMT is the TGF-β1-Smad3 signal pathway. Signaling-associated inhibitors had been proven to provide protection in animal models. The data suggest that MMT may be a therapeutic target for preventing renal fibrosis.
Author contributions
JW and XY designed the article. JW and ZX drafted the article. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, Fogo AB, et al. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet (2017) 390(10105):1888–917. doi: 10.1016/S0140-6736(17)30788-2
2. Lv JC, Zhang LX. Prevalence and disease burden of chronic kidney disease. Adv Exp Med Biol (2019) 1165:3–15. doi: 10.1007/978-981-13-8871-2_1
3. Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol (2018) 80:309–26. doi: 10.1146/annurev-physiol-022516-034227
4. Yang M, Liu JW, Zhang YT, Wu G. The role of renal macrophage, AIM, and TGF-beta1 expression in renal fibrosis progression in IgAN patients. Front Immunol (2021) 12:646650. doi: 10.3389/fimmu.2021.646650
5. Liu B, Jiang J, Liang H, Xiao P, Lai X, Nie J, et al. Natural killer T cell/IL-4 signaling promotes bone marrow-derived fibroblast activation and M2 macrophage-to-myofibroblast transition in renal fibrosis. Int Immunopharmacol (2021) 98:107907. doi: 10.1016/j.intimp.2021.107907
6. Feng YL, Wang WB, Ning Y, Chen H, Liu P. Small molecules against the origin and activation of myofibroblast for renal interstitial fibrosis therapy. BioMed Pharmacother (2021) 139:111386. doi: 10.1016/j.biopha.2021.111386
7. Djudjaj S, Boor P. Cellular and molecular mechanisms of kidney fibrosis. Mol Aspects Med (2019) 65:16–36. doi: 10.1016/j.mam.2018.06.002
8. Li L, Xu L, Wen S, Yang Y, Li X, Fan Q. The effect of lncRNA-ARAP1-AS2/ARAP1 on high glucose-induced cytoskeleton rearrangement and epithelial-mesenchymal transition in human renal tubular epithelial cells. J Cell Physiol (2020) 235(7-8):5787–95. doi: 10.1002/jcp.29512
9. Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest (2011) 121(2):468–74. doi: 10.1172/jci44595
10. Lovisa S, Fletcher-Sananikone E, Sugimoto H, Hensel J, Lahiri S, Hertig A, et al. Endothelial-to-mesenchymal transition compromises vascular integrity to induce myc-mediated metabolic reprogramming in kidney fibrosis. Sci Signal (2020) 13(635). doi: 10.1126/scisignal.aaz2597
11. Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ. Diverse origins of the myofibroblast-implications for kidney fibrosis. Nat Rev Nephrol (2015) 11(4):233–44. doi: 10.1038/nrneph.2014.246
12. Meng XM, Wang S, Huang XR, Yang C, Xiao J, Zhang Y, et al. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis (2016) 7(12):e2495. doi: 10.1038/cddis.2016.402
13. LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med (2013) 19(8):1047–53. doi: 10.1038/nm.3218
14. Wang YY, Jiang H, Pan J, Huang XR, Wang YC, Huang HF, et al. Macrophage-to-Myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. J Am Soc Nephrol (2017) 28(7):2053–67. doi: 10.1681/ASN.2016050573
15. Yang J, Lin SC, Chen G, He L, Hu Z, Chan L, et al. Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J Am Soc Nephrol (2013) 24(10):1644–59. doi: 10.1681/ASN.2013030217
16. Pan B, Liu G, Jiang Z, Zheng D. Regulation of renal fibrosis by macrophage polarization. Cell Physiol Biochem (2015) 35(3):1062–9. doi: 10.1159/000373932
17. Puthumana J, Thiessen-Philbrook H, Xu L, Coca SG, Garg AX, Himmelfarb J, et al. Biomarkers of inflammation and repair in kidney disease progression. J Clin Invest (2021) 131(3). doi: 10.1172/JCI139927
18. Torres A, Munoz K, Nahuelpan Y, AP RS, Mendoza P, Jara C, et al. Intraglomerular Monocyte/Macrophage infiltration and macrophage-myofibroblast transition during diabetic nephropathy is regulated by the A2B adenosine receptor. Cells (2020) 9(4). doi: 10.3390/cells9041051
19. Nikolic-Paterson DJ, Wang S, Lan HY. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int Suppl (2011) (2014) 4(1):34–8. doi: 10.1038/kisup.2014.7
20. Liu Y, Su YY, Yang Q, Zhou T. Stem cells in the treatment of renal fibrosis: a review of preclinical and clinical studies of renal fibrosis pathogenesis. Stem Cell Res Ther (2021) 12(1):333. doi: 10.1186/s13287-021-02391-w
21. Samarakoon R, Overstreet JM, Higgins SP, Higgins PJ. TGF-beta1 –> SMAD/p53/USF2 –> PAI-1 transcriptional axis in ureteral obstruction-induced renal fibrosis. Cell Tissue Res (2012) 347(1):117–28. doi: 10.1007/s00441-011-1181-y
22. Chen J, Xia Y, Lin X, Feng XH, Wang Y. Smad3 signaling activates bone marrow-derived fibroblasts in renal fibrosis. Lab Invest (2014) 94(5):545–56. doi: 10.1038/labinvest.2014.43
23. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol (2016) 12(6):325–38. doi: 10.1038/nrneph.2016.48
24. Chung AC, Huang XR, Zhou L, Heuchel R, Lai KN, Lan HY. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol Dial Transplant (2009) 24(5):1443–54. doi: 10.1093/ndt/gfn699
25. Yang Y, Feng X, Liu X, Wang Y, Hu M, Cao Q, et al. Fate alteration of bone marrow-derived macrophages ameliorates kidney fibrosis in murine model of unilateral ureteral obstruction. Nephrol Dial Transplant (2019) 34(10):1657–68. doi: 10.1093/ndt/gfy381
26. Wang S, Meng XM, Ng YY, Ma FY, Zhou S, Zhang Y, et al. TGF-beta/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget (2016) 7(8):8809–22. doi: 10.18632/oncotarget.6604
27. Meng XM, Zhang Y, Huang XR, Ren GL, Li J, Lan HY. Treatment of renal fibrosis by rebalancing TGF-beta/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget (2015) 6(35):36984–97. doi: 10.18632/oncotarget.6100
28. Tang PM, Zhang YY, Xiao J, Tang PC, Chung JY, Li J, et al. Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage-myofibroblast transition. Proc Natl Acad Sci U.S.A. (2020) 117(34):20741–52. doi: 10.1073/pnas.1917663117
29. Tang PM, Zhou S, Li CJ, Liao J, Xiao J, Wang QM, et al. The proto-oncogene tyrosine protein kinase src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int (2018) 93(1):173–87. doi: 10.1016/j.kint.2017.07.026
30. Yan Y, Ma L, Zhou X, Ponnusamy M, Tang J, Zhuang MA, et al. Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int (2016) 89(1):68–81. doi: 10.1038/ki.2015.293
31. Cheng Z, Liu L, Wang Z, Cai Y, Xu Q, Chen P. Hypoxia activates src and promotes endocytosis which decreases MMP-2 activity and aggravates renal interstitial fibrosis. Int J Mol Sci (2018) 19(2). doi: 10.3390/ijms19020581
32. Yan J, Zhang Z, Yang J, Mitch WE, Wang Y. JAK3/STAT6 stimulates bone marrow-derived fibroblast activation in renal fibrosis. J Am Soc Nephrol (2015) 26(12):3060–71. doi: 10.1681/ASN.2014070717
33. Jiao B, An C, Du H, Tran M, Wang P, Zhou D, et al. STAT6 deficiency attenuates myeloid fibroblast activation and macrophage polarization in experimental folic acid nephropathy. Cells (2021) 10(11). doi: 10.3390/cells10113057
34. Jiao B, An C, Tran M, Du H, Wang P, Zhou D, et al. Pharmacological inhibition of STAT6 ameliorates myeloid fibroblast activation and alternative macrophage polarization in renal fibrosis. Front Immunol (2021) 12:735014. doi: 10.3389/fimmu.2021.735014
35. Liang H, Zhang Z, Yan J, Wang Y, Hu Z, Mitch WE, et al. The IL-4 receptor alpha has a critical role in bone marrow-derived fibroblast activation and renal fibrosis. Kidney Int (2017) 92(6):1433–43. doi: 10.1016/j.kint.2017.04.021
36. Ma Z, Jin X, He L, Wang Y. CXCL16 regulates renal injury and fibrosis in experimental renal artery stenosis. Am J Physiol Heart Circ Physiol (2016) 311(3):H815–21. doi: 10.1152/ajpheart.00948.2015
37. Xia Y, Entman ML, Wang Y. Critical role of CXCL16 in hypertensive kidney injury and fibrosis. Hypertension (2013) 62(6):1129–37. doi: 10.1161/HYPERTENSIONAHA.113.01837
38. Xia Y, Yan J, Jin X, Entman ML, Wang Y. The chemokine receptor CXCR6 contributes to recruitment of bone marrow-derived fibroblast precursors in renal fibrosis. Kidney Int (2014) 86(2):327–37. doi: 10.1038/ki.2014.64
Keywords: macrophage, macrophage-to-myofibroblast transition (MMT), renal fibrosis, myofibroblast, inflammation
Citation: Wei J, Xu Z and Yan X (2022) The role of the macrophage-to-myofibroblast transition in renal fibrosis. Front. Immunol. 13:934377. doi: 10.3389/fimmu.2022.934377
Received: 02 May 2022; Accepted: 13 July 2022;
Published: 05 August 2022.
Edited by:
Qiulun Lu, Nanjing Medical University, ChinaReviewed by:
Yanlin Wang, University of Connecticut, United StatesJiexiong Feng, Huazhong University of Science and Technology, China
Copyright © 2022 Wei, Xu and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Wei, dr.weijia@zju.edu.cn; Xiang Yan, yanxiang@zju.edu.cn