 Kim Ngan Luu Hoang
Kim Ngan Luu Hoang Joanne E. Anstee
Joanne E. Anstee James N. Arnold
James N. Arnold- Faculty of Life Sciences and Medicine, School of Cancer and Pharmaceutical Sciences, King’s College London, London, United Kingdom
Heme oxygenase-1 (HO-1) is an inducible intracellular enzyme that is expressed in response to a variety of stimuli to degrade heme, which generates the biologically active catabolites carbon monoxide (CO), biliverdin and ferrous iron (Fe2+). HO-1 is expressed across a range of cancers and has been demonstrated to promote tumor progression through a variety of mechanisms. HO-1 can be expressed in a variety of cells within the tumor microenvironment (TME), including both the malignant tumor cells as well as stromal cell populations such as macrophages, dendritic cells and regulatory T-cells. Intrinsically to the cell, HO-1 activity provides antioxidant, anti-apoptotic and cytoprotective effects via its catabolites as well as clearing toxic intracellular heme. However, the catabolites of heme degradation can also diffuse outside of the cell to extrinsically modulate the wider TME, influencing cellular functionality and biological processes which promote tumor progression, such as facilitating angiogenesis and metastasis, as well as promoting anti-inflammation and immune suppression. Pharmacological inhibition of HO-1 has been demonstrated to be a promising therapeutic approach to promote anti-tumor immune responses and inhibit metastasis. However, these biological functions might be context, TME and cell type-dependent as there is also conflicting reports for HO-1 activity facilitating anti-tumoral processes. This review will consider our current understanding of the role of HO-1 in cancer progression and as a therapeutic target in cancer.
Introduction
The heme oxygenase (HO) family of enzymes serve as the rate-limiting step in the degradation of heme which is released by dying cells and yields the biologically active catabolites biliverdin, ferrous iron (Fe2+) and carbon monoxide (CO) (1). The HO-1 isoform has been demonstrated to be expressed in a wide variety of cancers and has become implicated in a diverse range of biological processes which can be exploited by the tumor to facilitate disease progression and metastasis. The pivotal role of HO-1 in tumoral immune suppression in some preclinical models suggests it could represent an immunotherapy target and innate immune checkpoint (2). There are two isoforms of HO expressed in humans and mice; HO-1 which is inducible (3), and HO-2 which is expressed at basal levels in all cells (4). There is also a third non-catalytically active isoform, HO-3, that has been identified in rats (5). The HO-1 isoform is induced by a variety of external stimuli including; oxidative stress, cytokines and prostaglandins (6). Heme is also a potent inducer of HO-1 expression and acts as a cytoprotective measure against the pro-oxidant properties of free-form heme (7). As such, it is not surprising that HO-1 is expressed in a variety of cancers including; bladder (8), breast (2), colorectal (9), glioblastoma (10), head and neck (11), leukemia (12), lung (13), melanoma (14), neuroblastoma (15), prostate (16) and renal (17) cancer. HO-1 in the TME has also been associated with poor prognosis for patients (2, 15, 18). HO-1 is a 32kDa protein that predominantly localizes to the endoplasmic reticulum (ER) (19, 20) but can also localize to the caveolae (21), mitochondria (22), and nucleus (23). HO-1 has also been detected in the plasma and its concentration has been demonstrated to be elevated in prostate cancer (24). At the cellular level, HO-1 has most frequently been reported to be expressed by tumor cells (12, 25) and tumor associated macrophages (TAMs) (2, 26, 27) within the TME. However, HO-1 can also be expressed by endothelial cells (28), dendritic cells (DCs) (29, 30) and regulatory T-cells (Tregs) (31, 32). This review will discuss the current knowledge and controversies on the biological roles of HO-1 in the TME and as a therapeutic target in cancer.
Biochemical properties of HO-1
Enzymatically active HO-1 predominantly localizes to the ER within the cell where it forms the microsomal heme oxygenase system (19, 21) (Figure 1). Heme is pro-oxidative and cytotoxic, due to the protein being involved in lipid peroxidation and catalyzing the production of free radicals (34, 35). Furthermore, heme can also trigger cell damage and apoptosis by inhibiting the proteasome and causing mitochondrial dysfunction (36–38). In the first stage of oxidative heme degradation, HO-1 forms a complex with heme and NADPH-cytochrome-P450 reductase (39) (Figure 1). NADPH acts as the electron donor while molecular oxygen binds to the complex, yielding CO, chelatable Fe2+ and biliverdin (40). Subsequently, biliverdin is converted to bilirubin by the enzyme NADPH-biliverdin reductase (BVR) using NADPH (1, 33, 41). The catabolites of heme degradation are biologically active and go on to elicit a variety of cell intrinsic and extrinsic effects (42). A truncated variant of HO-1 has also been identified in malignant cells which localizes to the nucleus and appears to be associated with a cellular response to hypoxia (23, 43). Nuclear localization of HO-1 requires the proteolytic cleavage of the C-terminal 23 amino acids of the protein and then chromosomal maintenance-1 (CRM1)-mediated nuclear-cytoplasmic shuttling to the nucleus (23). The truncated HO-1 variant, although not catalytically active, appears to be a response specifically of malignant cells rather than the non-malignant stromal cells within the TME (16). Within the nucleus the truncated HO-1 variant has been demonstrated to play a role in transcriptional regulation, down-regulating NF-κB and SP-1 DNA binding activity, while up-regulating oxidant-responsive transcription factors, such as activator protein-1 (AP-1), AP-2, Brn-3 and core-binding factor (23). The biological importance of truncated HO-1 in relation to its impact on cancer progression still needs further investigation, however it has now been observed in a range of cancers, but most well characterized in prostate cancer, where it has been associated with disease progression (11, 44). The non-catalytic function of HO-1 provides an intriguing additional layer of functionality to the protein.
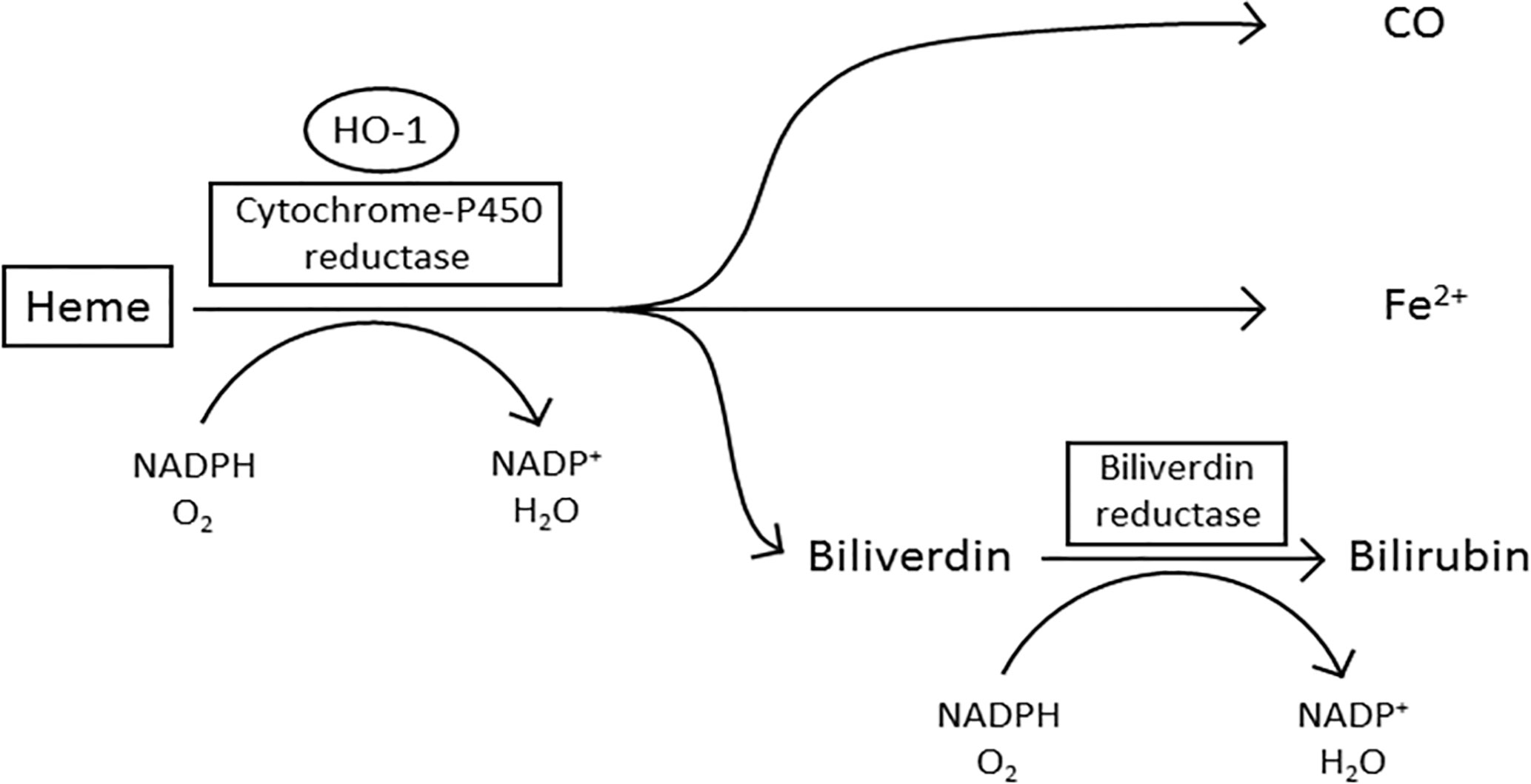
Figure 1 The oxidative degradation of heme by HO-1. In the first step of heme degradation, the ER membrane bound HO-1 interacts with the electron donor NADPH-cytochrome P450 reductase, and an oxygen molecule. The complex degrades heme to biliverdin, carbon monoxide (CO), and a ferrous iron (Fe2+). NADPH-biliverdin reductase competitively binds to HO-1 to reduce biliverdin to bilirubin, by using NADPH as an electron donor (1, 33).
The Sources of HO-1 in the TME
In the TME, HO-1 can be expressed by both the malignant tumor cells and stroma (2, 27, 45). Several stromal cell populations have been described to express HO-1 (45). TAMs, which and are a prevalent stromal cell type in the TME, and are a major stromal source of HO-1 in both murine models (2, 26, 27) and human disease (2, 27). In some TMEs, TAMs represent the major tumoral source of HO-1 (2, 14, 26, 27, 46). TAMs are a highly plastic stromal cell type which are capable of responding to the TME to adopt a variety of distinct phenotypes, creating a spectrum of possible polarization pro- and anti-inflammatory phenotypes that differ in their cytokine profiles and gene expression (47–50). HO-1 is not a pan-TAM marker per se, and is instead selectively up-regulated by a subset of these cells, suggesting its expression is restricted to certain polarization states (2, 14, 26, 27, 46, 51, 52). HO-1 has been associated with TAMs with a polarization program similar to the ‘M2’ tumor-promoting macrophage phenotype, and there is also evidence to suggest that HO-1 may play a direct role in skewing the macrophage program (53). In further support of this, inhibition of HO-1 activity in macrophages has been demonstrated in vitro to revert their phenotype to a more inflammatory state, marked by an increase in inducible nitric oxide synthase (iNOS) expression (51). A subset of HO-1 expressing TAMs can be identified, in certain TMEs, to co-express fibroblast activation protein alpha (FAP), a surface protease associated with fibroblast populations (26, 27, 54). In subcutaneous Lewis lung carcinoma (LL2) tumors, the FAP+ TAM subset accounted for 10% of the total TAM population and represented the major source of HO-1 in those tumors (26). FAP+ TAMs have also been identified in human breast cancer (55) and this subset also expresses HO-1 (27). In orthotopic 4T1 tumors a FAP+ HO-1+ TAM subset was also identified and, in this model, was associated with a macrophage polarization response to IL-6 in a TME rich in collagen (the substrate for FAP’s protease activity) (27). 4T1 tumors displayed a ‘healing wound’-like TME and, as such, it was not surprising that FAP+ HO-1+ macrophages were also present in the granulation tissue of a cutaneous healing wound (27). This observation highlights how tumors exploit TAM polarization from fundamental biological responses of these cells. Interestingly, the relationship between TAMs and HO-1 does not always promote tumor progression. In an A549 lung carcinoma model the presence of CD86+ myeloid cells, which are regarded as a pro-inflammatory M1-like macrophage phenotype, were required to mediate the anti-tumor growth effect of exogenously administered CO to tumor bearing mice (56).
DCs, which represent a professional antigen presenting cell type that can reside in the TME have been observed to express HO-1. HO-1 expression by DCs can also influence the activity of these cells, suppressing their immunogenicity and antigen presentation capabilities (29, 30). The immunomodulatory CD4+ CD25+ Foxp3+ Treg population has also been described to express HO-1 in humans (31, 32) but not mice (57), and may be directly linked to the Foxp3-regulated gene expression program in humans (58). As the catabolites of heme degradation can diffuse extracellularly to influence non-HO-1 expressing cells, the importance of the specific cell-type expressing HO-1 in relation to prognosis remains to be fully resolved, where location and prevalence could be the most important variables to consider. The widespread expression of HO-1 in cancer also highlights the fundamental relationship between the enzyme and the disease.
Signals In The TME for HMOX1 Gene expression
The gene for HO-1, HMOX1, is regulated by several transcription factors which allow HO-1 to be expressed in response to a variety of stimuli which are relevant to the TME (20, 53, 59, 60) (Figure 2). Induction of HO-1 protein expression is a cytoprotective measure in both cancer and normal tissue in response to an increase in reactive oxygen species (ROS) levels (61). Tumor cells are under constant oxidative stress, generating high levels of ROS (62). Nuclear factor erythroid 2-related factor 2 (Nrf2) is a major transcription factor for the HMOX1 gene. Nrf2 controls over 200 genes associated with the antioxidant response, which includes HO-1 (63), and has been considered as a therapeutic target in cancer, and even proposed as an oncogene (63). Nrf2-regulated HMOX1 gene transcription is, however, tightly regulated. Under basal redox conditions, Keap1, a repressor protein, binds to the Nrf2 transcription factor preventing its nuclear translocation and activity, through promoting its ubiquitination and proteasomal degradation (64, 65). However, under conditions of oxidative stress, Keap1 undergoes oxidation of its sulfhydryl groups, leading to the release of Nrf2 which permits stabilization and nuclear translocation (66) (Figure 2). In the nucleus, Nrf2 hetero-dimerizes with small Maf (sMaf) proteins to mediate HMOX1 expression through binding the antioxidant response element (ARE) site in the promoter region of the gene. However, access to the ARE site is further regulated by Bach1, a transcriptional repressor, which also hetero-dimerizes with sMaf proteins to compete for ARE elements, sterically preventing the access of Nrf2 to the site (67). Cellular heme levels regulate HMOX1 transcription through directly binding to Bach1, causing its release from the DNA (68), which in turn promotes Nrf2 access to the ARE to drive the expression of HMOX1 (69). Heme levels have been demonstrated to be increased in cancer (70, 71). As such, cellular heme exquisitely regulates the expression of HO-1 in a substrate-dependent manner. Several cellular stimuli activate the Nrf2 pathway, and Nrf2-dependent HO-1 activation, such as sphingosine-1-phosphate (S1P) which is released from apoptotic cells within the TME which engages with the sphingosine-1-phosphate receptor (S1PR), a G protein-coupled receptor (53).
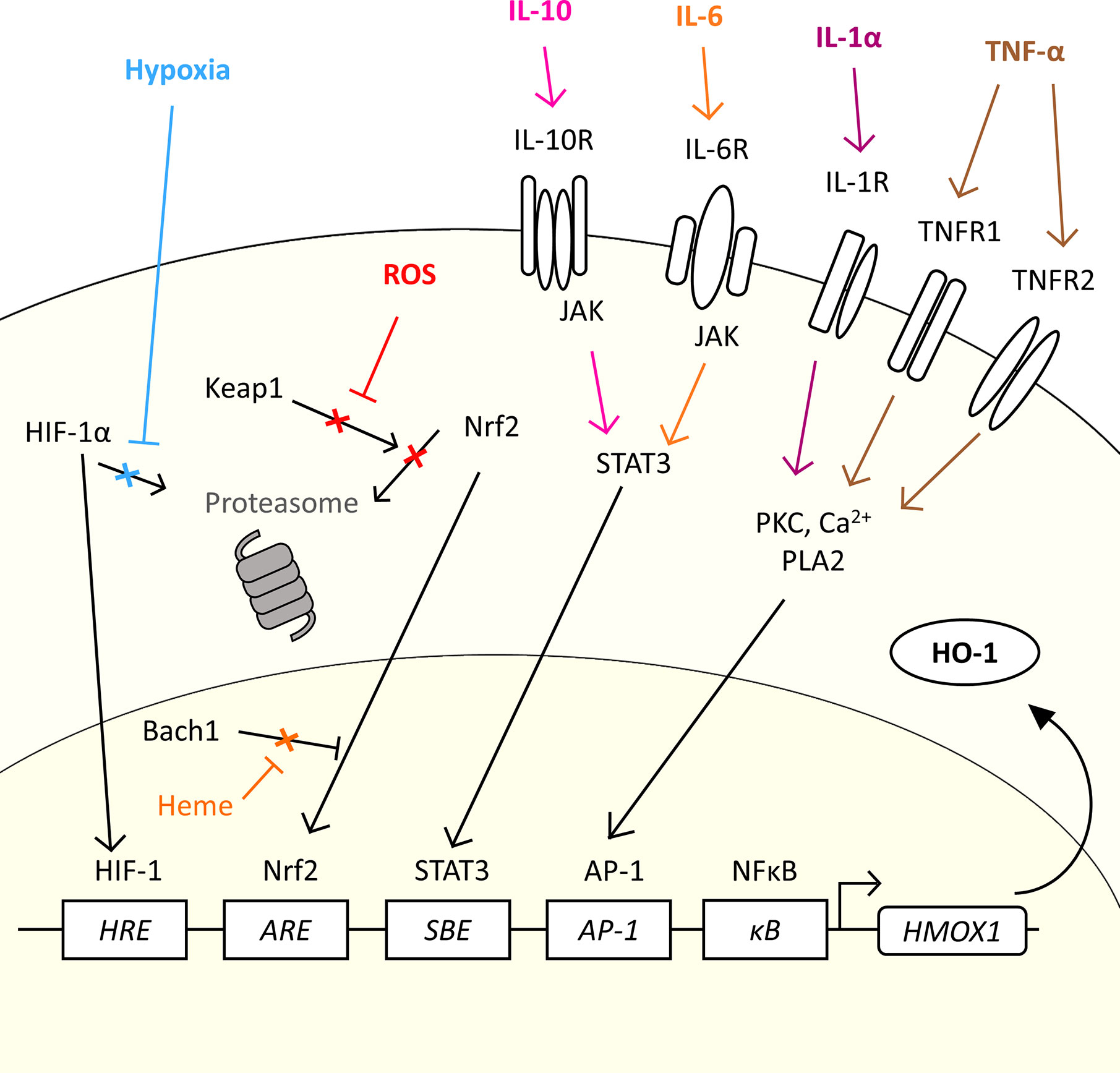
Figure 2 The diverse range of signals in the TME which could induce HMOX1 expression. HMOX1 mRNA expression is induced by a range of molecular and physical signals. The figure highlights common physical characteristics (hypoxia), metabolic by-products (ROS) and cytokines commonly associated with the TME which could induce the expression of HO-1 in cancer. In normal conditions, the transcription factor Nrf2 is inhibited by Keap1 which prevents nuclear translocation and promotes its proteasomal degradation. ROS generated by oxidative stress inhibit the interaction between Keap1 and Nrf2, allowing Nrf2 to translocate to the nucleus and bind to the ARE of the HMOX1 gene. In the presence of heme, Bach1 is prevented from inhibiting Nrf2 access to the ARE allowing the expression of HMOX1. Under conditions of hypoxia, the transcription factor HIF-1α escapes proteasomal degradation and translocates into the nucleus where it forms a complex with HIF-1β and binds to HRE to up-regulate HMOX1 gene expression. Cytokines such as IL-10 and IL-6 can induce expression via the JAK/STAT3 pathway, while IL-1α and TNF-α induced HMOX1 expression through a PKC, Ca2+ signaling, and PLA2 dependent pathway and AP-1 element in the promoter region.
There are other transcription factor binding elements with the promoter region which have been demonstrated to regulate HMOX1 gene expression, including a hypoxia-responsive element (HRE) which make HO-1 part of the cellular response to hypoxia. Hypoxia is a common characteristic of the TME where proliferation of the malignant tumor cells outstrips angiogenesis leading to inefficient vascular networks and poor perfusion of the tumor tissue (72, 73). Hypoxia, stabilizes the transcription factor HIF-1α in the cytoplasm allowing it to escape proteasomal degradation and permitting its translocation to the nucleus where it complexes with HIF-1β and initiates expression of the HMOX1 gene (74, 75) (Figure 2). In addition, the HMOX1 promoter contains several transcription factor binding sites that allow a variety of both pro-and anti-inflammatory cytokines to regulate HMOX1 gene expression (Figure 2), allowing HO-1 to play an important role in both the response and resolution phase of inflammation. The tumor stroma is a rich source of cytokines, soluble proteins which act as signaling molecules between cells, creating a complex crosstalk within the stroma and tumor cells (76). The cytokine milieu is different between cancers (27, 77), which contributes to the heterogeneity of the TME through influencing recruitment and/or polarization of cells such as is observed with TAM (27) and Treg (78) populations. Specifically in relation to HO-1 expression, IL-1α and TNF-α, which have pro-inflammatory effects, have been demonstrated to induce HMOX1 gene expression through a pathway involving protein kinase C (PKC), Ca2+ signaling and phospholipase A2 (PLA2) (79), which directs HMOX1 expression through the transcription factor AP-1 binding the AP-1 binding site in the HMOX1 promoter (80). The HMOX1 promoter also contains a NF-κB binding element (81) which further facilitates HMOX1 to be induced in response to pro-inflammatory signals. Anti-inflammatory cytokines also induce HMOX1 expression, including IL-6 (27, 60) and IL-10 (82) via the JAK/STAT3 pathway and the STAT binding element (SBE) located in the promoter region of the HMOX1 gene (60). The diversity of stimuli which induce HMOX1 gene expression highlight the fundamental role of this enzyme within inflammation and stress responses which are exploited in the TME to facilitate tumor progression.
Although, the expression of HO-1 is heavily influenced by the TME, there are instances where HO-1 can be constitutively expressed by tumor cells due to oncogenes driving the HMOX1 promoter (12) or genetic mutations within the promoter itself (25). The human HMOX1 gene promoter has a 5′-flanking region containing GT microsatellite repeats, which vary in number between individuals and influences basal HMOX1 transcription and inducibility of the gene in response to stimuli (83, 84). Polymorphisms in the number of GT repeats in the HMOX1 promoter have also been linked to a susceptibility to cancer (85), where longer GT repeats, which result in lower basal levels of HO-1 expression, increase the likelihood of the individual developing gastric, lung and oral squamous cancer (85). HMOX1 mRNA can also be modulated post-transcriptionally by microRNA-378, a small non-coding RNA that specifically targets and destabilizes HMOX1 mRNA, preventing its translation (86). The broad range of stimuli and genetic aberrations that lead to HO-1 in cancer highlight the importance of this enzyme in modulating the inflammatory response and cancer progression.
The influence of HO-1 on cell fate
HO-1 is a well-established cytoprotective protein which confers its cytoprotective effects via the anti-apoptotic and antioxidant properties of the catabolites of heme degradation (42) (Figure 3). CO can inhibit the activity of pro-apoptotic K+ channels, as well as activate p38 MAPK and PI3K/Akt pathways to protect cells from apoptosis (28, 87, 88). CO generated from a HO-1-expressing endothelial cell line has also been demonstrated to be capable of conferring a survival signal to non-HO-1-expressing cells, highlighting the influence that CO can have on cell-cell communication (28). However, HO-1 activity has also been demonstrated to have an anti-proliferative/pro-apoptotic function in some contexts (89–91). Interestingly, CO’s role in dictating cell fate provides an interesting dichotomy in the context of the TME. For example, CO modulates the activity of a variety of kinases in the cell, in particular p38 MAPK, which can prevent apoptosis in endothelial cells (28) but conversely inhibits proliferation and enhances cell death in tumor cells (16, 92). The differences in response have been suggested to be a result of the underlying differences in the metabolism between malignant and healthy cells (56). Malignant cells fuel their proliferative capacity through adapting their metabolism to preferentially uptake and process glucose anaerobically, in a process known as the ‘Warburg’ effect (93). Treatment of tumor cells with CO has been demonstrated to result in their metabolic exhaustion and apoptosis due to switching their metabolic state to an increase in oxygen consumption in an ‘anti-Warburg’ type effect (16). This metabolic switch of the cancer cell to oxidative metabolism by CO has been demonstrated to decrease nucleotide and amino acid synthesis pathways, arrest the cell cycle and then ultimately lead to cell death due to the resulting intense mitochondrial stress and mitochondrial-dependent generation of ROS (16). Although a similar response has been observed in macrophages, the outcome is survival (94). Mechanistically, a role of CO in promoting DNA repair processes in healthy cells is also believed to contribute to outcome (28, 95). These observations have been exploited for therapeutic-gain, and supplying exogenous CO to mice bearing prostate or lung cancers, protects healthy tissue while targeting the malignant cells, resulting in tumor control in these models (16).
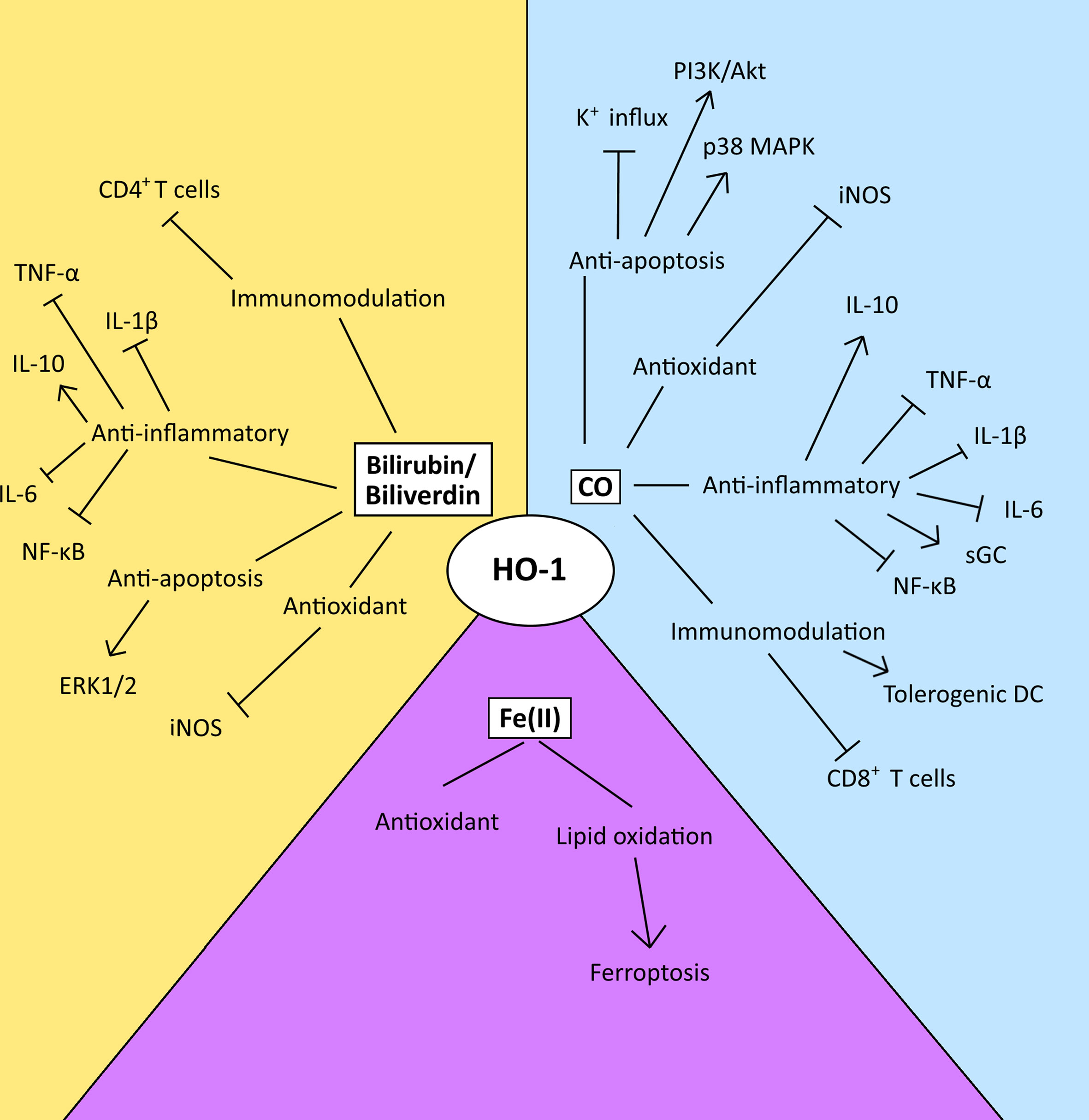
Figure 3 The diverse range of possible modulatory effects of the heme catabolites generated by the enzyme activity of HO-1 in the TME. This figure summarizes the possible modulatory effects of the heme catabolites in the TME. However, the relative utilization and biological importance of each pathway in disease progression or control is TME-dependent and key observations relating to these pathways are discussed within the text. CO and biliverdin/bilirubin mediate their anti-apoptotic effects via the modulation of signal transduction, and the inhibition of K+ influx. Their antioxidant effects are mediated via the inhibition of iNOS. Both CO and biliverdin/bilirubin inhibit the activity of pro-inflammatory cytokines, the NF-κB pathway and increase production of IL-10, and CO also activates sGC. CO induces the maturation of tolerogenic DCs and inhibits the activity and proliferation of CD8+ T-cells, while bilirubin inhibits the activation of CD4+ T-cells. Ferrous iron [Fe (II)] enacts antioxidant effects via ferritin, however, an imbalance between ROS levels and iron overload can lead to lipid oxidation and ferroptosis.
Although yet to be established in cancer, biliverdin/bilirubin has been demonstrated to also play a cytoprotective role, decreasing nitric oxide (NO) production by inhibiting iNOS expression (Figure 3) (96, 97) and mediating anti-apoptotic effects through the ERK1/2 pathway during hypoxia (98) in non-cancer models. Also, free chelatable Fe2+ can have a pro-oxidant role, by inducing lipid oxidation and ferroptosis. Ferroptosis is a form of non-apoptotic cell death that is caused by an overload of iron and is accompanied by lipid peroxidation (99) (Figure 3). Interestingly, experimental over-expression of the HO-1 protein can also cause a reversal of its cytoprotective role, as a result of the increase in reactive iron levels. Treatment with an iron-chelator significantly decreased signs of cellular injury including protein oxidation and lactate dehydrogenase (100). Ferritin is a protein which naturally sequesters free iron as a means to protects against oxidative stress (101). However, when HO-1 is highly expressed, the levels of ferritin become insufficient to neutralize the oxidative effects of reactive iron (102). The ferroptotic role of HO-1 has now been demonstrated in a range of tumor cells including in breast, lung and fibrosarcoma tumor cell lines (103, 104).
HO-1 expression is also induced by exposure to cytotoxic chemotherapies (CCTs) (105, 106) and provides a mechanism of resistance to CCTs such as; etoposide, doxorubicin, gemcitabine and cisplatin (45, 106–108), as well as other classes of anti-cancer drugs such as sapatinib, a pan-Her family kinase inhibitor (109). HO-1 can also indirectly affect the efficacy of CCTs through suppressing the immunomodulatory actions of these agents within the TME (2, 110). CCTs can both prime T-cell responses and elicit T-cell infiltration into the TME, which can turn immunologically ‘cold’ tumors ‘hot’ (2, 110). The immunomodulatory actions of CCTs are becoming increasingly understood and may actually underpin a significant proportion of their clinical efficacy (110–114). The immune suppressive actions of HO-1 activity within the TME (discussed below) can suppress CCT-elicited T-cell responses which indirectly effect the therapeutic efficacy of these agents (2). Interestingly, when tumor cells in vitro or tumor bearing mice in vivo are exposed to CO, CO can also play a contradictory role and increase the sensitivity of tumor cells to CCTs when exogenously delivered to the system, exacerbating the effect of CO-targeting mitochondrial function in malignant tumor cells to promote tumor cell death (16). In fact, PC3 prostate cancer cells exposed to exogenous CO became 1000-fold more sensitive to DNA damaging CCTs camptothecin and doxorubicin (16).
HO-1 activity has also been implicated as a tumor cell resistance mechanism for radiotherapy (115). The generation of ROS is a key event that underpins the anti-cancer effects of ionizing radiation (116), and pharmacological inhibition of HO-1 can enhance the radio-sensitivity of malignant cells (115, 117).
Interestingly, as these studies highlight, the role of HO-1 and cell fate is context and cell type-dependent which require careful considering in predicting and understanding how to most effectively exploit the HO-1 axis for therapeutic gain in the treatment of cancer.
The Effect of HO-1 on Tumor Infiltrating T-Cells
HO-1 has potent immunomodulatory effects within the TME, influencing several cell types which underpin the anti-tumor immune response (Figure 4). Work in preclinical models of cancer have solidified the importance of HO-1, in some TMEs, to be pivotal to immune suppression which prevents efficient anti-tumor immunity as HO-1 activity suppresses T-cell effector function (2, 26). In mice bearing subcutaneous LL2 tumors which were rendered immunogenic through the expression of ovalbumin (LL2/OVA), the FAP+ TAM subset was the major tumoral source of HO-1. Specific conditional depletion of FAP+ TAMs, using diphtheria toxin in a bone marrow chimera of a FAP/diphtheria toxin receptor (DTR) transgenic mouse (54), permitted immunological control of tumor growth (26). Pharmacologically targeting HO activity using tin mesoporphyrin (SnMP) in mice bearing LL2/OVA tumors also permitted efficient immunological control of tumor growth (26). This study highlighted the biological significance of both FAP+ TAMs and their expression of HO-1 in tumoral immune suppression in the TME. In an aggressive spontaneous murine model of breast cancer (MMTV-PyMT), in which TAMs were also the major tumoral source of HO-1, an influx of T-cells into the TME were generated using the immune-stimulating CCT 5-fluorouracil (5-FU) which synergized with pharmacological inhibition of HO using SnMP to alleviate immune suppression and permit CD8+ T-cell dependent control of tumor growth (2). Although, small molecule inhibitors can have off target effects, SnMP which inhibits both HO-1 and HO-2, has no identified off-target effects to date (118–121). Furthermore, genetic inactivation of HO-1 in the myeloid lineage did not affect the prevalence of TAMs or CD8+ T-cells in the TME, but did improve the proportion of cytotoxic T-cells capable of producing the effector molecules granzyme-B, IFN-γ and TNF-α (2) (Figure 4). Due to poor infiltration of T-cells in these MMTV-PyMT tumors, genetic inactivation of Hmox1 in TAMs neither affected the latency of tumor onset nor the kinetics of tumor growth, however, when 5-FU was administered to elicit a T-cell influx into the TME, tumor control was achieved (27).
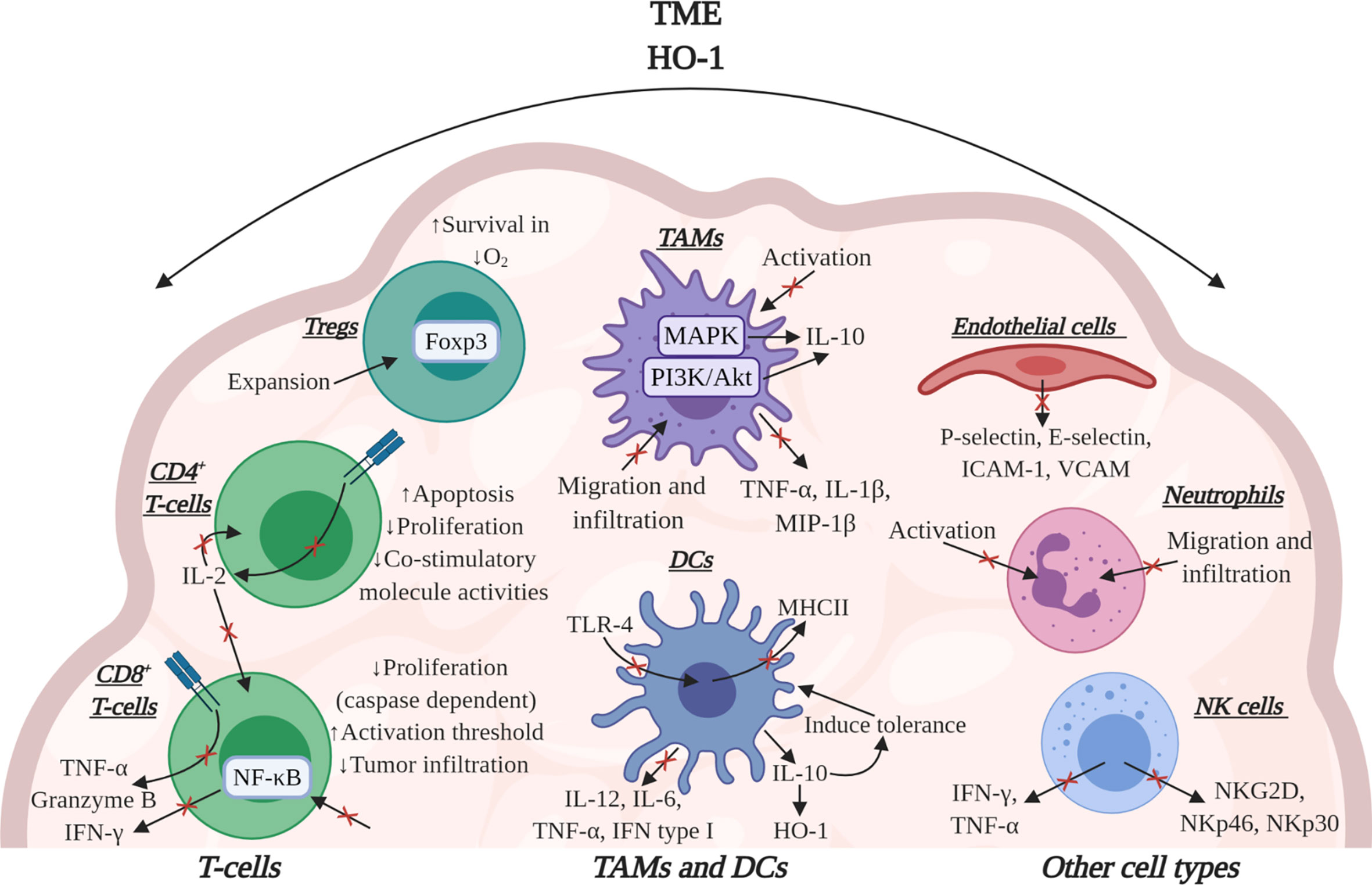
Figure 4 The effects of HO-1 activity on immune cells in the TME. HO-1 activity may affect several immune cell types to suppress the anti-tumor immune response. HO-1 reduces the effector functions of CD8+ T-cells by suppressing their expression of TNF-α, granzyme-B and IFN-γ. Specifically, CO suppresses the effector functions mediated via TCR engagement. HO-1 has also been implicated in suppressing CD8+ T-cell proliferation, tumor infiltration and increasing their activation threshold. In CD4+ T-cells CO can block TCR-dependent IL-2 production, thus influencing the proliferation of CD8+ and CD4+ T-cells. Bilirubin/Biliverdin can block CD4+ T-cell activity by inducing apoptosis, inhibiting proliferation and suppressing co-stimulatory molecule activities. In Tregs, HO-1 has been associated with the expansion of the population and HO-1-derived CO can improve Treg survival in conditions of hypoxia. HO-1 can also cause DCs to secrete IL-10 which maintains these cells in a tolerogenic state alongside up-regulating their expression of HO-1. HO-1 activity also down-regulates the expression of pro-inflammatory cytokines in these cells. Additionally, both CO and bilirubin can down-regulate MHCII expression in DCs. In TAMs, CO inhibits activation while also suppressing the release of pro-inflammatory cytokines and concurrently eliciting an increase in IL-10 production. In endothelial cells, CO can suppress the expression of adhesion molecules, affecting the ability of neutrophils to attach. HO-1 may also inhibit neutrophil activation itself and CO and biliverdin can inhibit migration and infiltration of both neutrophils and TAMs. Additionally, in NK cells HO-1 activity can decrease their cytotoxicity by suppressing their ability to secrete pro-inflammatory cytokines and express activatory cell receptors. Image was created using BioRender software.
The role of HO-1 expressing TAMs in immune suppression was further supported by a study using immunogenic OVA-expressing murine thymic lymphoma cells (EG7-OVA) (52). In this study, myeloid-specific HO-1 inactivation in mice increased proliferation, tumor infiltration and cytotoxic activity of CD8+ T-cells (52). CO has been demonstrated in vitro to be capable of modulating CD8+ T-cell effector function (26). This is likely achieved through CO’s ability to modulate cellular signaling, such as STAT1/3 (87) and NF-κB (122), which has the potential to directly compromise the anti-tumor response. NF-κB in particular, has been demonstrated to be vital for CD8+ T-cell effector function (123). However, it should be noted that bilirubin/biliverdin have also been demonstrated in non-cancer settings to be capable of suppressing NF-κB signaling (124–127).
Bilirubin has been demonstrated to block CD4+ T-cell activity by inducing apoptosis, inhibiting proliferation and suppressing co-stimulatory molecule activities (126). The immune suppressive actions mediated by human CD4+ CD25+ Foxp3+ Tregs have also been linked to HO-1 activity (58), however, the extent that HO-1 contributes to the immune suppressive actions of these cells remains debated in the literature (57, 128), and requires further investigation specifically in the context of cancer. CO has been demonstrated to improve Treg survival in hypoxic regions of the TME (129). Loss of HO-1 activity is associated with a loss of Tregs, whereas over-expression of HO-1 results in their accumulation (130) (Figure 4). Specifically, Tregs were demonstrated to induce tolerance in allografts when supplied with bilirubin or CO (131). In an elegant study which placed tumor bearing mice on a fasting mimicking-diet (FMD), the authors observed lowered HO-1 expression by the tumor cells, which rendered tumors more sensitive to doxorubicin and cyclophosphamide in the 4T1 model of breast cancer, and to doxorubicin in the B16 melanoma model, which resulted in CD8+ T-cell mediated tumor control associated with a loss of Tregs (130).
Several in vitro studies have demonstrated that HO-1 activity has the potential to suppress the expansion of T-cell populations (128, 132, 133). CO facilitates this through blocking CD4+ T-cell production of IL-2, a cytokine required for T-cell entry to the cell cycle (132) (Figure 4). In CD8+ T-cells, CO suppresses the expression and activity of caspase-3 and -8 through an up-regulation of a cyclin-dependent kinase inhibitor, p21cip1, a potent inhibitor against cell cycle progression (133). Interestingly, T-cells become insensitive to CO post-TCR signaling, highlighting the importance of HO-1/CO in facilitating a tolerogenic state in tissues through increasing the threshold for activation. The role of HO-1 activity in suppressing T-cell proliferation is also not just restricted to effector T-cell populations but also has been associated with the expansion of the immunomodulatory Treg population (128).
The pivotal role of HO-1 in modulating anti-tumor immune responses supports the consideration of HO-1 representing an innate immune checkpoint (2). Immune checkpoints are a family of regulatory proteins that act to suppress immune responses and T-cell activation/activity which are exploited in cancer to facilitate immune evasion. Therapeutically blocking the signaling of immune checkpoint molecules PD-1 and CTLA-4 has delivered unprecedented clinical responses in patients with cancer (134). Combining these therapies also improves the overall clinical response compared to the monotherapies (135), highlighting the redundency within this family of receptors. In a side-by-side comparison using 5-FU to elicit an anti-tumor immune response in MMTV-PyMT tumors, pharmacological inhibition of HO activity using SnMP displayed superior immunological control of tumor growth when compared to anti-PD-1 neutralizing antibodies (2). This observation demonstrated that, in some TMEs, HO-1 could be hierachically more important as an immune checkpoint than the clinically targeted PD-1, highlighting HO-1 as a potentially immportant immunotherapy target.
The Broad Immunomodulatory Roles of HO-1 in the TME
HO-1 has been demonstrated to influence a variety of cell responses which contribute to tumor progression and enforce the immune suppressive and tissue protective characteristics of the TME. The catabolites of HO-1 activity can interfere with the phenotype, activation states and cytokine profile of several stromal cell populations which facilitates immune evasion by the tumor (42). DCs are important in activating the inflammatory response and developing T-cell immune responses against cancer (136) (Figure 4). The expression of HO-1 inhibits the maturation of pro-inflammatory DCs and ROS production maintaining DCs in a tolerogenic state (29, 30). This is mediated through a direct effect of CO on these cells (30), as well as indirectly through modulating their cytokine profile, decreasing the expression of IL-12, IL-6, TNF-α and type I IFNs while up-regulating IL-10 production (29, 30) (Figure 4). IL-10 has been demonstrated to block DC maturation (15) and can induce HO-1 protein expression (82, 137), creating a positive feedback loop. Both CO and bilirubin have been demonstrated to also down-regulate MHCII expression in DCs (30, 126, 138) which compromises the ability of these cells to present antigens to CD4+ T-cells (Figure 4). Toll-like receptor-4 (TLR-4) signaling is an important maturation signal for DCs. Endogenous (non-pathogen associated) TLR-4 ligands have been identified in the TME and have broadly become regarded as damage associated molecular patterns (DAMPs), such as high-mobility group box 1 (HMGB1) protein and heat shock protein 90 (110). Although the mechanism still needs to be studied in the cancer setting, CO has the potential to compromise the generation of anti-tumor immune responses through modulating TLR-4 signaling. CO can regulate the interaction of TLR-4 with caveolin-1, the principle structural protein of the plasmalemmal caveolae which regulates inflammatory signals from the cell membrane (139, 140) (Figure 4). Macrophage polarization can also be directly modulated by CO which has been demonstrated in vitro to contribute to skewing an anti-inflammatory phenotype of these cells (141). Macrophage exposure to CO can modulate their cytokine expression profile through an effect on MAPK signaling, down regulating pro-inflammatory cytokines such as TNF-α, IL-1β, and macrophage inflammatory protein 1-β (MIP-1β), while conversely upregulating IL-10 production (137) (Figure 4). Biliverdin has also been demonstrated to upregulate IL-10 production by macrophages through a PI3K-Akt dependent pathway (142). Furthermore, CO can suppress macrophage activation to a pro-inflammatory state through modulating TLR signaling, iNOS expression and the release of HMGB1, leading to the inhibition of macrophage activation (143, 144). However, there is also in vivo evidence using A549 tumors that exogenous CO exposure can polarize macrophages to a pro-inflammatory anti-tumor phenotype which are directly involved in facilitating a control of tumor growth (56).
Neutrophil and macrophage migration and infiltration can be inhibited by both CO and biliverdin which has been demonstrated in non-cancer inflammation and ischemia studies respectively (96, 145) (Figure 4). Studying the effect of CO in allograft rejection also revealed a decreased infiltration of macrophages and T-cells (145). This could be a result of a role of HO-1/CO on modulating endothelial cell activation (26), which can influence the attachment and extravasation event from the blood to the TME, and requires investigation in the context of cancer. CO has been demonstrated to suppress the activation of endothelial cells in response to cytokines in vitro and reduced their expression of key adhesion molecules P- and E-selectin, ICAM-1 and VCAM that prevented neutrophil adhesion (146) (Figure 4). HO-1 activity may also suppress neutrophil (147, 148) and natural killer (NK) cell (149, 150) activation and effector function (Figure 4). HO-1 activity has been reported to suppress NK cell activation through suppressing their expression of activatory receptors such as NKG2D, NKp46 and NKp30, as well as compromising their ability to secrete IFN-γ and TNF-α (149) (Figure 4). These intriguing observations highlight the need to further explore the role of HO-1 on neutrophils and NK cell function in the context of the TME. However, it is clear that HO-1 activity has the ability to influence the anti-tumor immune response both directly, indirectly and at different stages of its development, allowing HO-1 to be exploited by cancer to facilitate in immune evasion.
HO-1 and Angiogenesis in Cancer
Neo-angiogenesis is pivotal to tumor progression and HO-1 has been demonstrated to modulate the process (42, 100, 151–153). In vitro models, not specific to cancer, have implicated a role for CO in promoting endothelial cell proliferation (154). Vascular endothelial growth factor (VEGF) is an important pro-angiogenic protein to cancer progression eliciting endothelial cell proliferation and vessel sprouting, and CO can act as a stimulus for inducing VEGF expression (155–157), which is mediated by a HIF-1α-dependent pathway (158, 159). However, there are conflicting reports in the literature that also suggest HO-1 over-expression can conversely elicit a down-regulation of VEGF and HIF-1α (92, 157), highlighting a potentially ‘tuning’ effect by CO that is dose dependent. Alongside these pro-proliferative roles of CO on vascular forming cells, CO has also been demonstrated to prevent endothelial cell apoptosis through NF-κB and p38 MAPK pathways and therefore indirectly influence angiogenesis, an effect which is reversed by inhibiting HO activity using SnMP (28, 137, 160). Pharmacological inhibition of HO-1 activity in LL2/OVA tumors also resulted in an elevation of tissue factor expression by endothelial cells in the TME. Tissue factor is a protein which initiates blood coagulation and is indicative of endothelial cell activation. This suggests that HO-1 activity may play a role in suppressing the activation state of these cells (26), which could influence the recruitment of immune cells to the TME (as discussed above). These diverse roles in which HO-1 activity has been implicated in facilitating angiogenesis provide an opportunity to target HO-1 as an anti-angiogenic strategy, however, it should be noted that in several preclinical models using pharmacological inhibition or genetic inactivation of HO-1 did not result in tumor control (2, 26, 27), suggesting that HO-1 could play a modulatory rather than non-redundant role in this process which may also be TME-dependent.
HO-1 in Metastasis
Metastasis, the ability of tumor cells to colonize sites distal to that of the primary tumor, accounts for 90% of cancer-related deaths and there is an emerging role of HO-1 in the process. However, the role of HO-1 in relation to metastasis still remains debated and may rely on variables which have yet to be fully elucidated. There are several studies supporting the pro-metastatic role of HO-1, where over-expression of the enzyme has correlated with an increase in metastatic potential (152, 161–163). In human advanced colorectal cancer, HO-1 is correlated to lymph node metastasis and a shorter disease-free survival time (164). In a loss of function example, pharmacological inhibition of HO-1 activity using SnMP in mice bearing orthotopic 4T1 mammary tumors, despite not controlling growth of the primary tumor, suppressed the number of pulmonary metastasis (27). Interestingly, SnMP did not affect the pulmonary seeding of intravenously injected 4T1 cells suggesting that the mechanism of action was potentially occurring at the primary tumor site. Ex vivo studies demonstrated that HO-1 activity and CO were able to facilitate transendothelial migration of tumor cells, implicating a potential role for HO-1 in facilitating the intravasation event via a mechanism that was independent of vascular leakiness (27). Tumor cells are capable of adopting a more motile mesenchymal-like phenotype, referred to as epithelial to mesenchymal transition (EMT), which facilitates the metastatic potential of the cells. In a model of human glioma, CO was demonstrated to be capable of increasing tumor cell migration (165). There is also direct evidence that HO-1 activity can contribute to EMT (166). In xenograft models of PC3 in which HO-1 was inactivated in the TAMs, E-cadherin expression was inhibited, accompanied by the up-regulation of mesenchymal markers Twist-1 and Snail in the tumor cells, indicating evidence of EMT (51). Matrix metalloproteinases (MMPs), are a family of matrix degrading proteases required to breakdown the extracellular matrix to allow tumor cell migration through the tissue. In an anti-metastatic role of the HO-1 axis, CO has also been demonstrated to down-regulate the expression of MMP9 to conversely decrease metastasis in models of breast cancer (86, 167).
Several studies also highlight a role for HO-1 within the metastatic niche in the lung, which represent sites to which tumor cells preferentially colonize. Myeloid expression of HO-1 in the lung has been demonstrated to promote lung metastatic colonization in vivo (168). In agreement, studies using intravenously injected B16 melanoma cells also observed an increase in lung colonization when HO-1 was over-expressed (152, 169). Further investigation is required in relation to the role of HO-1 and its role in metastasis and the metastatic niche, to elucidate the biological rules which predict the outcome of HO-1 activity in the TME.
Summary
Due to the breadth of the tumor promoting roles of HO-1, it is unsurprising that HO-1 is so widely expressed in cancer. These pathways are enacted by the biologically active catabolites of heme degradation to promote progression of the disease through anti-apoptotic, -oxidant, -inflammatory effects alongside pro-angiogenic and -metastatic effects. However, there are examples in the literature where HO-1 can play a converse anti-tumoral role. These contradicting roles of HO-1 highlight the complexity of the HO-1 axis in cancer. It is clear that in some cases the biological landscape of the specific TME may play a role in dictating the overall outcome of pharmacologically targeting HO-1 which can be dependent on the quality of the anti-tumor immune response and the degree of T-cell infiltration into the TME (2, 26). As the metabolic state of a cell can dictate the response to CO, such as is observed by its pro-apoptotic effects in malignant cells and anti-apoptotic effects in healthy cells (16). Also, the observation that in malignant tumor cells, HO-1 can become truncated and localized to the nucleus to elicit a transcriptional-regulatory role provides an intriguing additional layer to the functionality of this protein, but highlights the need to consider the cellular localization of HO-1 rather than just its presence or absence in the TME (11, 16, 23, 44). Also, there is scope for further studying the role of HO-1 expression level (92, 157) or catabolite concentration in dictating the response outcome in cancer (170, 171). Further in vitro investigations of the biological effects of endogenous cell derived heme catabolites produced by HO-1-expressing cells communicating to cells which do not express HO-1 is required to supplement the insight gained from exogenously supplied catabolites in such systems which could be at supra-physiological concentrations. In relation to exploiting our knowledge of the heme catabolites and HO-1 for therapeutic gain, the preclinical evidence demonstrating that exogenous CO exposure can deliver anti-tumor control provides a compelling translational avenue (16). In the context of boosting anti-tumor immunity, pharmacologically targeting HO activity as an immunotherapy approach may be equally attractive (2). However, no doubt as our understanding of the complexity of the TME increases, further roles for HO-1 will emerge in the years ahead.
Author Contributions
KNLH, JEA, and JNA wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work has been broadly supported by the Cancer Research UK King’s Health Partners Centre studentship and Experimental Cancer Medicine Centre at King’s College London, and the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. JEA is supported by the UK Medical Research Council (MRC/N013700/1) and is a member of the KCL MRC Doctoral Training Partnership in Biomedical Sciences.
References
1. Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA (1968) 61(2):748–55. doi: 10.1073/pnas.61.2.748
2. Muliaditan T, Opzoomer JW, Caron J, Okesola M, Kosti P, Lall S, et al. Repurposing Tin Mesoporphyrin as an Immune Checkpoint Inhibitor Shows Therapeutic Efficacy in Preclinical Models of Cancer. Clin Cancer Res An Off J Am Assoc Cancer Res (2018) 24(7):1617–28. doi: 10.1158/1078-0432.CCR-17-2587
3. Maines MD, Trakshel GM, Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem (1986) 261(1):411–9. doi: 10.1016/S0021-9258(17)42488-4
4. Trakshel GM, Kutty RK, Maines MD. Purification and characterization of the major constitutive form of testicular heme oxygenase. The noninducible isoform. J Biol Chem (1986) 261(24):11131–7. doi: 10.1016/S0021-9258(18)67358-2
5. Hayashi S, Omata Y, Sakamoto H, Higashimoto Y, Hara T, Sagara Y, et al. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene (2004) 336(2):241–50. doi: 10.1016/j.gene.2004.04.002
6. Jozkowicz A, Huk I, Nigisch A, Weigel G, Weidinger F, Dulak J. Effect of prostaglandin-J(2) on VEGF synthesis depends on the induction of heme oxygenase-1. Antioxid Redox Signaling (2002) 4(4):577–85. doi: 10.1089/15230860260220076
7. Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci U S A (2009) 106(37):15837–42. doi: 10.1073/pnas.0903419106
8. Miyata Y, Kanda S, Mitsunari K, Asai A, Sakai H. Heme oxygenase-1 expression is associated with tumor aggressiveness and outcomes in patients with bladder cancer: a correlation with smoking intensity. Trans Res J Lab Clin Med (2014) 164(6):468–76. doi: 10.1016/j.trsl.2014.06.010
9. Yin H, Fang J, Liao L, Maeda H, Su Q. Upregulation of heme oxygenase-1 in colorectal cancer patients with increased circulation carbon monoxide levels, potentially affects chemotherapeutic sensitivity. BMC Cancer (2014) 14:436. doi: 10.1186/1471-2407-14-436
10. Deininger MH, Meyermann R, Trautmann K, Duffner F, Grote EH, Wickboldt J, et al. Heme oxygenase (HO)-1 expressing macrophages/microglial cells accumulate during oligodendroglioma progression. Brain Res (2000) 882(1-2):1–8. doi: 10.1016/S0006-8993(00)02594-4
11. Gandini NA, Fermento ME, Salomon DG, Blasco J, Patel V, Gutkind JS, et al. Nuclear localization of heme oxygenase-1 is associated with tumor progression of head and neck squamous cell carcinomas. Exp Mol Pathol (2012) 93(2):237–45. doi: 10.1016/j.yexmp.2012.05.001
12. Mayerhofer M, Florian S, Krauth MT, Aichberger KJ, Bilban M, Marculescu R, et al. Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res (2004) 64(9):3148–54. doi: 10.1158/0008-5472.CAN-03-1200
13. Degese MS, Mendizabal JE, Gandini NA, Gutkind JS, Molinolo A, Hewitt SM, et al. Expression of heme oxygenase-1 in non-small cell lung cancer (NSCLC) and its correlation with clinical data. Lung Cancer (2012) 77(1):168–75. doi: 10.1016/j.lungcan.2012.02.016
14. Torisu-Itakura H, Furue M, Kuwano M, Ono M. Co-expression of thymidine phosphorylase and heme oxygenase-1 in macrophages in human malignant vertical growth melanomas. Jpn J Cancer Res (2000) 91(9):906–10. doi: 10.1111/j.1349-7006.2000.tb01033.x
15. Fest S, Soldati R, Christiansen NM, Zenclussen ML, Kilz J, Berger E, et al. Targeting of heme oxygenase-1 as a novel immune regulator of neuroblastoma. Int J Cancer (2016) 138(8):2030–42. doi: 10.1002/ijc.29933
16. Wegiel B, Gallo D, Csizmadia E, Harris C, Belcher J, Vercellotti GM, et al. Carbon Monoxide Expedites Metab Exhaustion Inhibit Tumor Growth. Cancer Res (2013) 73(23):7009–21. doi: 10.1158/0008-5472.CAN-13-1075
17. Goodman AI, Choudhury M, da Silva JL, Schwartzman ML, Abraham NG. Overexpression of the heme oxygenase gene in renal cell carcinoma Proceedings of the Society for Experimental Biology and Medicine. Soc Exp Biol Med (1997) 214(1):54–61. doi: 10.3181/00379727-214-44069
18. Jozkowicz A, Was H, Dulak J. Heme oxygenase-1 in tumors: is it a false friend? Antioxid Redox Signaling (2007) 9(12):2099–117. doi: 10.1089/ars.2007.1659
19. Shibahara S, Muller R, Taguchi H, Yoshida T. Cloning and expression of cDNA for rat heme oxygenase. Proc Natl Acad Sci USA (1985) 82(23):7865–9. doi: 10.1073/pnas.82.23.7865
20. Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA (1989) 86(1):99–103. doi: 10.1073/pnas.86.1.99
21. Kim HP, Wang X, Galbiati F, Ryter SW, Choi AM. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J (2004) 18(10):1080–9. doi: 10.1096/fj.03-1391com
22. Bindu S, Pal C, Dey S, Goyal M, Alam A, Iqbal MS, et al. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J Biol Chem (2011) 286(45):39387–402. doi: 10.1074/jbc.M111.279893
23. Lin Q, Weis S, Yang G, Weng YH, Helston R, Rish K, et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem (2007) 282(28):20621–33. doi: 10.1074/jbc.M607954200
24. Blann AD, Balakrishnan B, Ryan P, Lip GY. Increased levels of plasma haemoxygenase-1 in prostate cancer. Prostate Cancer Prostatic Dis (2011) 14(2):114–7. doi: 10.1038/pcan.2010.56
25. Vashist YK, Uzungolu G, Kutup A, Gebauer F, Koenig A, Deutsch L, et al. Heme oxygenase-1 germ line GTn promoter polymorphism is an independent prognosticator of tumor recurrence and survival in pancreatic cancer. J Surg Oncol (2011) 104(3):305–11. doi: 10.1002/jso.21926
26. Arnold JN, Magiera L, Kraman M, Fearon DT. Tumoral immune suppression by macrophages expressing fibroblast activation protein-alpha and heme oxygenase-1. Cancer Immunol Res (2014) 2(2):121–6. doi: 10.1158/2326-6066.CIR-13-0150
27. Muliaditan T, Caron J, Okesola M, Opzoomer JW, Kosti P, Georgouli M, et al. Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nat Commun (2018) 9(1):2951. doi: 10.1038/s41467-018-05346-7
28. Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, et al. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med (2000) 192(7):1015–26. doi: 10.1084/jem.192.7.1015
29. Chauveau C, Remy S, Royer PJ, Hill M, Tanguy-Royer S, Hubert FX, et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood (2005) 106(5):1694–702. doi: 10.1182/blood-2005-02-0494
30. Remy S, Blancou P, Tesson L, Tardif V, Brion R, Royer PJ, et al. Carbon monoxide inhibits TLR-induced dendritic cell immunogenicity. J Immunol (2009) 182(4):1877–84. doi: 10.4049/jimmunol.0802436
31. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
32. Pae HO, Oh GS, Choi BM, Chae SC, Chung HT. Differential expressions of heme oxygenase-1 gene in CD25- and CD25+ subsets of human CD4+ T cells. Biochem Biophys Res Commun (2003) 306(3):701–5. doi: 10.1016/S0006-291X(03)01037-4
33. Kutty RK, Maines MD. Purification and characterization of biliverdin reductase from rat liver. J Biol Chem (1981) 256(8):3956–62. doi: 10.1016/S0021-9258(19)69552-9
34. Fenton H. LXXIII.—Oxidation of tartaric acid in presence of iron. J Chem Soc Trans (1894) 65:899–910. doi: 10.1039/CT8946500899
35. Gutteridge JM, Smith A. Antioxidant protection by haemopexin of haem-stimulated lipid peroxidation. Biochem J (1988) 256(3):861–5. doi: 10.1042/bj2560861
36. Higdon AN, Benavides GA, Chacko BK, Ouyang X, Johnson MS, Landar A, et al. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. Am J Physiol Heart Circ Physiol (2012) 302(7):H1394–409. doi: 10.1152/ajpheart.00584.2011
37. Santoro AM, Lo Giudice MC, D’Urso A, Lauceri R, Purrello R, Milardi D. Cationic porphyrins are reversible proteasome inhibitors. J Am Chem Soc (2012) 134(25):10451–7. doi: 10.1021/ja300781u
38. Vallelian F, Deuel JW, Opitz L, Schaer CA, Puglia M, Lonn M, et al. Proteasome inhibition and oxidative reactions disrupt cellular homeostasis during heme stress. Cell Death Differ (2015) 22(4):597–611. doi: 10.1038/cdd.2014.154
39. Yoshida T, Kikuchi G. Features of the reaction of heme degradation catalyzed by the reconstituted microsomal heme oxygenase system. J Biol Chem (1978) 253(12):4230–6. doi: 10.1016/S0021-9258(17)34708-7
40. Yoshida T, Noguchi M, Kikuchi G. Oxygenated form of heme. heme oxygenase complex and requirement for second electron to initiate heme degradation from the oxygenated complex. J Biol Chem (1980) 255(10):4418–20. doi: 10.1016/S0021-9258(19)85506-0
41. Wang J, de Montellano PR. The binding sites on human heme oxygenase-1 for cytochrome p450 reductase and biliverdin reductase. J Biol Chem (2003) 278(22):20069–76. doi: 10.1074/jbc.M300989200
42. Was H, Dulak J, Jozkowicz A. Heme oxygenase-1 in tumor biology and therapy. Curr Drug Targ (2010) 11(12):1551–70. doi: 10.2174/1389450111009011551
43. Schaefer B, Behrends S. Translocation of heme oxygenase-1 contributes to imatinib resistance in chronic myelogenous leukemia. Oncotarget (2017) 8(40):67406–21. doi: 10.18632/oncotarget.18684
44. Sacca P, Meiss R, Casas G, Mazza O, Calvo JC, Navone N, et al. Nuclear translocation of haeme oxygenase-1 is associated to prostate cancer. Br J Cancer (2007) 97(12):1683–9. doi: 10.1038/sj.bjc.6604081
45. Nitti M, Piras S, Marinari UM, Moretta L, Pronzato MA, Furfaro AL. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants (2017) 6(2):26. doi: 10.3390/antiox6020029
46. Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, Puig-Kroger A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology (2010) 215(9-10):788–95. doi: 10.1016/j.imbio.2010.05.020
47. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/S1471-4906(02)02302-5
48. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
49. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
50. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front Immunol (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
51. Nemeth Z, Li M, Csizmadia E, Dome B, Johansson M, Persson JL, et al. Heme oxygenase-1 in macrophages controls prostate cancer progression. Oncotarget (2015) 6(32):33675–88. doi: 10.18632/oncotarget.5284
52. Alaluf E, Vokaer B, Detavernier A, Azouz A, Splittgerber M, Carrette A, et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight (2020) 5(11):e133929. doi: 10.1172/jci.insight.133929
53. Weis N, Weigert A, von Knethen A, Brune B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol Biol Cell (2009) 20(5):1280–8. doi: 10.1091/mbc.e08-10-1005
54. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science (2010) 330(6005):827–30. doi: 10.1126/science.1195300
55. Tchou J, Zhang PJ, Bi Y, Satija C, Marjumdar R, Stephen TL, et al. Fibroblast activation protein expression by stromal cells and tumor-associated macrophages in human breast cancer. Hum Pathol (2013) 44(11):2549–57. doi: 10.1016/j.humpath.2013.06.016
56. Nemeth Z, Csizmadia E, Vikstrom L, Li M, Bisht K, Feizi A, et al. Alterations of tumor microenvironment by carbon monoxide impedes lung cancer growth. Oncotarget (2016) 7(17):23919–32. doi: 10.18632/oncotarget.8081
57. Zelenay S, Chora A, Soares MP, Demengeot J. Heme oxygenase-1 is not required for mouse regulatory T cell development and function. Int Immunol (2007) 19(1):11–8. doi: 10.1093/intimm/dxl116
58. Choi BM, Pae HO, Jeong YR, Kim YM, Chung HT. Critical role of heme oxygenase-1 in Foxp3-mediated immune suppression. Biochem Biophys Res Commun (2005) 327(4):1066–71. doi: 10.1016/j.bbrc.2004.12.106
59. Murphy BJ, Laderoute KR, Short SM, Sutherland RM. The identification of heme oxygenase as a major hypoxic stress protein in Chinese hamster ovary cells. Br J Cancer (1991) 64(1):69–73. doi: 10.1038/bjc.1991.241
60. Tron K, Samoylenko A, Musikowski G, Kobe F, Immenschuh S, Schaper F, et al. Regulation of rat heme oxygenase-1 expression by interleukin-6 via the Jak/STAT pathway in hepatocytes. J Hepatol (2006) 45(1):72–80. doi: 10.1016/j.jhep.2005.12.019
61. Han Z, Varadharaj S, Giedt RJ, Zweier JL, Szeto HH, Alevriadou BR. Mitochondria-derived reactive oxygen species mediate heme oxygenase-1 expression in sheared endothelial cells. J Pharmacol Exp Ther (2009) 329(1):94–101. doi: 10.1124/jpet.108.145557
62. Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell (2002) 9(5):1031–44. doi: 10.1016/S1097-2765(02)00520-8
63. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the Hallmarks of Cancer. Cancer Cell (2018) 34(1):21–43. doi: 10.1016/j.ccell.2018.03.022
64. Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev (1999) 13(1):76–86. doi: 10.1101/gad.13.1.76
65. Katoh Y, Iida K, Kang MI, Kobayashi A, Mizukami M, Tong KI, et al. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys (2005) 433(2):342–50. doi: 10.1016/j.abb.2004.10.012
66. Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA (2002) 99(18):11908–13. doi: 10.1073/pnas.172398899
67. Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J (2002) 21(19):5216–24. doi: 10.1093/emboj/cdf516
68. Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J (2001) 20(11):2835–43. doi: 10.1093/emboj/20.11.2835
69. Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem (2005) 280(17):16891–900. doi: 10.1074/jbc.M500166200
70. Fukuda Y, Wang Y, Lian S, Lynch J, Nagai S, Fanshawe B, et al. Upregulated heme biosynthesis, an exploitable vulnerability in MYCN-driven leukemogenesis. JCI Insight (2017) 2(15):e92409. doi: 10.1172/jci.insight.92409
71. Fiorito V, Chiabrando D, Petrillo S, Bertino F, Tolosano E. The Multifaceted Role of Heme in Cancer. Front Oncol (2019) 9:1540. doi: 10.3389/fonc.2019.01540
72. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (2015) 3:83–92. doi: 10.2147/HP.S93413
73. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis (2018) 7(1):10. doi: 10.1038/s41389-017-0011-9
74. Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem (1997) 272(9):5375–81. doi: 10.1074/jbc.272.9.5375
75. Berra E, Roux D, Richard DE, Pouyssegur J. Hypoxia-inducible factor-1 alpha (HIF-1 alpha) escapes O(2)-driven proteasomal degradation irrespective of its subcellular localization: nucleus or cytoplasm. EMBO Rep (2001) 2(7):615–20. doi: 10.1093/embo-reports/kve130
76. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy Nature reviews. Cancer (2004) 4(1):11–22. doi: 10.1038/nrc1252
77. Dong Y, Liu Y, Bai H, Jiao S. Systematic assessment of the clinicopathological prognostic significance of tissue cytokine expression for lung adenocarcinoma based on integrative analysis of TCGA data. Sci Rep (2019) 9(1):6301. doi: 10.1038/s41598-019-42345-0
78. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res (2013) 73(3):1128–41. doi: 10.1158/0008-5472.CAN-12-2731
79. Terry CM, Clikeman JA, Hoidal JR, Callahan KS. TNF-alpha and IL-1alpha induce heme oxygenase-1 via protein kinase C, Ca2+, and phospholipase A2 in endothelial cells. Am J Physiol (1999) 276(5):H1493–501. doi: 10.1152/ajpheart.1999.276.5.H1493
80. Terry CM, Clikeman JA, Hoidal JR, Callahan KS. Effect of tumor necrosis factor-alpha and interleukin-1 alpha on heme oxygenase-1 expression in human endothelial cells. Am J Physiol (1998) 274(3):H883–91. doi: 10.1152/ajpheart.1998.274.3.H883
81. Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci USA (1994) 91(13):5987–91. doi: 10.1073/pnas.91.13.5987
82. Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med (2002) 8(3):240–6. doi: 10.1038/nm0302-240
83. Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radical Biol Med (2004) 37(8):1097–104. doi: 10.1016/j.freeradbiomed.2004.07.008
84. Taha H, Skrzypek K, Guevara I, Nigisch A, Mustafa S, Grochot-Przeczek A, et al. Role of heme oxygenase-1 in human endothelial cells: lesson from the promoter allelic variants. Arteriosclerosis Thrombosis Vasc Biol (2010) 30(8):1634–41. doi: 10.1161/ATVBAHA.110.207316
85. Lo SS, Lin SC, Wu CW, Chen JH, Yeh WI, Chung MY, et al. Heme oxygenase-1 gene promoter polymorphism is associated with risk of gastric adenocarcinoma and lymphovascular tumor invasion. Ann Surg Oncol (2007) 14(8):2250–6. doi: 10.1245/s10434-006-9290-7
86. Skrzypek K, Tertil M, Golda S, Ciesla M, Weglarczyk K, Collet G, et al. Interplay between heme oxygenase-1 and miR-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis. Antioxid Redox Signaling (2013) 19(7):644–60. doi: 10.1089/ars.2013.5184
87. Zhang X, Shan P, Alam J, Fu XY, Lee PJ. Carbon monoxide differentially modulates STAT1 and STAT3 and inhibits apoptosis via a phosphatidylinositol 3-kinase/Akt and p38 kinase-dependent STAT3 pathway during anoxia-reoxygenation injury. J Biol Chem (2005) 280(10):8714–21. doi: 10.1074/jbc.M408092200
88. Al-Owais MM, Scragg JL, Dallas ML, Boycott HE, Warburton P, Chakrabarty A, et al. Carbon monoxide mediates the anti-apoptotic effects of heme oxygenase-1 in medulloblastoma DAOY cells via K+ channel inhibition. J Biol Chem (2012) 287(29):24754–64. doi: 10.1074/jbc.M112.357012
89. Hill M, Pereira V, Chauveau C, Zagani R, Remy S, Tesson L, et al. Heme oxygenase-1 inhibits rat and human breast cancer cell proliferation: mutual cross inhibition with indoleamine 2,3-dioxygenase. FASEB J (2005) 19(14):1957–68. doi: 10.1096/fj.05-3875com
90. Andrés NC, Fermento ME, Gandini NA, Romero AL, Ferro A, Donna LG, et al. Heme oxygenase-1 has antitumoral effects in colorectal cancer: involvement of p53. Exp Mol Pathol (2014) 97(3):321–31. doi: 10.1016/j.yexmp.2014.09.012
91. Gandini NA, Alonso EN, Fermento ME, Mascaro M, Abba MC, Colo GP, et al. Heme oxygenase-1 has an antitumor role in breast cancer. Antioxid Redox Signal (2019) 30(18):2030–49. doi: 10.1089/ars.2018.7554
92. Vitek L, Gbelcova H, Muchova L, Vanova K, Zelenka J, Konickova R, et al. Antiproliferative effects of carbon monoxide on pancreatic cancer. Digestive Liver Dis Off J Ital Soc Gastroenterol Ital Assoc Study Liver (2014) 46(4):369–75. doi: 10.1016/j.dld.2013.12.007
93. DeBerardinis RJ, Chandel NS. We need to talk about the Warburg effect. Nat Metab (2020) 2(2):127–9. doi: 10.1038/s42255-020-0172-2
94. Chin BY, Jiang G, Wegiel B, Wang HJ, Macdonald T, Zhang XC, et al. Hypoxia-inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc Natl Acad Sci U States A (2007) 104(12):5109–14. doi: 10.1073/pnas.0609611104
95. Otterbein LE, Hedblom A, Harris C, Csizmadia E, Gallo D, Wegiel B. Heme oxygenase-1 and carbon monoxide modulate DNA repair through ataxia-telangiectasia mutated (ATM) protein. Proc Natl Acad Sci USA (2011) 108(35):14491–6. doi: 10.1073/pnas.1102295108
96. Fondevila C, Shen XD, Tsuchiyashi S, Yamashita K, Csizmadia E, Lassman C, et al. Biliverdin therapy protects rat livers from ischemia and reperfusion injury. Hepatology (2004) 40(6):1333–41. doi: 10.1002/hep.20480
97. Wang WW, Smith DL, Zucker SD. Bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats. Hepatology (2004) 40(2):424–33. doi: 10.1002/hep.20334
98. Song S, Wang S, Ma J, Yao L, Xing H, Zhang L, et al. Biliverdin reductase/bilirubin mediates the anti-apoptotic effect of hypoxia in pulmonary arterial smooth muscle cells through ERK1/2 pathway. Exp Cell Res (2013) 319(13):1973–87. doi: 10.1016/j.yexcr.2013.05.015
99. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
100. Suttner DM, Dennery PA. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J (1999) 13(13):1800–9. doi: 10.1096/fasebj.13.13.1800
101. Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, et al. Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem (1992) 267(25):18148–53. doi: 10.1016/S0021-9258(19)37165-0
102. Chiang S-K, Chen S-E, Chang L-C. A dual role of heme oxygenase-1 in cancer cells. Int J Mol Sci (2019) 20(1):39. doi: 10.3390/ijms20010039
103. Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget (2015) 6(27):24393–403. doi: 10.18632/oncotarget.5162
104. Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett (2018) 416:124–37. doi: 10.1016/j.canlet.2017.12.025
105. Berberat PO, Dambrauskas Z, Gulbinas A, Giese T, Giese N, Kunzli B, et al. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin Cancer Res (2005) 11(10):3790–8. doi: 10.1158/1078-0432.CCR-04-2159
106. Tan Q, Wang H, Hu Y, Hu M, Li X, Aodengqimuge, et al. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci (2015) 106(8):1023–32. doi: 10.1111/cas.12712
107. Miyake M, Fujimoto K, Anai S, Ohnishi S, Nakai Y, Inoue T, et al. Inhibition of heme oxygenase-1 enhances the cytotoxic effect of gemcitabine in urothelial cancer cells. Anticancer Res (2010) 30(6):2145–52. doi: 10.3892/or.2010.1125
108. Tang QF, Sun J, Yu H, Shi XJ, Lv R, Wei HC, et al. The Zuo Jin Wan Formula Induces Mitochondrial Apoptosis of Cisplatin-Resistant Gastric Cancer Cells via Cofilin-1. Evidence-Based Complementary Altern Med eCAM (2016) 2016:8203789. doi: 10.1155/2016/8203789
109. Tracey N, Creedon H, Kemp AJ, Culley J, Muir M, Klinowska T, et al. HO-1 drives autophagy as a mechanism of resistance against HER2-targeted therapies. Breast Cancer Res Treat (2020) 179(3):543–55. doi: 10.1007/s10549-019-05489-1
110. Opzoomer JW, Sosnowska D, Anstee JE, Spicer JF, Arnold JN. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front Immunol (2019) 10:1654. doi: 10.3389/fimmu.2019.01654
111. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med (2005) 202(12):1691–701. doi: 10.1084/jem.20050915
112. Geary SM, Lemke CD, Lubaroff DM, Salem AK. The combination of a low-dose chemotherapeutic agent, 5-fluorouracil, and an adenoviral tumor vaccine has a synergistic benefit on survival in a tumor model system. PloS One (2013) 8(6):e67904. doi: 10.1371/journal.pone.0067904
113. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ (2014) 21(1):15–25. doi: 10.1038/cdd.2013.67
114. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade. Ther Immun (2016) 44(2):343–54. doi: 10.1016/j.immuni.2015.11.024
115. Chen N, Wu L, Yuan H, Wang J. ROS/Autophagy/Nrf2 Pathway Mediated Low-Dose Radiation Induced Radio-Resistance in Human Lung Adenocarcinoma A549 Cell. Int J Biol Sci (2015) 11(7):833–44. doi: 10.7150/ijbs.10564
116. Caputo F, Vegliante R, Ghibelli L. Redox modulation of the DNA damage response. Biochem Pharmacol (2012) 84(10):1292–306. doi: 10.1016/j.bcp.2012.07.022
117. Zhang W, Qiao T, Zha L. Inhibition of heme oxygenase-1 enhances the radiosensitivity in human nonsmall cell lung cancer a549 cells. Cancer Biother Radiopharmaceut (2011) 26(5):639–45. doi: 10.1089/cbr.2010.0939
118. Meffert MK, Haley JE, Schuman EM, Schulman H, Madison DV. Inhibition of hippocampal heme oxygenase, nitric oxide synthase, and long-term potentiation by metalloporphyrins. Neuron (1994) 13(5):1225–33. doi: 10.1016/0896-6273(94)90060-4
119. Valaes T, Petmezaki S, Henschke C, Drummond GS, Kappas A. Control of jaundice in preterm newborns by an inhibitor of bilirubin production: studies with tin-mesoporphyrin. Pediatrics (1994) 93(1):1–11.
120. Lutton JD, Abraham NG, Drummond GS, Levere RD, Kappas A. Zinc porphyrins: potent inhibitors of hematopoieses in animal and human bone marrow. Proc Natl Acad Sci USA (1997) 94(4):1432–6. doi: 10.1073/pnas.94.4.1432
121. Di Noia MA, Van Driesche S, Palmieri F, Yang LM, Quan S, Goodman AI, et al. Heme oxygenase-1 enhances renal mitochondrial transport carriers and cytochrome C oxidase activity in experimental diabetes. J Biol Chem (2006) 281(23):15687–93. doi: 10.1074/jbc.M510595200
122. Megias J, Busserolles J, Alcaraz MJ. The carbon monoxide-releasing molecule CORM-2 inhibits the inflammatory response induced by cytokines in Caco-2 cells. Br J Pharmacol (2007) 150(8):977–86. doi: 10.1038/sj.bjp.0707184
123. Clavijo PE, Frauwirth KA. Anergic CD8+ T lymphocytes have impaired NF-kappaB activation with defects in p65 phosphorylation and acetylation. J Immunol (2012) 188(3):1213–21. doi: 10.4049/jimmunol.1100793
124. Gibbs PE, Maines MD. Biliverdin inhibits activation of NF-kappaB: reversal of inhibition by human biliverdin reductase. Int J Cancer (2007) 121(11):2567–74. doi: 10.1002/ijc.22978
125. Kim HS, Loughran PA, Billiar TR. Carbon monoxide decreases the level of iNOS protein and active dimer in IL-1beta-stimulated hepatocytes. Nitric Oxide (2008) 18(4):256–65. doi: 10.1016/j.niox.2008.02.002
126. Liu Y, Li P, Lu J, Xiong W, Oger J, Tetzlaff W, et al. Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J Immunol (2008) 181(3):1887–97. doi: 10.4049/jimmunol.181.3.1887
127. Wei Y, Chen P, de Bruyn M, Zhang W, Bremer E, Helfrich W. Carbon monoxide-releasing molecule-2 (CORM-2) attenuates acute hepatic ischemia reperfusion injury in rats. BMC Gastroenterol (2010) 10:42. doi: 10.1186/1471-230X-10-42
128. Biburger M, Theiner G, Schadle M, Schuler G, Tiegs G. Pivotal Advance: Heme oxygenase 1 expression by human CD4+ T cells is not sufficient for their development of immunoregulatory capacity. J Leukocyte Biol (2010) 87(2):193–202. doi: 10.1189/jlb.0508280
129. Dey M, Chang AL, Wainwright DA, Ahmed AU, Han Y, Balyasnikova IV, et al. Heme oxygenase-1 protects regulatory T cells from hypoxia-induced cellular stress in an experimental mouse brain tumor model. J Neuroimmunol (2014) 266(1-2):33–42. doi: 10.1016/j.jneuroim.2013.10.012
130. Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng CW, et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell (2016) 30(1):136–46. doi: 10.1016/j.ccell.2016.06.005
131. Lee SS, Gao W, Mazzola S, Thomas MN, Csizmadia E, Otterbein LE, et al. Heme oxygenase-1, carbon monoxide, and bilirubin induce tolerance in recipients toward islet allografts by modulating T regulatory cells. FASEB J (2007) 21(13):3450–7. doi: 10.1096/fj.07-8472com
132. Pae HO, Oh GS, Choi BM, Chae SC, Kim YM, Chung KR, et al. Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. J Immunol (2004) 172(8):4744–51. doi: 10.4049/jimmunol.172.8.4744
133. Song R, Mahidhara RS, Zhou Z, Hoffman RA, Seol DW, Flavell RA, et al. Carbon monoxide inhibits T lymphocyte proliferation via caspase-dependent pathway. J Immunol (2004) 172(2):1220–6. doi: 10.4049/jimmunol.172.2.1220
134. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348(6230):56–61. doi: 10.1126/science.aaa8172
135. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi: 10.1056/NEJMoa1302369
136. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell (2016) 30(2):324–36. doi: 10.1016/j.ccell.2016.06.003
137. Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med (2000) 6(4):422–8. doi: 10.1038/74680
138. Chora AA, Fontoura P, Cunha A, Pais TF, Cardoso S, Ho PP, et al. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest (2007) 117(2):438–47. doi: 10.1172/JCI28844
139. Ding JL, Li Y, Zhou XY, Wang L, Zhou B, Wang R, et al. Potential role of the TLR4/IRAK-4 signaling pathway in the pathophysiology of acute pancreatitis in mice. Inflammation Res Off J Eur Histamine Res Soc (2009) 58(11):783–90. doi: 10.1007/s00011-009-0048-0
140. Botto S, Gustin JK, Moses AV. The Heme Metabolite Carbon Monoxide Facilitates KSHV Infection by Inhibiting TLR4 Signaling in Endothelial Cells. Front Microbiol (2017) 8:568. doi: 10.3389/fmicb.2017.00568
141. Lee TS, Tsai HL, Chau LY. Induction of heme oxygenase-1 expression in murine macrophages is essential for the anti-inflammatory effect of low dose 15-deoxy-Delta 12,14-prostaglandin J2. J Biol Chem (2003) 278(21):19325–30. doi: 10.1074/jbc.M300498200
142. Wegiel B, Baty CJ, Gallo D, Csizmadia E, Scott JR, Akhavan A, et al. Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J Biol Chem (2009) 284(32):21369–78. doi: 10.1074/jbc.M109.027433
143. Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med (2006) 203(10):2377–89. doi: 10.1084/jem.20060845
144. Tsoyi K, Nizamutdinova IT, Jang HJ, Mun L, Kim HJ, Seo HG, et al. Carbon monoxide from CORM-2 reduces HMGB1 release through regulation of IFN-beta/JAK2/STAT-1/INOS/NO signaling but not COX-2 in TLR-activated macrophages. Shock (2010) 34(6):608–14. doi: 10.1097/SHK.0b013e3181e46f15
145. Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med (2003) 9(2):183–90. doi: 10.1038/nm817
146. Nassour I, Kautza B, Rubin M, Escobar D, Luciano J, Loughran P, et al. Carbon monoxide protects against hemorrhagic shock and resuscitation-induced microcirculatory injury and tissue injury. Shock (2015) 43(2):166–71. doi: 10.1097/SHK.0000000000000264
147. Graca-Souza AV, Arruda MA, de Freitas MS, Barja-Fidalgo C, Oliveira PL. Neutrophil activation by heme: implications for inflammatory processes. Blood (2002) 99(11):4160–5. doi: 10.1182/blood.V99.11.4160
148. Porto BN, Alves LS, Fernandez PL, Dutra TP, Figueiredo RT, Graca-Souza AV, et al. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J Biol Chem (2007) 282(33):24430–6. doi: 10.1074/jbc.M703570200
149. Gómez-Lomelí P, Bravo-Cuellar A, Hernández-Flores G, Jave-Suárez LF, Aguilar-Lemarroy A, Lerma-Díaz JM, et al. Increase of IFN-gamma and TNF-alpha production in CD107a + NK-92 cells co-cultured with cervical cancer cell lines pre-treated with the HO-1 inhibitor. Cancer Cell Int (2014) 14(1):100. doi: 10.1186/s12935-014-0100-1
150. Furfaro AL, Ottonello S, Loi G, Cossu I, Piras S, Spagnolo F, et al. HO-1 downregulation favors BRAF(V600) melanoma cell death induced by Vemurafenib/PLX4032 and increases NK recognition. Int J Cancer (2020) 146(7):1950–62. doi: 10.1002/ijc.32611
151. Sunamura M, Duda DG, Ghattas MH, Lozonschi L, Motoi F, Yamauchi J-I, et al. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis (2003) 6(1):15–24. doi: 10.1023/A:1025803600840
152. Was H, Cichon T, Smolarczyk R, Rudnicka D, Stopa M, Chevalier C, et al. Overexpression of heme oxygenase-1 in murine melanoma: increased proliferation and viability of tumor cells, decreased survival of mice. Am J Pathol (2006) 169(6):2181–98. doi: 10.2353/ajpath.2006.051365
153. Miyake M, Fujimoto K, Anai S, Ohnishi S, Kuwada M, Nakai Y, et al. Heme oxygenase-1 promotes angiogenesis in urothelial carcinoma of the urinary bladder. Oncol Rep (2011) 25(3):653–60. doi: 10.3892/or.2010.1125
154. Li Volti G, Sacerdoti D, Sangras B, Vanella A, Mezentsev A, Scapagnini G, et al. Carbon monoxide signaling in promoting angiogenesis in human microvessel endothelial cells. Antioxid Redox Signaling (2005) 7(5-6):704–10. doi: 10.1089/ars.2005.7.704
155. Hirai K, Sasahira T, Ohmori H, Fujii K, Kuniyasu H. Inhibition of heme oxygenase-1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int J Cancer (2007) 120(3):500–5. doi: 10.1002/ijc.22287
156. Sass G, Leukel P, Schmitz V, Raskopf E, Ocker M, Neureiter D, et al. Inhibition of heme oxygenase 1 expression by small interfering RNA decreases orthotopic tumor growth in livers of mice. Int J Cancer (2008) 123(6):1269–77. doi: 10.1002/ijc.23695
157. Ferrando M, Gueron G, Elguero B, Giudice J, Salles A, Leskow FC, et al. Heme oxygenase 1 (HO-1) challenges the angiogenic switch in prostate cancer. Angiogenesis (2011) 14(4):467–79. doi: 10.1007/s10456-011-9230-4
158. Choi YK, Kim CK, Lee H, Jeoung D, Ha KS, Kwon YG, et al. Carbon monoxide promotes VEGF expression by increasing HIF-1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein. J Biol Chem (2010) 285(42):32116–25. doi: 10.1074/jbc.M110.131284
159. Cheng C-C, Guan S-S, Yang H-J, Chang C-C, Luo T-Y, Chang J, et al. Blocking heme oxygenase-1 by zinc protoporphyrin reduces tumor hypoxia-mediated VEGF release and inhibits tumor angiogenesis as a potential therapeutic agent against colorectal cancer. J Biomed Sci (2016) 23(1):18. doi: 10.1186/s12929-016-0219-6
160. Brouard S, Berberat PO, Tobiasch E, Seldon MP, Bach FH, Soares MP. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-kappa B to protect endothelial cells from tumor necrosis factor-alpha-mediated apoptosis. J Biol Chem (2002) 277(20):17950–61. doi: 10.1074/jbc.M108317200
161. Tertil M, Skrzypek K, Florczyk U, Weglarczyk K, Was H, Collet G, et al. Regulation and novel action of thymidine phosphorylase in non-small cell lung cancer: crosstalk with Nrf2 and HO-1. PloS One (2014) 9(5):e97070–0. doi: 10.1371/journal.pone.0097070
162. Huang S-M, Lin C, Lin H-Y, Chiu C-M, Fang C-W, Liao K-F, et al. Brain-derived neurotrophic factor regulates cell motility in human colon cancer. Endocrine-related Cancer (2015) 22(3):455–64. doi: 10.1530/ERC-15-0007
163. Tertil M, Golda S, Skrzypek K, Florczyk U, Weglarczyk K, Kotlinowski J, et al. Nrf2-heme oxygenase-1 axis in mucoepidermoid carcinoma of the lung: Antitumoral effects associated with down-regulation of matrix metalloproteinases. Free Radical Biol Med (2015) 89:147–57. doi: 10.1016/j.freeradbiomed.2015.08.004
164. Kimura S, Aung NY, Ohe R, Yano M, Hashimoto T, Fujishima T, et al. Increasing heme oxygenase-1-expressing macrophages indicates a tendency of poor prognosis in advanced colorectal cancer. Digestion (2020) 101(4):401–10. doi: 10.1159/000500225
165. Castruccio Castracani C, Longhitano L, Distefano A, Di Rosa M, Pittalà V, Lupo G, et al. Heme Oxygenase-1 and Carbon Monoxide Regulate Growth and Progression in Glioblastoma Cells. Mol Neurobiol (2020) 57(5):2436–46. doi: 10.1007/s12035-020-01869-7
166. Zhao Z, Zhao J, Xue J, Zhao X, Liu P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am J Cancer Res (2016) 6(10):2162–77.
167. Lin CW, Shen SC, Hou WC, Yang LY, Chen YC. Heme oxygenase-1 inhibits breast cancer invasion via suppressing the expression of matrix metalloproteinase-9. Mol Cancer Ther (2008) 7(5):1195–206. doi: 10.1158/1535-7163.MCT-07-2199
168. Lin HH, Chiang MT, Chang PC, Chau LY. Myeloid heme oxygenase-1 promotes metastatic tumor colonization in mice. Cancer Sci (2015) 106(3):299–306. doi: 10.1111/cas.12604
169. Liu Q, Wang B, Yin Y, Chen G, Wang W, Gao X, et al. Overexpressions of HO-1/HO-1G143H in C57/B6J mice affect melanoma B16F10 lung metastases rather than change the survival rate of mice-bearing tumours. Exp Biol Med (Maywood) (2013) 238(6):696–704. doi: 10.1177/1535370213490628
170. Józkowicz A, Huk I, Nigisch A, Weigel G, Dietrich W, Motterlini R, et al. Heme oxygenase and angiogenic activity of endothelial cells: stimulation by carbon monoxide and inhibition by tin protoporphyrin-IX. Antioxid Redox Signaling (2003) 5(2):155–62. doi: 10.1089/152308603764816514
Keywords: heme oxygenase-1 (HO-1), cancer, cytoprotection, tumor immunology, angiogenesis, tumor associated macrophages (TAMs), metastasis
Citation: Luu Hoang KN, Anstee JE and Arnold JN (2021) The Diverse Roles of Heme Oxygenase-1 in Tumor Progression. Front. Immunol. 12:658315. doi: 10.3389/fimmu.2021.658315
Received: 25 January 2021; Accepted: 15 March 2021;
Published: 31 March 2021.
Edited by:
Giovanna Schiavoni, National Institute of Health (ISS), ItalyReviewed by:
Barbara Wegiel, Beth Israel Deaconess Medical Center and Harvard Medical School, United StatesLeo E. Otterbein, Beth Israel Deaconess Medical Center and Harvard Medical School, United States
Copyright © 2021 Luu Hoang, Anstee and Arnold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James N. Arnold, james.n.arnold@kcl.ac.uk