 Lisa Haas
Lisa Haas Anna C. Obenauf
Anna C. Obenauf- Research Institute of Molecular Pathology (IMP), Vienna Biocenter (VBC), Vienna, Austria
For decades, cancer was considered a disease driven by genetic mutations in tumor cells, therefore afflicting a single cell type. This simplified view was slowly replaced by the understanding that interactions between malignant cells and neighboring stromal and immune cells—the tumor microenvironment (TME)—profoundly shape cancer progression. This understanding paved the way for an entirely new form of therapy that targets the immune cell compartment, which has revolutionized the treatment of cancer. In particular, agents activating T lymphocytes have become a key focus of these therapies, as they can induce durable responses in several cancer types. However, T cell targeting agents only benefit a fraction of patients. Thus, it is crucial to identify the roles of other immune cell types in the TME and understand how they influence T cell function and/or whether they present valuable therapeutic targets themselves. In this review, we focus on the myeloid compartment of the TME, a heterogeneous mix of cell types with diverse effector functions. We describe how distinct myeloid cell types can act as enemies of cancer cells by inducing or enhancing an existing immune response, while others act as strong allies, supporting tumor cells in their malignant growth and establishing an immune evasive TME. Specifically, we focus on the role of myeloid cells in the response and resistance to immunotherapy, and how modulating their numbers and/or state could provide alternative therapeutic entry-points.
Introduction
Myeloid cells are a diverse group of cells belonging to the innate immune system that are prone to adapt their phenotype to their tissue of residence (1). Thus, in cancer, they exist in a vast amount of different states and exert a range of distinct functions (Figure 1). Among those myeloid cells, macrophages, dendritic cells (DCs), and myeloid-derived suppressor cells (MDSCs) have received much attention in the last decades, due to their ability to both initiate or suppress an anti-tumor immune response (Figure 2) (2). In the following, we will specifically focus on those three groups of myeloid cells and provide an overview of their roles as cancer cell allies or enemies.
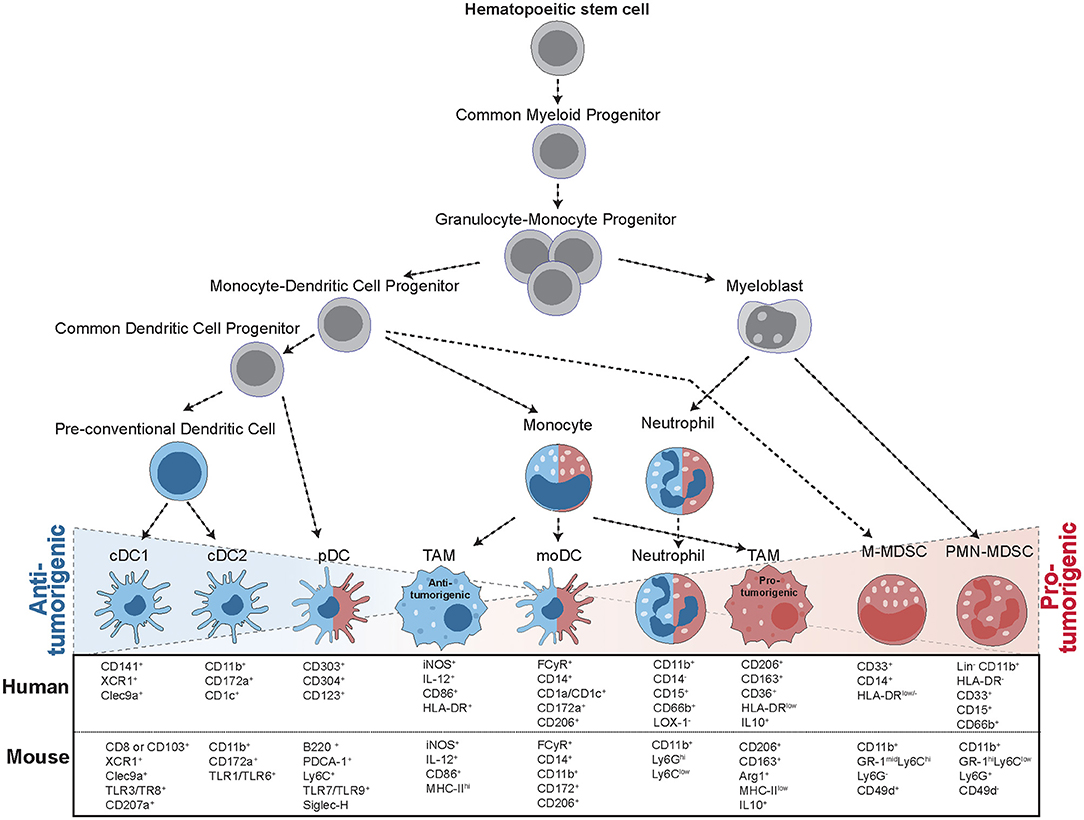
Figure 1. Progression from hematopoietic stem cells (HSC) to tumor-infiltrating myeloid cells. The formation of tumor-infiltrating myeloid cells occurs in a step-wise process: In the bone marrow, HSCs give rise to common myeloid progenitors (CMP), which give rise to granulocyte-monocyte progenitors (GMP). GMPs then further specify into myeloblasts (MB) and monocyte-dendritic cell progenitors (MDP). These precursors then differentiate into a range of different cell types with anti-tumorigenic (blue) and pro-tumorigenic (red) capacities. MDPs can give rise to a common dendritic cell progenitor (CDP), further leading to the formation of conventional DCs (cDCs) or plasmacytoid (pDCs). MDPs also form monocytes, giving rise to monocytic DCs (moDCs) or differentiating into tumor-associated macrophages (TAMS) upon instruction by the tumor. Macrophages can display multiple different activation states, ranging from anti-tumorigenic to pro-tumorigenic subsets. MBs give rise to mature neutrophils or polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs), while monocytic MDSCs (M-MDSCs) arise from MDPs upon instruction by inflammatory signals. All of these cell types are characterized by specific surface marker expression, indicated for both human and mouse cells.
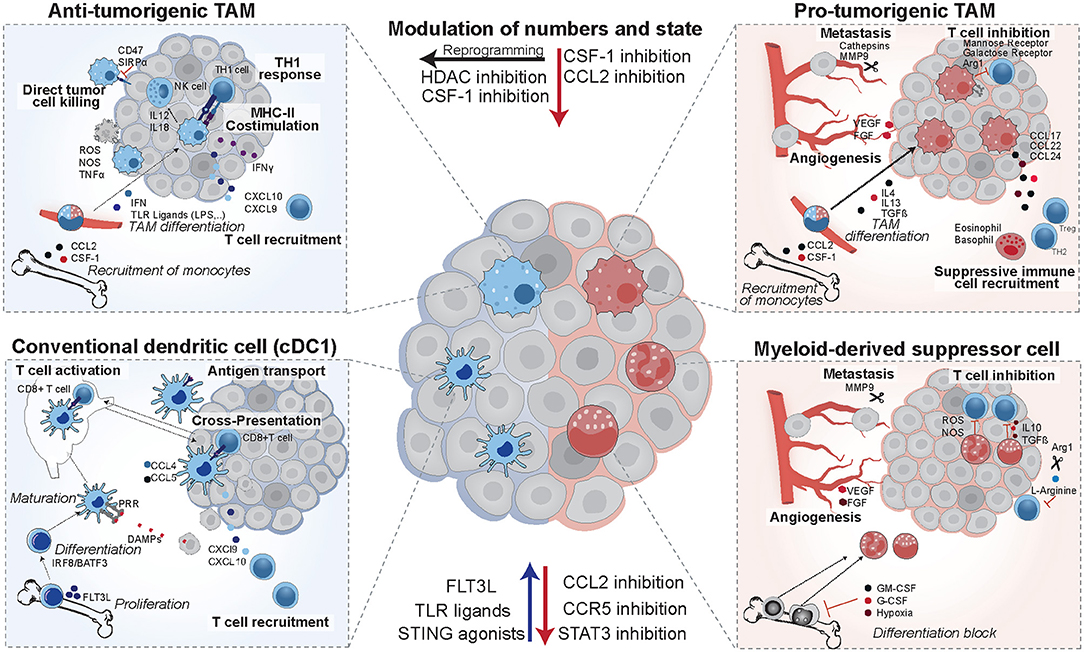
Figure 2. Opposing functions of tumor-infiltrating myeloid cells. Anti-tumorigenic TAMs arise from circulating monocytes in response to TLR ligands and interferon. They are characterized by high expression of costimulatory molecules and MHCII. In mouse models they were shown to induce potent TH1 responses and augment NK cells responses. cDC1 dendritic cells differentiate in response to FLT3L, mature upon recognition of danger associated molecular patterns (DAMPs), and then induce T cell activation via antigen presentation on MHCI. They establish a favorable cytokine environment in the tumor (CXCL9, CXCL10) and murine studies revealed that they are recruited in response to CCL4 and CCL5. In patients, they have positive prognostic value, correlate with T cell infiltration and are enriched in immunotherapy responders. Their numbers and maturation state can be enhanced by FLT3L, TLR ligands, or STING agonists. Pro-tumorigenic TAMs arise from circulating monocytes in response to IL4, IL13, and TGFβ, and establish an immune suppressive environment via recruitment of eosinophils, basophils, Tregs, and TH2 cells. They are pro-metastatic and induce angiogenesis, and their recruitment can be reduced by CSF-1 and CCL2 inhibitors in pre-clinical models. In addition, mouse models identified that they can be re-educated to an anti-tumorigenic state using HDAC inhibitors. MDSCs form from immature myeloid progenitors upon stimulation by the tumor and suppress T cell activity via IL10, TGFβ, and production of reactive oxygen and nitrogen species (ROS and NOS). They deplete intracellular L-arginine pools and hamper T cell proliferation in murine models and in patients their presence is a negative prognostic factor.
Dendritic Cells
Since their identification in mice in 1973 by Steinman and Cohn, DCs have become widely accepted as important players in the network of phagocytizing and antigen presenting cells (APCs) that sculpt immune outcomes (3). In tumor immunity, DCs have predominantly an anti-tumorigenic role. DCs arise from a common bone marrow (BM) progenitor—the common dendritic cell progenitor (CDP)—and then differentiate into plasmacytoid (pDCs) and precursors for conventional dendritic cells (cDCs) (Figure 1). These immature DCs subsequently migrate out of the bone marrow and colonize peripheral tissues, where they encounter antigens (4–8). The maturation of DCs represents a critical step in their life-cycle, allowing them to gain full APC capacities. Maturation is initiated upon recognition of danger-associated molecular patterns (DAMPs) via pattern recognition receptors (PRRs), where different DC subsets express different PRRs, further contributing to their functional specification. Upon maturation, DCs upregulate their antigen presentation machinery and costimulatory molecules, transforming themselves into potent T cell activators and thus bridging innate and adaptive immunity (9, 10). DCs can license anti-tumor immune responses by processing and cross-presenting exogenous antigens via MHC class I molecules to CD8 T cells, presenting antigens via MHC class II molecules to CD4 T cells, and secreting immune-stimulatory cytokines. In this capacity, they have become an integral part of the cancer immunity cycle and are attractive targets for immunotherapy (11, 12).
cDCs Are Potent Activators of Anti-tumor Immunity
cDCs differentiate into two subsets—cDC1 and cDC2—which are distinguished by their differential marker expression (Figure 1), transcription factor (TF) dependency, and functions. The differentiation into cDC1s or cDC2s is instructed by different chemokines and single cell sequencing studies in mice revealed distinct gene signatures that become evident early after the differentiation from CDPs (Figure 1): cDC1s are instructed by FLT3L and express the TFs IRF8, BATF3, and ID2, cDC2s are instructed by GM-CSF and are dependent on the TF IRF4, Notch2, and RelB (4, 8, 13, 14).
The role of cDC1 cells in anti-tumor immunity is well-established (15, 16). cDC1s are present as both lymph node resident (CD8+) and migratory (CD103+) populations. Lymph node resident DCs sample antigens in blood and lymph fluid, and migratory cDC1s transport antigens from the peripheral tissue to lymph nodes and spleen. This is indicated by the ability of CD103+ cDC1s to transport tumor-derived fluorescent proteins to the lymph node in a CCR7-dependent manner (17, 18). A substantial fraction of intratumoral CD103+ cDC1s does not migrate to the lymph node, yet they still play a crucial role in anti-tumor immunity. In mouse models those intratumoral, non-migratory CD103+ cDC1s were shown to mediate their effects via direct antigen presentation and establishment of a favorable chemokine environment and were found necessary for tumor control in a lymph node-independent manner (13, 17). They are an important source of CXCL9 and CXCL10 in tumors, which makes them indispensable for the infiltration of both naïve and pre-activated T cells. In patients, the levels of CD103+ transcripts correlate with the levels of CXCL9 and CXCL10, and degree of T cell infiltration (19, 20). The crucial role of CD103+ cDC1s has been further substantiated using BATF3−/− mice devoid of CD103+ cDC1s, which fail to reject immunogenic cancer cell lines and are unresponsive to immune checkpoint inhibition (17, 19, 21, 22).
In contrast to cDC1s, the role of the more heterogeneous population of CD11b+ cDC2s in anti-tumor immunity is less well-explored. They are superior to cDC1s in the induction of CD4 T cell responses via antigen presentation on MHCII, and have been shown to activate TH17 cells, a cell type with controversial roles in cancer that produces high levels of pro-inflammatory cytokines (23, 24). Compared to cDC1s, cDC2s fail to deliver antigen to lymph nodes. Because they have lower levels of endocytic receptors, higher levels of lysosomal enzymes and a lower phagosomal pH, it was hypothesized that antigens are directly degraded during migration instead of being further processed and presented on the surface (8, 13, 25). However, reduced antigen presentation of cDC2s may (also) be due to a lack of appropriate stimuli in the tumor and if stimulated, cDC2s may still play an important role in anti-tumor immunity. This is supported by studies showing that immune responses induced by the TLR7 agonist R848, acting on cDC2s, or anthracyclines, also induce protection in BATF3-deficient mice (26, 27).
pDCs and moDCs Have Antagonistic Roles in Cancer Immunity
DCs display high functional plasticity and despite having largely anti-tumorigenic capacities, they can under certain circumstances for example when present in an immature state, act immune suppressive. This is illustrated by the complex role of pDCs in tumor immunity. pDCs express MHCII, costimulatory molecules, and a narrow set of TLR receptors and have been identified as the main producers of Type I IFN upon activation by DAMPs (1, 28). Despite their capacity to produce Type I IFN, the presence of pDCs is a poor prognostic marker in breast cancer, melanoma, and ovarian cancer in human and animal models (29–32). This could be due to the poor activation of pDCs in the TME and an active instruction of pDCs by the tumor to fulfill immune-suppressive functions, such as production of IDO, IL10, or OX40 expression (33). Monocytic DCs (moDCs or inflammatory DCs) have a different origin and differentiate from Ly6Chigh monocytes in the context of cancer or inflammation (Figure 1). They are efficient in the uptake and processing of antigens and correlate with CD8+ T cell infiltration in several tumor models (34). Yet in direct contrast, they can also display an immune-suppressive phenotype, based on high expression of iNOS, TNF-α, IL-6, IL-10, and their capacity to hamper T cell proliferation in vitro, as it was shown using moDCs isolated from murine lung cancer models (23, 34). Thus, further investigation is needed to understand the pro- vs. anti-tumorigenic functions of this complex cell type, the tumor-derived signals that skew them, and particularly how this plays out in patient settings.
Tumors Inhibit DC Functionality on Multiple Levels
In addition to the diverse effects of DCs on tumor cells, in return, the tumors can interfere with DC functionality, either by affecting their differentiation or by suppressing their activation and maturation at the tumor site. Many tumor-secreted factors affect DC differentiation. For example, IL-6 and CSF-1 promote lineage commitment toward suppressive monocytes (35), and vascular endothelial growth factor (VEGF) inhibits DC maturation by suppressing NFκB signaling in hematopoietic progenitors (36). In addition, secreted factors can also directly inhibit the anti-tumor activity of DCs, such as TGF-β, which can inhibit antigen uptake in vitro and it was shown that inhibiting TGF-β signaling synergizes with immunotherapy in pre-clinical mouse models (15, 37, 38). In the local TME, metabolic dysfunction can hamper DC activity. For example, high levels of lactic acids were shown to interfere with DC activation and antigen presentation (39). Studies in mouse models showed that lipid peroxidation byproducts can induce continuous activation of the TF XBP1 in DCs, resulting in abnormal lipid accumulation and DC dysfunctionality (40). Recently, it became clear that the TME is strongly influenced by the oncogenic pathways driving cancer progression, which have a profound impact on the immune cell infiltrate (41, 42). In the context of DCs, upregulated beta-catenin signaling reduces infiltration of cDC1s via reducing the production of CCL4, among other chemokines (43). Elevated COX activity in tumor cells results in production of prostaglandin E2, which reduces NK cell infiltration and thus reduced the cDC1 recruitment factors XCL1 and CCL5 in the tumor microenvironment. Consequently, tumors with high prostaglandin E2 displayed reduced cDC1 levels, contributing to reduced effector T cell infiltration (44).
DCs Promote Response to Immunotherapy
CD8 T cell priming against tumor-specific antigens requires cross-presentation of the antigen on an MHC I complex by DCs and marks a crucial step for mounting a functional T cell response (45). Indeed, the presence of cDC1s in human tumors correlates with T cell infiltration levels and increased survival in breast, lung, and head and neck cancer patients (13). Moreover, murine studies using cDC1-deficient BATF3−/− mice highlighted their crucial importance for the response to immunotherapy (13, 17, 19, 46). Recently, a systematic comparison of biopsies from patients responding vs. non-responding to immunotherapy identified intratumoral abundance of cDC1s (CD141+ in humans) as predictive for immunotherapy success (47). This is in line with a second study that characterized IL-12 producing BATF3+ DCs as crucial for immunotherapy success in mice and showed that IL-12 activates lymphocyte effector functions in patients (48).
Targeting DCs as a Therapeutic Strategy
The central role of DCs in the initiation of immunity and their positive effect on patient survival provide a strong rationale to harness DCs and boost an endogenous anti-tumor immune response. To this end, different approaches are being explored, including: (1) increasing intra-tumoral DC numbers, (2) boosting DC maturation and function, and/or (3) alleviating tumor cell-mediated DC repression (8, 49). Vaccination strategies to increase DC numbers using both non-targeting and targeting vaccines represented a first wave of therapies that was initiated more than two decades ago (50). Non- targeting vaccines composed of peptides together with adjuvant agents showed limited clinical success and were later improved to contain patients' antigenic peptides in combination with the chemokine GM-CSF, resulting in clinical responses (51, 52). In addition, GVAX—a vaccine containing cancer cells overexpressing GM-CSF—was shown to attract and activate DCs in patients, and later to have some clinical activity (53). There remains however a big discrepancy between the capacity of these vaccinations to induce DC activation and their actual clinical efficacy. This could be due to a suppressive TME and exhaustion of T cells, and thus combination therapies may be the key to their success and are being actively explored in pre-clinical and clinical studies (12, 50, 53, 54). In 2012, a combination trial of GVAX and checkpoint inhibition was shown to be clinically safe (55) and more recently in 2016, an overall response rate of 38% was achieved in patients receiving transfer of modified, autologous DCs with checkpoint inhibition (56). Intra-tumoral injection of FLT3L increases numbers of circulating cDC1s, mobilizes DCs to the TME and has been successful in murine studies in combination with Poly I:C induced maturation (17). In patients, injections of FLT3L resulted in an increase of circulating cDCs (57).
An alternative approach is the maturation of DCs, which results in high expression of chemokines, costimulatory molecules and antigen presentation (9). Different maturation cocktails, comprising proinflammatory cytokines or TLR ligands have been evaluated in clinics and were shown to induce robust T cell activation capacities in DCs (58). To reduce side effects a direct intra-tumoral administration of maturation stimuli may be preferred and direct and abscopal effects of the TLR ligands Poly I:C or CpG are being evaluated (59). The STING pathway, sensing cytoplasmic DNA and inducing prominent Type I interferon release from DCs, has been another focus of intense research and modified cyclic dinucleotides, mimicking the endogenous STING ligands, have progressed into clinical trials (60). In addition, in 2018 a small molecule STING agonist has been published to induce potent, long-lasting responses in mice bearing colon cancer (61). While many of these approaches focus on cDC1s, triggering the release of IFN by pDCs could be an alternative entry point, which is under active investigation in checkpoint inhibitor resistant melanoma patients (62, 63).
Tumor-Associated Macrophages (TAMs)
Macrophages are a heterogeneous population of myeloid cells and are highly abundant in many cancer types. Their heterogeneity is influenced by: (1) their developmental origin, (2) their tissue of residence, and (3) the environmental cues they are exposed to (64). This is reflected by the vast number of different activation states, ranging from anti-tumorigenic to strongly pro-tumorigenic phenotypes.
TAMs Are a Heterogeneous Population of Myeloid Cells With Different Developmental Origins
In tumors, it was predominantly believed that TAMs arise from circulating monocytes that are recruited from the BM or spleen via cytokines such as CCL2 and CSF-1. However, macrophages can also arise from embryonic precursors and develop into tissue-resident macrophages, such as microglia in the brain, alveolar macrophages in the lung, or Kupffer cells in the liver (65, 66). In recent years, it has become clear that both monocyte-derived and tissue-resident macrophages play a role in tumorigenesis. Lineage tracing studies in mouse brain tumors revealed that both tissue-resident and monocyte-derived macrophages populate brain tumors, and macrophages of dual origin were reported in pancreatic ductal adenocarcinoma (67–70). The identification of a unique marker to characterize these heterogeneous TAM populations has proven difficult. In murine macrophages the glycoprotein CD68 is fairly specific and in combination with F4/80 identifies the majority of TAMs. In humans, CD68 is less specific and also expressed on granulocytes, dendritic cells, endothelial cells, fibroblasts and some lymphoid subsets (71). Due to the lack of a specific marker, the scavenger receptor CD163 (in humans M130) is often used in combination with CD68 to identify TAMs in humans (2). Moreover, CD49D can be used as a discriminatory marker between bone-marrow derived macrophages recruited to the brain and tissue-resident microglia in both mouse and humans and CD45 expression levels allow to distinguish these cell types in murine tumors (67).
Due to their substantial heterogeneity, TAMs need further sub-classification. They are commonly divided into “classically activated” M1 and “alternatively activated” M2 macrophages, with M1 referring to anti-tumorigenic and M2 to pro-tumorigenic macrophages. However, this classification is an oversimplification and the M1/M2 activation states present the extremes of a large spectrum of different functional states with various features (72, 73). Pro- and anti-tumorigenic TAMs are instructed by different sets of stimuli: anti-tumorigenic TAMs arise in response to TLR ligands and IFN, whereas pro-tumorigenic TAMs expand in response to IL4, IL13, TGFβ, and glucocorticoids (73–76). TAMs with anti-tumorigenic potential produce IFNγ, have high levels of MHCII and costimulatory molecules and secrete TH1-recruiting chemokines such as CXCL9 and CXCL10. They are strong promoters of TH1 responses, which results in production of IFNγ and IL12, and induces a positive feedback loop. In addition, anti-tumorigenic macrophages augment NK cell responses by producing IL18 and IL22 (Figure 2) (2, 77–79). In contrast, TAMs acting in a pro-tumorigenic manner are more phagocytic, express higher levels of mannose and galactose receptors, and have a highly active arginase pathway (79). The depletion of arginine pools by Arg1, an enzyme converting L-arginine into L-ornithine, is detrimental to T cells and has been shown to drive their cycle arrest in murine models (80, 81). Additionally, pro-tumorigenic TAMs express a distinct set of chemokines, including CCL17, CCL22, and CCL24. This, in turn, recruits TH2 cells, regulatory T cells, eosinophils and basophils, and induces a more immune suppressive microenvironment (76). Bulk sequencing studies of breast and endometrial cancer patient-derived monocytes and TAMs published earlier this year, provided further insight into human TAMs and identified CCL8 as an additional pro-tumorigenic TAM effector molecule, inducing the expression of an invasive gene expression profile in the cancer cells (82).
Moreover, the spatial distribution of macrophages and the respective environmental conditions in different tumor areas has a profound impact on their function. At the leading edge of tumors, macrophages can drive invasive cellular states through a paracrine signaling loop involving CSF-1 and EGF (83). They act as a major source of matrix metalloproteinases, cathepsins, and serine proteases, which promote degradation of basement membranes and promote invasion and metastases (84–86). In growing tumors TAMs frequently accumulate in regions of hypoxia, where the hypoxic conditions could induce a switch to a pro-angiogenic, invasive phenotype, mediated via diverse range of angiogenic factors, such as TGFβ, VEGF, PDGF, and fibrin (83, 87).
TAM Activation Influences Patient Prognosis and Response to Immunotherapy
High levels of TAMs are associated with poor prognosis, such as in patients with breast, lung, head and neck cancer, as well as Hodgkin's lymphoma. However, high levels of CD68+ cells (consisting largely of TAMs but also granulocytes, dendritic cells and fibroblasts, which also express this marker) are reported to correlate with better prognosis in patients with colon, gastric, and endometrial cancer (2, 71, 88, 89). In consideration of the vast heterogeneity of this cell type, the activation state of TAMs may be a better prognostic marker than cell numbers. Especially the strong immune-suppressive effects of pro-tumorigenic TAMs and their expression of PD-L1, PD-L2, CD80, and CD86, which are ligands for the T cell checkpoints PD-1 and CTLA-4, would suggest TAM infiltration to have a negative effect on immunotherapy. Indeed, in several studies using mouse models, depletion or re-education of TAMs using HDAC inhibitors or blockade of CSF-1 signaling, has shown synergism with checkpoint inhibition (90, 91). However, clinical proof of this treatment modality has yet to be obtained (89). What is needed first, are better markers of the different activation states so that they can be characterized in patients.
Targeting TAMs as a Therapeutic Strategy
The recruitment of TAMs into the TME is strongly dependent on the CCL2 and CSF-1 signaling axes mediating their replenishment from circulating monocytes. Thus, multiple treatment strategies including mAbs, small-molecule inhibitors, and RNAi targeting these pathways have been developed (49). In pre-clinical pancreatic cancer models, CSF-1R signaling inhibition reduces both the numbers of tumor-infiltrating TAMs and their expression of immune-suppressive molecules and therefore acts synergistically with checkpoint inhibition (90). In 2017, a promising study reported response to immunotherapy in combination of CSF-1R and PD1 antagonists in pancreatic cancer patients and is now moving on to a phase II clinical trial (64). Conceptually similar, the humanized CSF-1R Ab emactuzumab reduces TAM infiltration and increases T cell infiltration, which was also confirmed in patients with diffuse type giant cell tumors (64). Several CCL2 blockade combination trials are underway and first results showed a 40% increase in chemotherapy response in pancreatic cancer patients (64, 92). Blockade of the CCL2 axis has however limitations, as it is rapidly compensated by granulocytes and cessation of the therapy induces a burst of monocytes from the bone marrow, increasing metastasis and invasion in a breast cancer mouse model, warranting caution (93).
Other than modulating TAM numbers, alternative strategies have focused on directly targeting immune suppressive TAM effector molecules, such as Arg1 inhibitors (94), or on reprogramming TAMs into an anti-tumorigenic population. In murine glioblastoma, inhibition of CSF-1R regressed established tumors and increased survival, which was attributed to a re-education from an M2 to an anti-tumorigenic phenotype (95). Loss of the receptor tyrosine kinase MERTK triggers a proinflammatory TAM phenotype and induces T cell activation (96, 97), while HDAC inhibition reprograms TAMs into highly phagocytic tumor suppressors (91). However, cancer cells can escape phagocytosis by expressing the membrane receptor CD47—the “don't eat me signal,” which binds to SIRPα on macrophages, inhibiting phagocytosis. Several clinical compounds targeting this suppressive axis are currently in clinical trials (98). Despite many encouraging results, TAM targeting still needs further investigation, since it was recently shown that classical monocytes and macrophages are required for better response to checkpoint inhibition in mouse models (99, 100) and that binding of antibodies to FC receptors of macrophages contributes to the success of several therapeutic responses (101). Thus, a depletion strategy specific for pro-tumorigenic TAMs or strategy to convert TAMs is needed, further highlighting the need to identify specific markers.
Myeloid-Derived Suppressor Cells
Soluble factors released into systemic circulation can cause a differentiation block in normal hematopoiesis and promote the expansion of immature myeloid precursors (IMCs), which fail to terminally differentiate. These so-called MDSCs are best characterized in the field of cancer, but also accumulate in infectious diseases, aging or obesity. They are distinct to terminally differentiated mature myeloid cells (e.g., DCs and TAMs), yet their distinction from neutrophils is often a topic of controversy. As evident from their name, these pathologically activated cells exhibit strong immune suppressive capacities and are crucial drivers of an immune-suppressive microenvironment.
MDSCs Are a Heterogeneous Population of Highly Immune Suppressive Cells
Myeloblasts give rise to neutrophils and myeloid-dendritic cell progenitors (MDPs) can specify into monocytes, however, upon tumor mediated instruction these fail to fully mature and form MDSCs (Figure 1). MDSCs arise in response to many tumor-derived factors and are further subdivided into two groups: monocytic (M) MDSCs (LY6G−/LY6Chigh), which are morphologically similar to monocytes, and polymorphonuclear (PMN) MDSCs (Ly6G+/LY6Clow), which are morphologically similar to neutrophils (102, 103). The distinction between PMD-MDSCs and neutrophils has proven difficult, as they share cellular origin and many phenotypic and morphological features. Thus, a few reports suggest the use of the term N1 and N2 neutrophils for describing different neutrophil activation states, where N2 refers to a more PMD-MDSC like phenotype (104, 105). While a few advances to delineate these cell types have been made, further knowledge and additional markers are needed to faithfully distinguish them (106).
MDSCs are mobilized from the bone marrow via G-CSF, GM-CSF, or hypoxia, and recruited to the tumor site, where inflammatory mediators such as IL-6, TNF-α, and prostaglandin E2 then further enhance their immune suppressive functions. PMN-MDSCs mainly inhibit T cell functions via production of reactive oxygen (ROS) and nitrogen species (NOS), inducing T cell apoptosis or anergy, and do so in an antigen-specific manner. Their relatively weak suppressive role has led to speculations about their contribution to immune suppression. However, their high prevalence in cancer patients and several reports showing improved immune responses upon PMN-MDSCs depletion in mouse models, indicate that further investigation is required to delineate their role. In contrast to PMD-MDSCs, M-MDSCs are considered more suppressive and inhibit both antigen-specific and non-specific T cell responses (1, 78, 106). They exert their suppressive functions via high expression of Arg1, driving T cell anergy by depleting arginine pools (80, 81). In addition, MDSCs can express high levels of IL-10 and TGF-β, and produce reactive nitrogen species, negatively affecting T cell recruitment and activation (1, 78, 107). They also harbor tumor-promoting functions that are independent of immune suppression, such as the promotion of metastasis and angiogenesis via the production of VEGF, bFGF, and MMP9 (Figure 2) (2, 108).
MDSC Levels Correlate With Poor Patient Prognosis and Resistance to Immunotherapy
In lung, breast, and colorectal cancer the abundance of MDSCs in the tumor has been correlated with advanced stage and decreased overall survival (2), also circulating MDSCs negatively influence patient outcome (109, 110). Circulating neutrophils in clusters with cancer cells were recently reported to promote cell cycle progression and metastatic potential in mouse models and patients (111). While high levels of neutrophils are often associated with poor clinical outcome, they can also have anti-tumorigenic functions, especially in early-stage, small-sized tumors, where they are capable of stimulating T cell responses and secreting proinflammatory mediators. Larger, more advanced tumors preferentially recruit immune suppressive MDSCs (104, 112, 113), which negatively correlates with immunotherapy response in melanoma (110, 114). In conclusion, despite difficulties to faithfully distinguish between PMD-MDSCs and neutrophils, these studies indicate that neutrophils can be both pro- and anti-tumorigenic, whereas MDSCs are exclusively supportive of tumor progression (115).
Targeting MDSCs as a Therapeutic Strategy
MDSCs can be modulated in several ways, by targeting (1) their formation in the bone marrow, (2) their recruitment to the tumor site, or (3) their immune suppressive activities. For targeting MDSC formation and inhibiting their expansion, all-trans retinoic acid was shown to differentiate MDSCs into mature DCs and macrophages and confirmed to reduce numbers of circulating MDSCs in patients (116, 117). MDSC formation is also reduced as an advantageous side-effect of several cancer cell-targeting therapies, such as the tyrosine kinase inhibitor sunitinib, via blockade of VEGF, and c-kit signaling (118, 119), or the cytotoxic drugs gemcitabine and 5-fluorouracil that induce selective apoptosis of MDSC in several tumor models, while leaving T cells, DCs, B cells and NK cells unharmed (120, 121). In order to inhibit the recruitment of MDSCs to the tumor, targeting of the CCL2 axis is being evaluated. Conceptually similar, antagonists for CCR5 are known to reduce MDSC recruitment (122). Targeting of effector functions can be achieved via inhibition of phosphodiesterase, reducing expression of Arg1 and iNOS (123, 124), similar to the HDAC inhibitor entinostat, which reduces expression of COX2, Arg1, and NOS2 in mouse models of melanoma and renal carcinoma (125). HDAC inhibition was shown to act synergistically with PD-1 inhibition in murine models and clinical trials are underway (126, 127). siRNA or decoy nucleotides targeting the TF STAT3, which drives the immune suppressive activities of MDSCs, represent another therapeutic approach to block immune suppressive features (122). Overall, targeting MDSCs is conceptually very attractive due to the wide range of immune suppressive effector molecules. However, due to their heterogeneous nature and the lack of highly specific surface markers, it remains a challenging task that requires further investigation.
Perspectives and Outlook
The recent success of T cell targeting agents has validated immune-cell based therapies as an innovative approach to treat cancer. However, immune suppressive mechanisms hampering their success are manifold and myeloid cells are crucial mediators of the suppressive TME. They are a heterogeneous population of cells and rapidly adapting their phenotype to the surrounding tissue. Tumors provide a unique, complex milieu with distinct oncogenic drivers, altered metabolism, hypoxia, and many secreted factors that drive the emergence of myeloid phenotypes unique to the disease. This induces a very complex situation, with many different cell types and activation states that need to be characterized in-depth to allow an understanding of their contribution to immunotherapy success and the development of new therapeutic tools. Technical advances and high dimensional-analytic tools with single cell resolution, ranging from sequencing to mass cytometry, now give us the opportunity to investigate these cell types at an unprecedented rate of detail during steady-state and disease conditions and in different phases of therapy. These tools need to be implemented in immune-oncology in both, patient samples from different cancer entities with clinical follow-up data available, and pre-clinical models that allow their perturbation and experimental testing of therapeutic targets. Together this will allow a deeper understanding of how activation state, localization, and phenotype of myeloid cells in the tumor shape the microenvironment and provide the basis for modulating the tumor microenvironment in targeted approaches, ultimately improving therapeutic outcomes of cancer patients treated with immunotherapy.
Author Contributions
LH and AO researched the data for article, contributed to the discussion of the content, wrote the manuscript, and reviewed and/or edited the manuscript before submission. The authors apologize that due to space limitations, only selective original articles could be cited.
Funding
LH and AO were supported through the ERC Starting Grant CombaTCancer #759590 and the Vienna Science and Technology Fund #LS16-063.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
2. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. (2016) 16:447–62. doi: 10.1038/nrc.2016.54
3. Steinman R, Cohn Z. Identification of a novel cell type in peripheral lymphoid organs of mice. J Exp Med. (1973) 137:1–21. doi: 10.1084/jem.137.5.1142
4. Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity. (2014) 40:642–56. doi: 10.1016/j.immuni.2014.04.016
5. Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, et al. In vivo analysis of dendritic cell development and homeostasis. Science. (2009) 324:392–7. doi: 10.1126/science.1170540
6. Breton G, Lee J, Zhou YJ, Schreiber JJ, Keler T, Puhr S, et al. Circulating precursors of human CD1c+and CD141+ dendritic cells. J Exp Med. (2015) 212:401–13. doi: 10.1084/jem.20141441
7. Onai N, Kurabayashi K, Hosoi-Amaike M, Toyama-Sorimachi N, Matsushima K, Inaba K, et al. A Clonogenic progenitor with prominent plasmacytoid dendritic cell developmental potential. Immunity. (2013) 38:943–57. doi: 10.1016/j.immuni.2013.04.006
8. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol. (2016) 37:855–65. doi: 10.1016/j.it.2016.09.006
9. Dudek A, Shaun M, Garg A, Agostinis P. Immature, semi-mature, and fully mature dendritic cells: toward a DC-cancer cells interface that augments anticancer immunity. Front Immunol. (2013) 4:438. doi: 10.3389/fimmu.2013.00438
10. Dalod M, Chelbi R, Malissen B, Lawrence T. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO J. (2014) 33:1104–16. doi: 10.1002/embj.201488027
11. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
12. Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. (2016) 27:74–95. doi: 10.1038/cr.2016.157
13. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. (2014) 26:638–52. doi: 10.1016/j.ccell.2014.09.007
14. Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HRB, Schreuder J, Lum J, et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol. (2015) 16:718–28. doi: 10.1038/ni.3200
15. Böttcher JP, Sousa CRE. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer. (2018) 4:784–92. doi: 10.1016/j.trecan.2018.09.001
16. Cancel J-C, Crozat K, Dalod M, Mattiuz R. Are conventional type 1 dendritic cells critical for protective antitumor immunity and how? Front Immunol. (2019) 10:9. doi: 10.3389/fimmu.2019.00009
17. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. (2016) 44:924–38. doi: 10.1016/j.immuni.2016.03.012
18. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. (2016) 30:324–36. doi: 10.1016/j.ccell.2016.06.003
19. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell Therapy. Cancer Cell. (2017) 31:711–23.e4. doi: 10.1016/j.ccell.2017.04.003
20. Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity. (2019) 50:1498–1512.e5. doi: 10.1016/j.immuni.2019.04.010
21. Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J Exp Med. (2011) 208:2005–16. doi: 10.1084/jem.20101159
22. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. (2008) 322:1097–100. doi: 10.1126/science.1164206
23. Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun. (2016) 7:13720. doi: 10.1038/ncomms13720
24. Asadzadeh Z, Mohammadi H, Safarzadeh E, Hemmatzadeh M, Mahdian-shakib A, Jadidi-Niaragh F, et al. The paradox of Th17 cell functions in tumor immunity. Cell Immunol. (2017) 322:15–25. doi: 10.1016/j.cellimm.2017.10.015
25. Haan den JMM, Bevan MJ. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8+and CD8− dendritic cells in vivo. J Exp Med. (2002) 196:817–27. doi: 10.1084/jem.20020295
26. Desch AN, Gibbings SL, Clambey ET, Janssen WJ, Slansky JE, Kedl RM, et al. Dendritic cell subsets require cis-activation for cytotoxic CD8 T-cell induction. Nat Commun. (1AD) 5:1–12. doi: 10.1038/ncomms5674
27. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. (2013) 38:729–41. doi: 10.1016/j.immuni.2013.03.003
28. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471–85. doi: 10.1038/nri3865
29. Mitchell D, Chintala S, Dey M. Plasmacytoid dendritic cell in immunity and cancer. J Neuroimmunol. (2018) 322:63–73. doi: 10.1016/j.jneuroim.2018.06.012
30. Sisirak V, Faget J, Vey N, Blay J-Y, Ménétrier-Caux C, Caux C, et al. Plasmacytoid dendritic cells deficient in IFNα production promote the amplification of FOXP3+ regulatory T cells and are associated with poor prognosis in breast cancer patients. Oncoimmunology. (2014) 2:e22338–3. doi: 10.4161/onci.22338
31. Pinto A, Rega A, Crother TR, Sorrentino R. Plasmacytoid dendritic cells and their therapeutic activity in cancer. OncoImmunology. (2014) 1:726–34. doi: 10.4161/onci.20171
32. Treilleux I, Blay J-Y, Bendriss-Vermare N, Ray-Coquard I, Bachelot T, Guastalla J-P, et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res. (2004) 10:7466–74. doi: 10.1158/1078-0432.CCR-04-0684
33. Aspord C, Leccia M-T, Charles J, Plumas J. Melanoma hijacks plasmacytoid dendritic cells to promote its own progression. OncoImmunology. (2014) 3:e27402–3. doi: 10.4161/onci.27402
34. Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol. (2017) 45:43–51. doi: 10.1016/j.coi.2017.01.002
35. Menetrier-Caux C, Montmain G. Inhibition of the differentiation of dendritic cells from CD34 progenitors by tumor cells: role of interleukin-6 and macrophage colony-stimulating factor. Blood. (1998) 92:4778–91. doi: 10.1182/blood.V92.12.4778
36. Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol. (1998) 160:1224–32.
37. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
38. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
39. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. (2006) 107:2013–21. doi: 10.1182/blood-2005-05-1795
40. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. (2015) 161:1527–38. doi: 10.1016/j.cell.2015.05.025
41. Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nature. (2018) 363:1–9. doi: 10.1038/nrc.2017.117
42. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:1–10. doi: 10.1038/s41591-018-0014-x
43. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
44. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–8.e14. doi: 10.1016/j.cell.2018.01.004
45. Pfirschke C, Siwicki M, Liao H-W, Pittet MJ. Tumor microenvironment: no effector T cells without dendritic cells. Cancer Cell. (2017) 31:614–5. doi: 10.1016/j.ccell.2017.04.007
46. Liu J, Rozeman EA, O'Donnell JS, Allen S, Fanchi L, Smyth MJ, et al. Batf3+ DCs and type I IFN are critical for the efficacy of neoadjuvant cancer immunotherapy. OncoImmunology. (2018) 8:e1546068. doi: 10.1080/2162402X.2018.1546068
47. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med. (2018) 24:1–21. doi: 10.1038/s41591-018-0085-8
48. Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. (2018) 49:1148–61.e7. doi: 10.1016/j.immuni.2018.09.024
49. Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn back the TIMe: targeting tumor infiltrating myeloid cells to revert cancer progression. Front Immunol. (2018) 9:1977. doi: 10.3389/fimmu.2018.01977
50. Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. (2013) 39:38–48. doi: 10.1016/j.immuni.2013.07.004
51. Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. (2005) 175:6169–76. doi: 10.4049/jimmunol.175.9.6169
52. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. (2012) 18:1254–61. doi: 10.1038/nm.2883
53. Le DT, Pardoll DM, Jaffee EM. Cellular vaccine approaches. Cancer J. (2010) 16:304–10. doi: 10.1097/PPO.0b013e3181eb33d7
54. Melief CJM. Cancer immunotherapy by dendritic cells. Immunity. (2008) 29:372–83. doi: 10.1016/j.immuni.2008.08.004
55. van den Eertwegh AJ, Versluis J, van den Berg HP, Santegoets SJ, van Moorselaar RJ, van der Sluis TM, et al. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. (2012) 13:509–17. doi: 10.1016/S1470-2045(12)70007-4
56. Wilgenhof S, Corthals J, Heirman C, van Baren N, Lucas S, Kvistborg P, et al. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) Plus Ipilimumab in patients with pretreated advanced melanoma. J Clin Oncol. (2016) 34:1330–8. doi: 10.1200/JCO.2015.63.4121
57. Fong L, Hou Y, Rivas A, Benike C, Yuen A, Fisher GA, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci USA. (2001) 98:8809–14. doi: 10.1073/pnas.141226398
58. Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1–polarizing program in dendritic cells. Nat Immunol. (2005) 6:769–76. doi: 10.1038/ni1223
59. Iribarren K, Bloy N, Buqué A, Cremer I, Eggermont A, Fridman W-H, et al. Trial watch: immunostimulation with Toll-like receptor agonists in cancer therapy. OncoImmunology. (2016) 5:e1088631. doi: 10.1080/2162402X.2015.1088631
60. Mullard A. Can innate immune system targets turn up the heat on “cold” tumours? Nat Rev Drug Discov. (2018) 17:3–5. doi: 10.1038/nrd.2017.264
61. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang S-Y, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. (2018) 564:1–16. doi: 10.1038/s41586-018-0705-y
62. Krieg AM. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene. (2008) 27:161–7. doi: 10.1038/sj.onc.1210911
63. Milhem M, Gonzales R, Medina T, Kirkwood JM, Buchbinder E, Mehmi I, et al. Abstract CT144: Intratumoral toll-like receptor 9 (TLR9) agonist, CMP-001, in combination with pembrolizumab can reverse resistance to PD-1 inhibition in a phase Ib trial in subjects with advanced melanoma. Cancer Res. (2018) 78:CT144. doi: 10.1158/1538-7445.AM2018-CT144
64. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nature. (2019) 7:1–14. doi: 10.1038/s41577-019-0127-6
65. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
66. Lahmar Q, Keirsse J, Laoui D, Movahedi K, Van Overmeire E, Van Ginderachter JA. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. BBA Rev Cancer. (2016) 1865:23–34. doi: 10.1016/j.bbcan.2015.06.009
67. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. (2016) 17:2445–59. doi: 10.1016/j.celrep.2016.10.052
68. Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, et al. The pancreas anatomy conditions the origin and properties of resident macrophages. J Exp Med. (2015) 212:1497–512. doi: 10.1084/jem.20150496
69. Zhu Y, Herndon JM, Sojka DK, Kim K-W, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. (2017) 47:323–38.e6. doi: 10.1016/j.immuni.2017.07.014
70. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. (2017) 77:2266–78. doi: 10.1158/0008-5472.CAN-16-2310
71. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. (2015) 27:462–72. doi: 10.1016/j.ccell.2015.02.015
72. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
73. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. (2011) 11:750–61. doi: 10.1038/nri3088
74. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
75. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
76. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
77. Osaki T, Hashimoto W, Gambotto A, Okamura H, Robbins PD, Kurimoto M, et al. Potent antitumor effects mediated by local expression of the mature form of the interferon-gamma inducing factor, interleukin-18 (IL-18). Gene Ther. (1999) 6:808–15. doi: 10.1038/sj.gt.3300908
78. Kiss M, Van Gassen S, Movahedi K, Saeys Y, Laoui D. Myeloid cell heterogeneity in cancer_ not a single cell alike. Cell Immunol. (2018) 330:188–201. doi: 10.1016/j.cellimm.2018.02.008
79. Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol. (2018) 8:49. doi: 10.3389/fonc.2018.00049
80. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. (2007) 109:1568–73. doi: 10.1182/blood-2006-06-031856
81. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–36.e13. doi: 10.1016/j.cell.2016.09.031
82. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. (2019) 35:588–602.e10. doi: 10.1016/j.ccell.2019.02.009.
83. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
84. Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. (2002) 2:289–300. doi: 10.1016/S1535-6108(02)00153-8
85. Lindahl C, Simonsson M, Bergh A, Thysell E, Antti H, Sund M, et al. Increased levels of macrophage-secreted cathepsin S during prostate cancer progression in TRAMP mice and patients. Cancer Genom Proteomics. (2009) 6:149–59.
86. Akkari L, Gocheva V, Kester JC, Hunter KE, Quick ML, Sevenich L, et al. Distinct functions of macrophage-derived and cancer cell-derived cathepsin Z combine to promote tumor malignancy via interactions with the extracellular matrix. Genes Dev. (2014) 28:2134–50. doi: 10.1101/gad.249599.114
87. Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. (2014) 5:75. doi: 10.3389/fphys.2014.00075
88. Zhang Q-W, Liu L, Gong C-Y, Shi H-S, Zeng Y-H, Wang X-Z, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE. (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
89. Cassetta L, Kitamura T. Targeting tumor-associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front Cell Dev Biol. (2018) 6:38. doi: 10.3389/fcell.2018.00038
90. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
91. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. (2017) 543:428–32. doi: 10.1038/nature21409
92. Nywening T, Wang-Gillam A, Sanford D, Belt B, Panni R, Cusworth B, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. (2016) 17:1–12. doi: 10.1016/S1470-2045(16)00078-4
93. Bonapace L, Coissieux M-M, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. (2014) 515:130–3. doi: 10.1038/nature13862
94. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. (2017) 5:1–18. doi: 10.1186/s40425-017-0308-4
95. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
96. Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest. (2013) 123:3231–42. doi: 10.1172/JCI67655
97. Crittenden MR, Baird J, Friedman D, Savage T, Uhde L, Alice A, et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget. (2016) 7:78653–66. doi: 10.18632/oncotarget.11823
98. Zhang X, Fan J, Ju D. Insights into CD47/SIRPα axis-targeting tumor immunotherapy. Antibody Therap. (2018) 1:27–32. doi: 10.1093/abt/tby006
99. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. doi: 10.1084/jem.20130579
100. Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. (2018) 24:144–53. doi: 10.1038/nm.4466
101. DiLillo DJ, Ravetch JV. Fc-receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunol Res. (2015) 3:704–13. doi: 10.1158/2326-6066.CIR-15-0120
102. Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, Mandruzzato S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:1–10. doi: 10.1038/ncomms12150
103. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. (2008) 111:4233–44. doi: 10.1182/blood-2007-07-099226
104. Mishalian I, Bayuh R, Levy L, Zolotarov L, Michaeli J, Fridlender ZG. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol Immunother. (2013) 62:1745–56. doi: 10.1007/s00262-013-1476-9
105. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
106. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. (2015) 125:3356–64. doi: 10.1172/JCI80005
107. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. (2015) 128:95–139. doi: 10.1016/bs.acr.2015.04.002
108. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
109. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. (2015) 21:1–12. doi: 10.1038/nm.3909
110. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. (2013) 63:247–57. doi: 10.1007/s00262-013-1508-5
111. Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. (2019) 566:553–7. doi: 10.1038/s41586-019-0915-y
112. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. (2014) 124:5466–80. doi: 10.1172/JCI77053
113. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol Immunother. (2008) 58:49–59. doi: 10.1007/s00262-008-0523-4
114. Sade-Feldman M, Kanterman J, Klieger Y, Ish-Shalom E, Olga M, Saragovi A, et al. Clinical significance of circulating CD33+ CD11b+ HLA-DR- myeloid cells in patients with stage IV melanoma treated with ipilimumab. Clin Cancer Res. (2016) 22:5661–72. doi: 10.1158/1078-0432.CCR-15-3104
115. Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC_ Their biological role and interaction with stromal cells. Semin Immunol. (2018) 35:19–28. doi: 10.1016/j.smim.2017.12.004
116. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all- transretinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. (2007) 67:11021–8. doi: 10.1158/0008-5472.CAN-07-2593
117. Tobin RP, Davis D, Jordan KR, McCarter MD. The clinical evidence for targeting human myeloid-derived suppressor cells in cancer patients. J Leukocyte Biol. (2017) 102:381–91. doi: 10.1189/jlb.5VMR1016-449R
118. Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T, Daemen T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. OncoImmunology. (2015) 4:e989764–11. doi: 10.4161/2162402X.2014.989764
119. Kao J, Ko EC, Eisenstein S, Sikora AG, Fu S, Chen S-H. Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit Rev Oncol Hematol. (2011) 77:12–9. doi: 10.1016/j.critrevonc.2010.02.004
120. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690
121. Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med. (2016) 14:1–12. doi: 10.1186/s12967-016-1037-z
122. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. (2018) 9:398. doi: 10.3389/fimmu.2018.00398
123. Shiyong L, Wang J. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am J Cancer Res. (2017) 7:41–52.
124. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. (2006) 203:2691–702. doi: 10.1084/jem.20061104
125. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res. (2017) 23:5187–201. doi: 10.1158/1078-0432.CCR-17-0741
126. Zheng H, Zhao W, Yan C, Watson CC, Messengill M, Xie M, et al. HDAC inhibitors enhance T cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res. (2016) 22:4119–32. doi: 10.1158/1078-0432.CCR-15-2584
Keywords: immunotherapy, cancer, myeloid cells, dendritic cells, macrophages, myeloid-derived suppressor cells, immune suppression, tumor microenvironment
Citation: Haas L and Obenauf AC (2019) Allies or Enemies—The Multifaceted Role of Myeloid Cells in the Tumor Microenvironment. Front. Immunol. 10:2746. doi: 10.3389/fimmu.2019.02746
Received: 15 April 2019; Accepted: 08 November 2019;
Published: 28 November 2019.
Edited by:
Nicola Giuliani, University of Parma, ItalyReviewed by:
Alessandro Poggi, San Martino Hospital (IRCCS), ItalyDaniela F. Quail, McGill University, Canada
Copyright © 2019 Haas and Obenauf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna C. Obenauf, anna.obenauf@imp.ac.at