 Kathleen R. Chirco
Kathleen R. Chirco Lawrence A. Potempa
Lawrence A. Potempa- 1Buck Institute for Research on Aging, Novato, CA, United States
- 2Roosevelt University College of Pharmacy, Schaumburg, IL, United States
Age-related macular degeneration (AMD) is a devastating neurodegenerative disease affecting millions worldwide. Complement activation, inflammation, and the loss of choroidal endothelial cells have been established as key factors in both normal aging and AMD; however, the exact mechanisms for these events have yet to be fully uncovered. Herein, we provide evidence that the prototypic acute phase reactant, C-reactive protein (CRP), contributes to AMD pathogenesis. We discuss serum CRP levels as a risk factor for disease, immunolocalization of distinct forms of CRP in the at-risk and diseased retina, and direct effects of CRP on ocular tissue. Furthermore, we discuss the complement system as it relates to AMD pathophysiology, provide a model for the role of CRP in this disease, and outline current therapies being developed and tested to treat AMD patients.
Introduction
Age-related macular degeneration (AMD) is a progressive neurodegenerative disease affecting more than 8% of the global population (1), with roughly 11 million cases in the United States alone (2). Inflammation and complement activation are recognized as prominent events in the manifestation and progression of AMD, with C-reactive protein (CRP) as a potential mediator in these processes. Here, we review AMD pathophysiology, the complement system, and the current evidence supporting CRP as a contributor to complement activation and inflammation in the context of AMD. Finally, we will present a model for the role of CRP in this disease and provide insight into future therapies for AMD patients.
AMD Pathophysiology
Early AMD is clinically identified by the presence of extracellular deposits, called drusen, and/or pigmentary changes within the macula, the central 6 mm of the retina. If the disease progresses, vision loss can occur either from atrophy of the retina (termed advanced dry AMD or atrophic AMD) or aberrant growth of vessels beneath the retina (termed wet AMD or neovascular AMD). The primary pathology in AMD includes dysfunction and/or loss of (1) the photoreceptor cells, which are the light-sensing cells of the retina, (2) the retinal pigment epithelium (RPE), which forms the outer blood–retinal barrier and consists of a single layer of epithelial cells that support photoreceptor cell function, and (3) the choriocapillaris, the capillary bed that lies just outside the RPE (and forms the innermost layer of the choroid) and provides oxygen and nutrients to both the RPE and photoreceptor cells.
While the pathologic changes that occur within the macula during AMD are incompletely understood, evidence supports complement-mediated changes and the loss of choroidal endothelial cells (CECs) as early events in both normal aging and disease (3, 4). CECs are believed to be lost early in disease progression, which may accompany or even precede dysfunction and loss of the RPE (4–9). Loss of these supporting cells eventually leads to loss of photoreceptor cells within the macula, causing significant and irreversible visual decline (10).
The Complement System in AMD
Although AMD is a complex, multifactorial disease, many genetic factors have been associated with disease risk. To date, 19 genetic loci have been identified, including genes involved in regulating complement activity (11). The discovery of a genetic link between AMD and the complement system supports previous histological and proteomic data identifying complement proteins as constituents of drusen in postmortem eyes (12–14).
The complement system, which is important for eliminating pathogens, cellular debris, and dying host cells, consists of three distinct arms: the classical pathway, the alternative pathway, and the lectin pathway. After pathway-specific initiation, followed by a series of cleavage events, the pathways continue, similarly, through to cleavage of C5 and entrance into a common terminal pathway. The terminal pathway culminates in formation of the membrane attack complex (MAC), which promotes cell lysis. Similar to other arms of the immune system, the complement system must be carefully regulated, and various serum- and tissue-derived complement proteins aid in the regulation of one or more pathways. One of the major genetic risk loci for AMD includes the complement factor H (CFH) gene, which encodes an alternative pathway regulator and harbors multiple disease-associated variants (15). Importantly, the Y402H single nucleotide polymorphism (SNP) in CFH is a common variant that significantly increases AMD risk (15–18). The Y402H substitution occurs in the SCR7 region of the complement factor H (FH) protein, resulting in impaired protein binding to various substrates, including proteoglycans (19–22) and CRP (23).
Another component of the complement system, the MAC, has been linked to AMD through various histological studies. MAC deposition becomes increasingly elevated within the choriocapillaris with advancing age in human postmortem eyes, and this phenomenon is specific to the eye (24). MAC accumulation is further elevated in CFH Y402H high-risk and AMD maculae beyond what is observed in normal aging (3). Compromised binding of the FH Y402H protein to its extracellular matrix substrates, such as heparan sulfate proteoglycans, may increase complement activation in the human retina (21), and may be one cause for elevated MAC deposition in the macula in AMD (3, 4). The potential consequences of MAC accumulation in the choriocapillaris are twofold. First, MAC formation on CECs in vitro results in significant cell lysis. Second, the cells that survive complement attack adopt an angiogenic phenotype (25). While complement activation and MAC accumulation in the choriocapillaris in AMD are well-established events (26), the precise details for how and why these events occur remain to be fully elucidated.
CRP in Complement Activation and Inflammation
C-reactive protein is an established component of drusen in human postmortem eyes (14, 27–29) and it plays a role in both complement activation and regulation (23, 30, 31), implicating CRP as an intriguing candidate for disease involvement.
Studies examining the role of CRP as a regulator of complement activation and other inflammatory response pathways have recently evolved to evaluate and distinguish the bioactivities of two distinct conformational forms of the protein. The widely appreciated serum-associated form is a hepatically synthesized pentameric discoid protein (pCRP) with high aqueous solubility and calcium-dependent phosphocholine (PC) binding affinity. pCRP can bind to PC groups exposed on disrupted plasma membranes, as occurs when lysophospatidyl choline is formed (32). Upon binding to disrupted cell membranes, as shown with activated platelets and apoptotic monocytes, pCRP undergoes rapid dissociation into monomeric form (mCRP) (33, 34) with distinctive solubility, antigenicity, tissue localization, binding ligands, and functions compared to pCRP (30, 35, 36). It is now known that mCRP rather than pCRP is an efficient activator of the classical complement pathway involving C1 (via binding to C1q), C4, C2, and C3 (37, 38). mCRP also acts as a regulator of the alternative complement pathway, via recruitment of FH to injured tissues. This binding to FH was shown to occur in a dose-dependent manner and at a site that does not interfere with mCRP binding to C1q (38). Importantly, the high-risk FH Y402H protein has reduced mCRP binding by up to 45% when compared to the FH Y402 protein (28, 39, 40).
In the context of atherosclerosis and coronary artery disease, mCRP has been shown to be strongly pro-inflammatory, differentiating monocytes toward a pro-inflammatory M1 phenotype (41), delaying neutrophil apoptosis, and stimulating leukocytes pro-inflammatory effector responses (42). In addition, Zouki and colleagues revealed a mCRP-mediated upregulation of CD11b/CD18 expression on the surface of human neutrophils, which results in enhanced adhesion of these cells to activated endothelium (43). Monomeric CRP can also activate human coronary artery endothelial cells in vitro, resulting in production of IL-8 and MCP-1 (44). Moreover, mCRP stimulates angiogenesis and promotes tube formation in brain microvasculature (45). While CRP has been widely studied outside the eye, the bioactivities of both forms of CRP are now beginning to be elucidated within ocular tissue in the context of AMD.
Serum-Associated CRP as a Risk Factor in AMD
Seddon and colleagues began examining a potential link between CRP and AMD, by assessing CRP levels in the serum of AMD and control patients. They found CRP levels to be higher in individuals with intermediate and advanced AMD compared to controls (46), and they discovered a positive correlation between serum CRP levels and AMD progression (47). Furthermore, they found the lowest risk of AMD progression associated with CRP levels <0.5 mg/L, little change to AMD risk for CRP levels within the range of 0.5–10.0 mg/L, and the highest risk for AMD progression when CRP levels reached at least 10 mg/L (47). Finally, Seddon’s group examined the relationship between serum CRP levels, CFH genotype, and AMD risk (48), and they found CRP and CFH genotype to be independently associated with AMD risk. In fact, both the CFH variant and high CRP levels have an additive effect on AMD risk.
Interestingly, others have gone on to assess the relationship between serum CRP levels and AMD status in individuals harboring variants in the promoter region of the CRP gene, all of which either increase or decrease CRP levels in the serum. However, regardless of whether the SNP increased or decreased CRP levels, the authors found no direct association between any of the variants and AMD status (49–51). While none of these variants result in an amino acid change to the CRP protein and, therefore, do not affect biological function of the protein, these studies suggest that serum CRP levels alone may not be enough to alter AMD risk.
CRP in the AMD Retina
The association between serum CRP levels and AMD risk may be important for defining disease biomarkers; however, the primary pathology of AMD occurs in the retina. Prior to the realization that CRP exists in more than one form (e.g., mCRP and pCRP), work done by Johnson and colleagues examined the presence of total CRP in the retina of postmortem eyes genotyped at the CFH locus (27). They showed that high-risk eyes (homozygous for the Y402H SNP) had more CRP immunoreactivity in the choroid compared to low-risk eyes (homozygous for Y402), especially in regions containing drusen-like deposits (27). A similar study aimed to determine differences in total CRP levels in the retina based on AMD status (52). CRP immunoreactivity differed based on disease status, with early and wet AMD eyes having more intense CRP immunolabeling compared to controls. However, in advanced dry AMD eyes with geographic atrophy (GA), CRP immunoreactivity in the non-atrophic area was similar to that of age-matched controls, with CRP levels significantly reduced within the atrophic lesions. CRP was primarily immunolabeling extending into Bruch’s membrane in early and wet AMD eyes (52). Interestingly, FH immunoreactivity was significantly reduced in the choroid in AMD eyes compared to controls, regardless of disease stage, suggesting an imbalance in CRP and FH levels, especially in early and wet AMD.
A recent study of human postmortem eyes examined CRP in the retina to determine which form of CRP is present in the tissue and to begin teasing out its precise role in AMD pathogenesis (29). Using monoclonal antibodies that clearly differentiate pCRP and mCRP antigens (53), the study found that mCRP is the primary form of CRP in the choroid, and it is predominantly localized to the choriocapillaris and Bruch’s membrane. Similar to previous work looking at total CRP (27), mCRP is more abundant in donor eyes with the high-risk CFH polymorphism compared to age-matched controls. Monomeric CRP also exhibits a direct effect on CECs in vitro, including increasing CEC migration and increasing monolayer permeability. Further-more, mCRP treatment of human RPE-choroid tissue ex vivo results in a significant upregulation of pro-inflammatory gene expression, including an increase in ICAM1, which has been associated with AMD previously (54). These data suggest a role for mCRP in promoting inflammation in the choroid, which may be especially true in individuals at risk for AMD development. This study revealed co-localization between mCRP and the MAC in the choriocapillaris of human postmortem eyes, providing evidence to support the hypothesis that mCRP increases complement activation in the choriocapillaris (29).
The work outlined above shows that mCRP primarily localizes to the choroid; however, other retinal cell types may be affected by the presence of this potent pro-inflammatory molecule within ocular tissue. For example, Molins et al. propose a role for mCRP in disruption of the outer blood–retinal barrier. They found that treatment with mCRP in vitro led to a disruption in tight junction integrity in ARPE-19 monolayers (55). Exposing ARPE-19 cells to mCRP also stimulates IL8 and CCL2 expression, two molecules that are important for leukocyte recruitment and blood–retinal barrier integrity (40, 56). Monomeric CRP binds necrotic RPE cells in vitro and enhances recruitment of FH to those cells by over 100% compared to FH recruitment without mCRP. In contrast, mCRP increases the recruitment of the Y402H variant of FH by just 50%. The mCRP-mediated recruitment of FH results in a 35% decrease in TNFα secretion from necrotic RPE cells, which is reduced to a 1% decrease for the high-risk FH molecule (28). These data indicate that without efficient recruitment of FH to RPE cells, the pro-inflammatory effects of mCRP may override the protective effects, leading to exacerbation of AMD pathology.
Model for the Role of CRP in the Pathogenesis of AMD
The amount of CRP that gets made during an acute phase response is up to 1,000-fold beyond baseline levels. While it is unlikely that all of the pCRP in serum gets into the tissue, it is possible that much of it does reach the target tissue and dissociate into monomers. Furthermore, local production of mCRP cannot be ruled out; however, more work is required to determine the precise source(s) of mCRP in the choriocapillaris. Regardless of the source, once mCRP reaches the choriocapillaris, it may presumably outnumber the regulatory FH molecules present, which could create an imbalance in complement regulation in favor of more complement activation. Furthermore, in the choriocapillaris, where complement injury is highest in AMD, FH is the primary regulator of the complement system. This is in contrast to the RPE, which has multiple complement regulators present and, therefore, may be better armed against complement-mediated injury (57, 58). In an individual who harbors the high-risk CFH polymorphism, their ability to control complement, at least the alternative pathway, is further limited due to altered binding capabilities of mutant FH to its tissue-associated ligands (e.g., glycosaminoglycans, such as heparan sulfate, within Bruch’s membrane). Since mCRP is shown to be present in these tissues, it is possible that mCRP may affect complement regulation resulting in the increased MAC deposition observed in the choriocapillaris with advanced age and disease. Together, these data propose a mechanism for CEC loss in AMD pathogenesis, via complement-mediated attack (Figure 1).
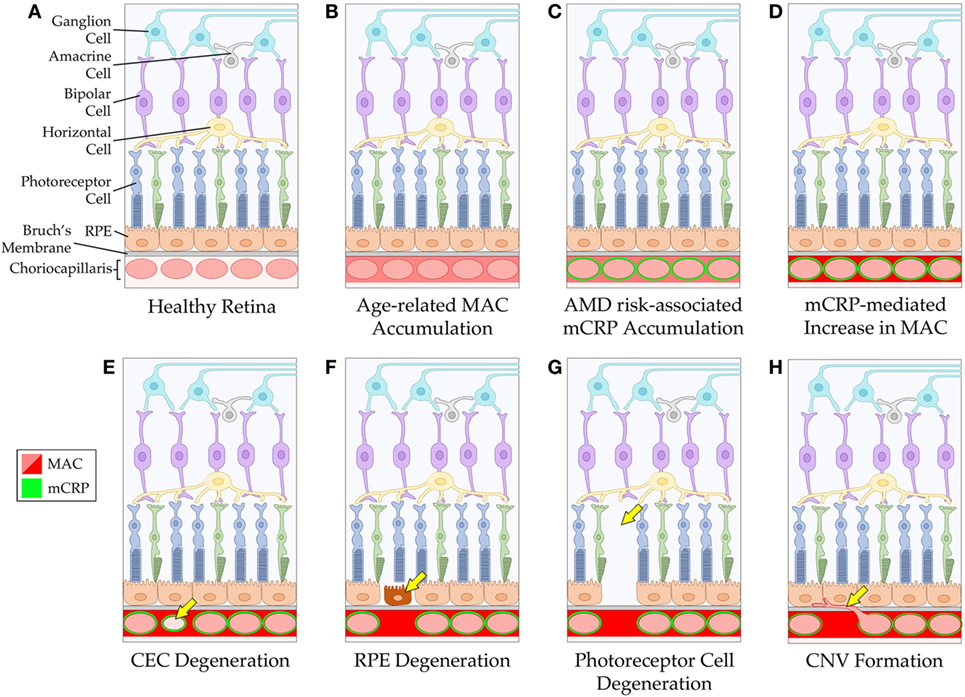
Figure 1. Schematic of mCRP-associated age-related macular degeneration (AMD) pathogenesis. The healthy retina and choriocapillaris is depicted in (A). With advancing age, the membrane attack complex (MAC) accumulates around the vessels of the choriocapillaris (B). In individuals with an increased genetic risk for AMD (via the CFH Y402H polymorphism), mCRP accumulates around the vessels of the choriocapillaris (C), and this may lead to increased complement activation and subsequent elevation in MAC levels in the tissue (D). The mCRP- and/or MAC-mediated changes to the tissue environment may result in CEC death and degeneration of the choriocapillaris (E). Loss of the vessels of the choriocapillaris can cause dysfunction and degeneration of the RPE (F), and eventually the photoreceptor cells (G). Alternatively, loss of choriocapillaris vessels can lead to choroidal neovascularization formation (H). RPE, retinal pigment epithelium; CEC, choroidal endothelial cell; CNV, choroidal neovascularization.
Evidence suggests that mCRP also plays a key role in promoting an inflammatory environment in AMD eyes. For example, mCRP increases ICAM1 mRNA and protein levels in human postmortem RPE-choroid tissue ex vivo (29). ICAM-1, which is constitutively expressed in the choriocapillaris with highest levels in the macula (59), acts to promote leukocyte recruitment in the choroid (60) and elevated levels of ICAM-1 have been associated with AMD (54). Ultimately, combining the evidence for (1) its localization to the choriocapillaris in high-risk and AMD patients, (2) its role in complement system activation and its interaction with AMD-associated complement proteins, (3) its ability to directly promote CEC activation in vitro and ex vivo, and (4) its pro-inflammatory effects on RPE-choroid tissue, mCRP is a promising target for the treatment of AMD.
Therapeutic Development to Target Inflammation and Complement Activation in AMD
Treatment options for AMD are currently limited, with the most effective therapies consisting of AREDS supplements (antioxidants plus zinc), which have been shown to reduce the risk of progression beyond early or intermediate AMD by about 25–30% over 5 years (61), and intravitreal anti-vascular endothelial growth factor injections, which can help ameliorate the symptoms of wet AMD (62). The absence of a more effective and universal therapy has been a driver for the continued pursuit of novel therapeutic targets. Based on the evidence for mCRP and complement activation as key players in AMD pathogenesis, future therapies may need to target both mCRP-mediated effects in addition to complement proteins to effectively treat the disease. Many clinical trials are already completed or underway to assess treatments that target inflammation or complement activation in individuals with AMD [Table 1; reviewed in Ref. (63, 64)]. For example, a handful of trials have aimed to target inflammation in participants with AMD using corticosteroids such as dexamethasone [NCT01162746] and fluocinolone acetonide (iluvien) [NCT00605423]. In addition to steroid therapies to reduce inflammation, various studies have taken aim at regulating complement system activation in AMD patients, including drugs targeting complement components C5, C3, and CFD.
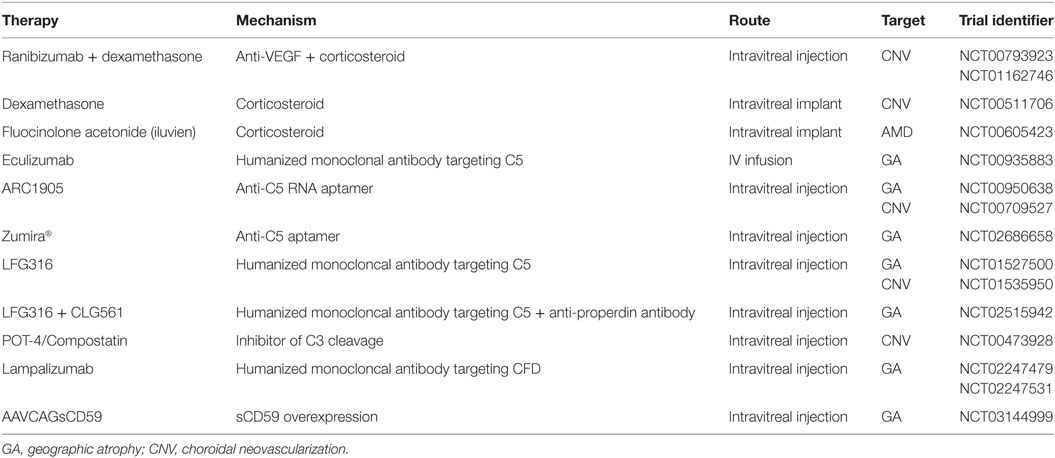
Table 1. Completed and ongoing clinical trials to reduce inflammation and complement activation in age-related macular degeneration (AMD).
Despite the promise these ongoing trials provide, many hurdles still exist in the therapeutic regulation of inflammation and complement in AMD patients, including delivery method, dose, and disease stage at time of treatment. The use of gene therapy to treat AMD may help resolve these current issues and provide a promising option for future treatments. Early studies to examine gene therapy-mediated treatments are already underway in mice [e.g., Cr2-fH fusion protein (65) and FH overexpression (66)] and in human clinical trials (AAVCAGsCD59; NCT03144999). Additional gene therapy options, such as CRISPR/Cas9-mediated gene editing, may be useful to correct high-risk variants in AMD patients, including the CFH Y402H polymorphism. Since advanced AMD pathology includes loss of multiple cell types within the macula, cell replacement therapy, possibly in combination with other gene editing or drug therapies, could provide the greatest promise for improved visual acuity in AMD patients. As we continue to advance our understanding of the key mediators in AMD pathogenesis, we continue to move closer to the development of life-changing treatments for millions of individuals.
Author Contributions
KC and LP wrote and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Robert Mullins and Dr. Deepak Lamba for their support and contributions to this work.
References
1. Wong WL, Su X, Li X, Cheung CMG, Klein R, Cheng C-Y, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health (2014) 2(2):e106–16. doi:10.1016/S2214-109X(13)70145-1
2. Pennington KL, Deangelis MM. Epidemiology of age-related macular degeneration (AMD): associations with cardiovascular disease phenotypes and lipid factors. Eye Vis (Lond) (2016) 3(1):34. doi:10.1186/s40662-016-0063-5
3. Mullins RF, Dewald AD, Streb LM, Wang K, Kuehn MH, Stone EM. Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp Eye Res (2011) 93(4):565–7. doi:10.1016/j.exer.2011.06.015
4. Mullins RF, Schoo DP, Sohn EH, Flamme-Wiese MJ, Workamelahu G, Johnston RM, et al. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol (2014) 184(11):3142–53. doi:10.1016/j.ajpath.2014.07.017
5. Sarks SH. Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol (1976) 60(5):324–41. doi:10.1136/bjo.60.5.324
6. Yuan X, Gu X, Crabb JS, Yue X, Shadrach K, Hollyfield JG, et al. Quantitative proteomics: comparison of the macular Bruch membrane/choroid complex from age-related macular degeneration and normal eyes. Mol Cell Proteomics (2010) 9(6):1031–46. doi:10.1074/mcp.M900523-MCP200
7. Mullins RF, Johnson MN, Faidley EA, Skeie JM, Huang J. Choriocapillaris vascular dropout related to density of drusen in human eyes with early age-related macular degeneration. Invest Ophthalmol Vis Sci (2011) 52(3):1606–12. doi:10.1167/iovs.10-6476
8. Whitmore SS, Braun TA, Skeie JM, Haas CM, Sohn EH, Stone EM, et al. Altered gene expression in dry age-related macular degeneration suggests early loss of choroidal endothelial cells. Mol Vis (2013) 19:2274–97.
9. Sohn EH, Khanna A, Tucker BA, Abràmoff MD, Stone EM, Mullins RF. Structural and biochemical analyses of choroidal thickness in human donor eyesanalyses of choroidal thickness in human donor eyes. Invest Ophthalmol Vis Sci (2014) 55(3):1352–60. doi:10.1167/iovs.13-13754
10. Biesemeier A, Taubitz T, Julien S, Yoeruek E, Schraermeyer U. Choriocapillaris breakdown precedes retinal degeneration in age-related macular degeneration. Neurobiol Aging (2014) 35(11):2562–73. doi:10.1016/j.neurobiolaging.2014.05.003
11. Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, et al. Seven new loci associated with age-related macular degeneration. Nat Genet (2013) 45(4):433–9. doi:10.1038/ng.2578
12. Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A (2002) 99(23):14682–7. doi:10.1073/pnas.222551899
13. Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res (2001) 73(6):887–96. doi:10.1006/exer.2001.1094
14. Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J (2000) 14(7):835–46. doi:10.1096/fasebj.14.7.835
15. Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A (2005) 102(20):7227–32. doi:10.1073/pnas.0501536102
16. Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science (2005) 308(5720):421–4. doi:10.1126/science.1110189
17. Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science (2005) 308(5720):419–21. doi:10.1126/science.1110359
18. Klein RJ, Zeiss C, Chew EY, Tsai J-Y, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science (2005) 308(5720):385–9. doi:10.1126/science.1109557
19. Clark SJ, Higman VA, Mulloy B, Perkins SJ, Lea SM, Sim RB, et al. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J Biol Chem (2006) 281(34):24713–20. doi:10.1074/jbc.M605083200
20. Prosser BE, Johnson S, Roversi P, Herbert AP, Blaum BS, Tyrrell J, et al. Structural basis for complement factor H linked age-related macular degeneration. J Exp Med (2007) 204(10):2277–83. doi:10.1084/jem.20071069
21. Clark SJ, Perveen R, Hakobyan S, Morgan BP, Sim RB, Bishop PN, et al. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch’s membrane in human retina. J Biol Chem (2010) 285(39):30192–202. doi:10.1074/jbc.M110.103986
22. Kelly U, Yu L, Kumar P, Ding JD, Jiang H, Hageman GS, et al. Heparan sulfate, including that in Bruch’s membrane, inhibits the complement alternative pathway: implications for age-related macular degeneration. J Immunol (2010) 185(9):5486–94. doi:10.4049/jimmunol.0903596
23. Yu J, Wiita P, Kawaguchi R, Honda J, Jorgensen A, Zhang K, et al. Biochemical analysis of a common human polymorphism associated with age-related macular degeneration. Biochemistry (2007) 46(28):8451–61. doi:10.1021/bi700459a
24. Chirco KR, Tucker BA, Stone EM, Mullins RF. Selective accumulation of the complement membrane attack complex in aging choriocapillaris. Exp Eye Res (2016) 146(C):393–7. doi:10.1016/j.exer.2015.09.003
25. Zeng S, Whitmore SS, Sohn EH, Riker MJ, Wiley LA, Scheetz TE, et al. Molecular response of chorioretinal endothelial cells to complement injury: implications for macular degeneration. J Pathol (2016) 238:446–56. doi:10.1002/path.4669
26. Mullins RF, Warwick AN, Sohn EH, Lotery AJ. From compliment to insult: genetics of the complement system in physiology and disease in the human retina. Hum Mol Genet (2017) 26(R1):R51–7. doi:10.1093/hmg/ddx181
27. Johnson PT, Betts KE, Radeke MJ, Hageman GS, Anderson DH, Johnson LV. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc Natl Acad Sci U S A (2006) 103(46):17456–61. doi:10.1073/pnas.0606234103
28. Lauer N, Mihlan M, Hartmann A, Schlötzer-Schrehardt U, Keilhauer C, Scholl HPN, et al. Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. J Immunol (2011) 187(8):4374–83. doi:10.4049/jimmunol.1002488
29. Chirco KR, Whitmore SS, Wang K, Potempa LA, Halder JA, Stone EM, et al. Monomeric C-reactive protein and inflammation in age-related macular degeneration. J Pathol (2016) 240(2):173–83. doi:10.1002/path.4766
30. Wu Y, Potempa LA, Kebir El D, Filep JG. C-reactive protein and inflammation: conformational changes affect function. Biol Chem (2015) 396(11):1181–97. doi:10.1515/hsz-2015-0149
31. Mold C, Gewurz H, Clos Du TW. Regulation of complement activation by C-reactive protein. Immunopharmacology (1999) 42(1–3):23–30. doi:10.1016/S0162-3109(99)00007-7
32. Braig D, Nero TL, Koch H-G, Kaiser B, Wang X, Thiele JR, et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat Commun (2017) 8:14188. doi:10.1038/ncomms14188
33. Eisenhardt SU, Habersberger J, Murphy A, Chen Y-C, Woollard KJ, Bassler N, et al. Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ Res (2009) 105(2):128–37. doi:10.1161/CIRCRESAHA.108.190611
34. Molins B, Peña E, la Torre de R, Badimon L. Monomeric C-reactive protein is prothrombotic and dissociates from circulating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc Res (2011) 92(2):328–37. doi:10.1093/cvr/cvr226
35. Potempa LA, Maldonado BA, Laurent P, Zemel ES, Gewurz H. Antigenic, electrophoretic and binding alterations of human C-reactive protein modified selectively in the absence of calcium. Mol Immunol (1983) 20(11):1165–75. doi:10.1016/0161-5890(83)90140-2
36. Potempa LA, Siegel JN, Fiedel BA, Potempa RT, Gewurz H. Expression, detection and assay of a neoantigen (Neo-CRP) associated with a free, human C-reactive protein subunit. Mol Immunol (1987) 24(5):531–41. doi:10.1016/0161-5890(87)90028-9
37. Berman S, Gewurz H, Mold C. Binding of C-reactive protein to nucleated cells leads to complement activation without cytolysis. J Immunol (1986) 136(4):1354–9.
38. Bíró A, Rovó Z, Papp D, Cervenak L, Varga L, Füst G, et al. Studies on the interactions between C-reactive protein and complement proteins. Immunology (2007) 121(1):40–50. doi:10.1111/j.1365-2567.2007.02535.x
39. Okemefuna AI, Nan R, Miller A, Gor J, Perkins SJ. Complement factor H binds at two independent sites to C-reactive protein in acute phase concentrations. J Biol Chem (2010) 285(2):1053–65. doi:10.1074/jbc.M109.044529
40. Molins B, Fuentes-Prior P, Adán A, Antón R, Arostegui JI, Yagüe J, et al. Complement factor H binding of monomeric C-reactive protein downregulates proinflammatory activity and is impaired with at risk polymorphic CFH variants. Sci Rep (2016) 6:22889. doi:10.1038/srep22889
41. Devaraj S, Jialal I. C-reactive protein polarizes human macrophages to an M1 phenotype and inhibits transformation to the M2 phenotype. Arterioscler Thromb Vasc Biol (2011) 31(6):1397–402. doi:10.1161/ATVBAHA.111.225508
42. Khreiss T, József L, Hossain S, Chan JSD, Potempa LA, Filep JG. Loss of pentameric symmetry of C-reactive protein is associated with delayed apoptosis of human neutrophils. J Biol Chem (2002) 277(43):40775–81. doi:10.1074/jbc.M205378200
43. Zouki C, Haas B, Chan JS, Potempa LA, Filep JG. Loss of pentameric symmetry of C-reactive protein is associated with promotion of neutrophil-endothelial cell adhesion. J Immunol (2001) 167(9):5355–61. doi:10.4049/jimmunol.167.9.5355
44. Khreiss T, József L, Potempa LA, Filep JG. Conformational rearrangement in C-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation (2004) 109(16):2016–22. doi:10.1161/01.CIR.0000125527.41598.68
45. Slevin M, Matou Nasri S, Turu M, Luque A, Rovira N, Badimon L, et al. Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol (2010) 20(1):151–65. doi:10.1111/j.1750-3639.2008.00256.x
46. Seddon JM, Gensler G, Milton RC, Klein ML, Rifai N. Association between C-reactive protein and age-related macular degeneration. JAMA (2004) 291(6):704–10. doi:10.1001/jama.291.6.704
47. Seddon JM, George S, Rosner B, Rifai N. Progression of age-related macular degeneration: prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch Ophthalmol (2005) 123(6):774–82. doi:10.1001/archopht.123.6.774
48. Seddon JM, Gensler G, Rosner B. C-reactive protein and CFH, ARMS2/HTRA1 gene variants are independently associated with risk of macular degeneration. Ophthalmology (2010) 117(8):1560–6. doi:10.1016/j.ophtha.2009.11.020
49. Cipriani V, Hogg RE, Sofat R, Moore AT, Webster AR, Yates JRW, et al. Association of C-reactive protein genetic polymorphisms with late age-related macular degeneration. JAMA Ophthalmol (2017) 135(9):909–16. doi:10.1001/jamaophthalmol.2017.2191
50. Schaumberg DA, Christen WG, Kozlowski P, Miller DT, Ridker PM, Zee RYL. A prospective assessment of the Y402H variant in complement factor H, genetic variants in C-reactive protein, and risk of age-related macular degeneration. Invest Ophthalmol Vis Sci (2006) 47(6):2336–40. doi:10.1167/iovs.05-1456
51. Despriet DD, Klaver CC, Witteman JC, Bergen AA, Kardys I, de Maat MP, et al. Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. JAMA (2006) 296(3):301–9. doi:10.1001/jama.296.3.301
52. Bhutto IA, Baba T, Merges C, Juriasinghani V, McLeod DS, Lutty GA. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br J Ophthalmol (2011) 95(9):1323–30. doi:10.1136/bjo.2010.199216
53. Ying SC, Gewurz H, Kinoshita CM, Potempa LA, Siegel JN. Identification and partial characterization of multiple native and neoantigenic epitopes of human C-reactive protein by using monoclonal antibodies. J Immunol (1989) 143(1):221–8.
54. Skeie JM, Fingert JH, Russell SR, Stone EM, Mullins RF. Complement component C5a activates ICAM-1 expression on human choroidal endothelial cells. Invest Ophthalmol Vis Sci (2010) 51(10):5336–42. doi:10.1167/iovs.10-5322
55. Molins B, Pascual A, Méndez , Llorenç V, Zarranz-Ventura J, Mesquida M, et al. C-reactive protein isoforms differentially affect outer blood-retinal barrier integrity and function. Am J Physiol Cell Physiol (2017) 312(3):C244–53. doi:10.1152/ajpcell.00057.2016
56. Wang Y, Bian Z-M, Yu W-Z, Yan Z, Chen W-C, Li X-X. Induction of interleukin-8 gene expression and protein secretion by C-reactive protein in ARPE-19 cells. Exp Eye Res (2010) 91(2):135–42. doi:10.1016/j.exer.2010.02.008
57. Yang P, Tyrrell J, Han I, Jaffe GJ. Expression and modulation of RPE cell membrane complement regulatory proteins. Invest Ophthalmol Vis Sci (2009) 50(7):3473–81. doi:10.1167/iovs.08-3202
58. McHarg S, Clark SJ, Day AJ, Bishop PN. Age-related macular degeneration and the role of the complement system. Mol Immunol (2015) 67(1):43–50. doi:10.1016/j.molimm.2015.02.032
59. Mullins RF, Skeie JM, Malone EA, Kuehn MH. Macular and peripheral distribution of ICAM-1 in the human choriocapillaris and retina. Mol Vis (2006) 12:224–35.
60. Liu G, Vogel SM, Gao X, Javaid K, Hu G, Danilov SM, et al. Src phosphorylation of endothelial cell surface intercellular adhesion molecule-1 mediates neutrophil adhesion and contributes to the mechanism of lung inflammation. Arterioscler Thromb Vasc Biol (2011) 31(6):1342–50. doi:10.1161/ATVBAHA.110.222208
61. Chew EY, Clemons TE, SanGiovanni JP, Danis R, Ferris FL, Elman M, et al. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the age-related eye disease study 2 (AREDS2) randomized clinical trial. JAMA (2013) 309(19):2005–15. doi:10.1001/jama.2013.4997
62. Folk JC, Stone EM. Ranibizumab therapy for neovascular age-related macular degeneration. N Engl J Med (2010) 363(17):1648–55. doi:10.1056/NEJMct1000495
63. Taskintuna I, Elsayed ME, Schatz P. Update on clinical trials in dry age-related macular degeneration. Middle East Afr J Ophthalmol (2016) 23(1):13–26. doi:10.4103/0974-9233.173134
64. Xu H, Chen M. Targeting the complement system for the management of retinal inflammatory and degenerative diseases. Eur J Pharmacol (2016) 787:94–104. doi:10.1016/j.ejphar.2016.03.001
65. Schnabolk G, Parsons N, Obert E, Annamalai B, Nasarre C, Tomlinson S, et al. Delivery of CR2-fH using AAV vector therapy as treatment strategy in the mouse model of choroidal neovascularization. Mol Ther Methods Clin Dev (2018) 9:1–11. doi:10.1016/j.omtm.2017.11.003
Keywords: C-reactive protein, age-related macular degeneration, inflammation, complement, complement factor H, membrane attack complex
Citation: Chirco KR and Potempa LA (2018) C-Reactive Protein As a Mediator of Complement Activation and Inflammatory Signaling in Age-Related Macular Degeneration. Front. Immunol. 9:539. doi: 10.3389/fimmu.2018.00539
Received: 15 January 2018; Accepted: 02 March 2018;
Published: 15 March 2018
Edited by:
Mark Slevin, Manchester Metropolitan University, United KingdomReviewed by:
Agustina Alaimo, Universidad de Buenos Aires, ArgentinaGabor Csanyi, Augusta University, United States
Copyright: © 2018 Chirco and Potempa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathleen R. Chirco, kchirco@buckinstitute.org