 Xinfang Huang1,2
Xinfang Huang1,2 Stephanie Dorta-Estremera
Stephanie Dorta-Estremera Nan Shen
Nan Shen Wei Cao
Wei Cao- 1Shanghai Institute of Rheumatology, Shanghai Renji Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China
- 2Department of Immunology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
- 3The University of Texas Graduate School of Biomedical Sciences, Houston, TX, USA
- 4Cellular Biomedicine Group Inc., Palo Alto, CA, USA
Plasmacytoid dendritic cells (pDCs), which are prominent type I interferon (IFN-I)-producing immune cells, have been extensively implicated in systemic lupus erythematosus (SLE). However, whether they participate critically in lupus pathogenesis remains unknown. Recent studies using various genetic and cell type-specific ablation strategies have demonstrated that pDCs play a pivotal role in the development of autoantibodies and the progression of lupus under diverse experimental conditions. The findings of several investigations highlight a notion that pDCs operate critically at the early stage of lupus development. In particular, pDCs have a profound effect on B-cell activation and humoral autoimmunity in vivo. This deeper understanding of the vital role of pDCs in lupus pathogenesis supports the therapeutic targeting of the pDC-IFN-I pathway in SLE.
Introduction
Patients with systemic lupus erythematosus (SLE) frequently have aberrant expression of genes that are stimulated by type 1 interferons (IFN-α, IFN-β, IFN-ω, IFN-τ; IFN-I), a family of pluripotent cytokines that are important for antiviral immune response, and this expression profile is correlated with anti-dsDNA antibody levels and disease severity (1, 2). Plasmacytoid dendritic cells (pDCs), a distinct subset of DCs, are capable of rapidly secreting large amounts of IFN-I in response to viral infection through endosomal Toll-like receptor (TLR) activation. In SLE patients, pDCs are believed to be a major cellular source of IFN-I, primarily because they readily produce IFN-I when exposed to SLE immune complexes or other lupus-related, nucleic acid-containing compounds (3–5). The implications IFN-I and pDCs have in clinical SLE have been extensively investigated and reviewed previously (3, 6–8).
In this perspective, we focus on the findings of recent studies that collectively illuminate the involvement of pDCs in systemic autoimmunity in vivo and their role in promoting SLE through IFN-I production in particular. Studies using various experimental lupus models have revealed that pDCs play an indispensable role in stimulating autoantibody response and facilitating lupus progression, which bolsters the rationale of targeting pDCs to alleviating SLE.
pDCs are Instrumental to Spontaneous Lupus Development
The development of pDCs from bone marrow progenitors is critically controlled by the transcription factors E2-2 (encoded by Tcf4) (9) and IRF8 (10, 11). In humans, naive pDCs, a distinct type of immune cells, abundantly express the cell type-specific receptor BDCA2, and in mice, they express PDCA1 and Siglec-H (3). Slc15a4, a peptide/histidine transporter, specifically facilitates endosomal TLR signaling and the production of IFN and other cytokines in pDCs (12). On the basis of these insights, researchers have developed several genetic systems for selectively deleting or disabling pDCs in vivo (Table 1). Consequently, several groups have investigated the contribution of pDCs to SLE in experimental lupus models.
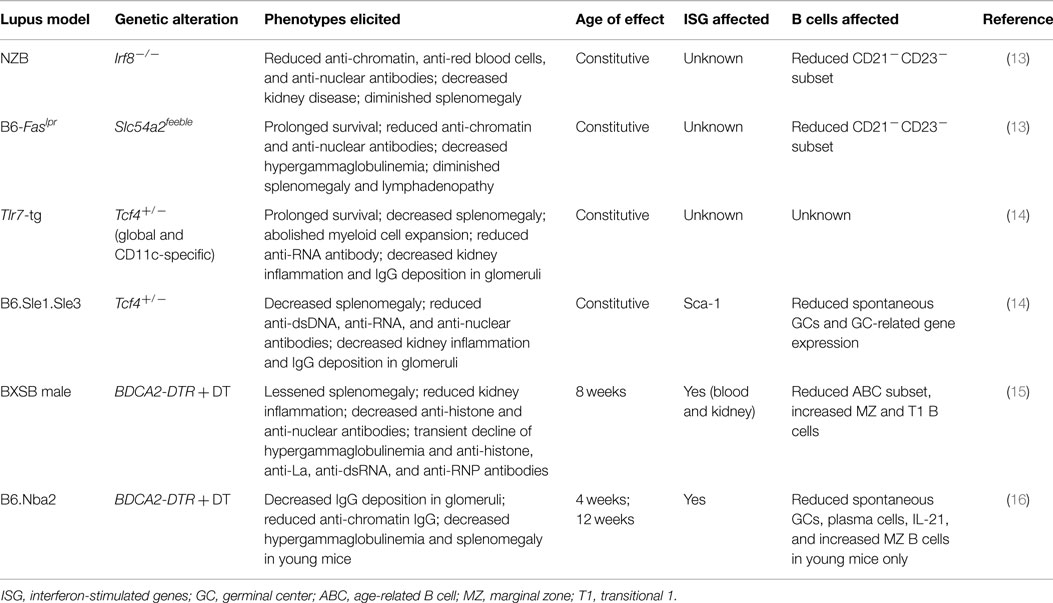
Table 1. Effects of pDC depletion in different spontaneous lupus models.
New Zealand Black (NZB) mice spontaneously develop elevated immunoglobulin levels, anti-DNA antibodies, hemolytic anemia, and circulating immune complexes that cause glomerulonephritis. Baccala et al. reported that Irf8 deficiency effectively abolished all the key autoimmune phenotypes in NZB mice, indicating that the IRF8-instructed program is essential to lupus development (13).
Mice homozygous for the lymphoproliferation spontaneous mutation (Faslpr) show systemic autoimmunity, which is associated with glomerulonephritis and lymphadenopathy. pDC-defective C57BL/6-Faslpr mice, which were generated by introducing the Slc15a4 mutation that selectively disrupts the pDC-mediated TLR response (13), have reduced autoantibodies and splenomegaly and prolonged survival compared with pDC-intact mice. Despite these intriguing observations, pDCs’ influence on lupus pathogenesis remains uncertain because both Irf8 and Slc15a4 affect the development or function of other immune cell types (13, 17–20).
Monoallelic loss of Tcf4 is sufficient to impair the innate immune function of pDCs in mice and humans (9). Sisirak et al. (14) examined the effects of Tcf4 haplodeficiency in autoimmune mice overexpressing the endosomal RNA sensor Tlr7 (Tlr7-tg) (21–23). Intriguingly, they found that both global and DC-specific Tcf4 haplodeficiency abolished splenomegaly and myeloid cell expansion and diminished anti-RNA autoantibody levels in the presence of Tlr7 overexpression, which suggests a profound involvement of pDCs in this lupus model.
B6.Sle1.Sle3 mice contain genomic intervals of two susceptibility loci from the lupus-prone NZM2410 strain on a C57BL/6 background (24). They spontaneously develop glomerulonephritis and high titers of autoantibodies against dsDNA and chromatin. Similar to Tlr7-tg, Tcf4 haplodeficiency effectively blocked the development of lupus, as indicated by the significantly reduced anti-DNA antibody levels and glomerulonephritis (14). Although Tcf4 also affects the development of B and T cells as well as a subset of conventional DCs (cDCs) (25–27), these data nevertheless suggest that pDCs are critically involved in lupus pathogenesis and autoantibody production.
In transgenic mice that express the diphtheria toxin (DT) receptor (DTR) under the control of the BDCA2 promoter, the administration of DT results in the selective and transient ablation of pDCs (28). BXSB is a recombinant inbred lupus-prone strain, in which male mice harbor the Y-linked autoimmune accelerator locus, which has a duplicated chromosome segment containing Tlr7 (21–23). After successfully backcrossing the BDCA2-DTR transgene into a BXSB background, Rowland et al. treated pre-autoimmune male BXSB.DTR mice with continuous DT injections for 3 weeks (15). Depletion of pDCs reduced the activation and expansion of immune cells, restricted autoantibody production, and minimized kidney inflammation. DT treatment also had a lasting regulatory effect on the serum levels of inflammatory factors, such as soluble VCAM-1, soluble CD40L, and chemokines (15). The finding that an early and transient depletion of pDCs was sufficient to ameliorate lupus further highlighted the predominant role pDCs have in lupus pathogenesis.
In another study, Davison and Jorgensen assessed the impact of transient pDC depletion in B6.Nba2 mice, which harbor IFN+ pDCs and develop spontaneous lupus-like disease (16). Nba2.BDCA2-DTR mice received continuous DT treatment, which significantly decreased antichromatin autoantibody levels and diminished the deposition of IgG immune complexes in kidney glomeruli.
Collectively, these studies demonstrate that pDCs play a pivotal role in promoting lupus-like autoimmunity in mice with diverse genetic backgrounds.
Activation of pDCs and Induction of IFN-I Instigate Humoral Autoimmunity
The identical twin concordance rate for lupus is less than 50% (29, 30), which suggests that factors in addition to genetics also control SLE. Indeed, infections, drugs, and chemical compounds have been shown to facilitate the development of systemic autoimmunity (29, 31–33). However, much less is known about the molecular determinants and cellular pathways critical to this process.
One particular agent implicated in systemic autoimmunity recently is terminally misfolded amyloid proteins, which contain extensive β-sheet structures (34, 35). More than two dozen aberrant polypeptides that deposit amyloid have been implicated in human pathological conditions, including Alzheimer disease, Parkinson disease, and type 2 diabetes (35–38). In human tissues, amyloid often contains non-proteinaceous cofactors (34, 39) because amyloid precursor proteins have an affinity for nucleic acids and glycosaminoglycans, and interactions with these molecules expedite the formation of amyloid (40). Intriguingly, nucleic acid-containing amyloid fibrils activate endosomal TLRs to induce the release of IFN-I from pDCs (41). Our group showed that injections of DNA-containing amyloid induced pDC infiltration and IFN-I production in mice. We also found that the inoculation of non-autoimmune mice with DNA-containing amyloid stimulated the development of antinuclear autoantibodies and led to the deposition of immunoglobulin G in the kidney glomeruli (41). The findings of recent studies of amyloid-triggered autoimmunity in mice are summarized in Table 2. Importantly, the transient antibody-mediated depletion of pDCs prior to amyloid inoculation abolished IFN-I production and subsequent autoantibody generation. These findings illustrate that the innate immune activation of pDCs can break immune tolerance and initiate autoimmune responses. Given that pDCs actively secrete IFN-I during the early stages of many viral infections (3, 28), the pDC-IFN-I pathway may also contribute to the production of broadly self-reactive antibodies that are aggravated by viral infections.

Table 2. Amyloid-triggered autoimmunity in mice.
Amyloid is not rare or intrinsically harmful. More prevalent than previously thought, functional amyloids participate in diverse cellular processes, including biofilm and spore assembly in bacteria, the storage of peptide hormones within mammalian secretory granules, and enhanced HIV infectivity during sexual transmission (43–48). Gallo et al. reported recently that curli, an amyloid expressed by Enterobacteriaceae, could irreversibly form fibers with bacterial DNA during biofilm formation. Curli-DNA composites stimulated high IFN-I and cytokine production in DCs in vitro. Interestingly, a bacterial amyloid–DNA complex and curli+ Escherichia coli and Salmonella triggered the production of autoantibodies in wild-type and lupus-prone mice (42). Although Gallo et al. did not analyze pDCs, DNA-containing bacterial amyloid shares key innate immune properties with mammalian amyloid. Therefore, the IFN-I induced by certain bacterial infections may also evoke humoral autoimmunity, an intriguing notion requiring further investigation.
Mechanism of pDC-Mediated Autoimmune Responses
The functional contribution of pDCs to the pathogenesis of chronic diseases has been difficult to establish because (1) pDCs are rare immune cells; (2) IFN-I production by pDCs is rapid but transient; and (3) activated pDCs, by downregulating specific surface receptors, are almost indistinguishable from cDCs. Fortunately, new genetic tools are enabling researchers to elucidate the mechanism by which pDCs impact lupus development.
First, pDCs play a crucial role in the initiation of lupus. In pre-autoimmune BXSB mice, pDC depletion rapidly and effectively diminished hypergammaglobulinemia, reduced the development of a wide spectrum of autoantibodies, and restrained kidney inflammation, which coincided with the decreased transcription of IFN-stimulated genes (15). This finding is consistent with an earlier report that an anti-IFNα/β receptor-blocking antibody had a protective effect only in young BXSB mice (49). Similarly, a prophylactic IFN receptor blockade in young MRL-Faslpr mice prevented the escalation of anti-RNP autoantibody titers and proteinuria transiently (49). Although the molecular entity that triggers IFN-I response in polygenic lupus-prone mice is unknown, the amyloid-induced autoimmunity explicitly illustrates that the activation of the pDC-IFN-I pathway can activate B cells, break immune tolerance, and induce antinuclear antibodies (41).
However, neither IFN-I nor pDCs are required for the regular antibody response to foreign antigens (13, 41, 50). Thus, the way in which the pDC-IFN-I axis promotes the positive selection, affinity maturation, and expansion of autoimmune B cells is enigmatic. The germinal center (GC) represents a unique lymphoid microenvironment where antigen-reactive B cells are expanded and diversified (51–53). Prevalent in the spleen of many lupus-prone mice, spontaneous GCs are crucial for the generation of autoantibodies (54–56). Interestingly, Tcf4-haplodeficient B6.Sle1.Sle3 mice displayed reduced spontaneous GC reaction and a decreased GC-associated gene expression signature (14). Similarly, B6.Nba2 mice with depleted pDCs had significantly fewer GCs, as well as fewer follicular helper T cells and plasma cells, a phenomenon only observed in DT-treated young mice (16).
However, B cells other than GCs also constitute autoreactive B-cell compartments in a number of lupus models (57, 58). The overexpression of IFN-α in vivo sustained the proliferation of B cells and stimulated the expansion of short-lived plasmablasts, suggesting that the IFN-I-mediated autoimmune B-cell response had a non-GC origin (59). Age-associated B cells (ABCs), which lack CD21 and CD23 expression but express myeloid cell-specific markers, are stimulated by TLR7 activation and have an increased presence in lupus-prone mice (60–62). The findings from studies of BXSB, NZB, and B6-Faslpr mice implied that ABCs diminish as a consequence of pDC ablation (13, 15). In contrast, the spleens of BXSB mice after DT injection contained increased numbers of marginal zone and transitional T1 B cells (15). Driven by Tlr7 overexpression, T1 B cells undergo significant expansion and proliferation and are reportedly responsible for the production of autoantibodies in Tlr7-tg mice (57). Therefore, the way in which pDCs differentially regulate ABC and T1 B subsets and the relative contribution of specific B-cell subsets in the pathogenesis of lupus-prone mice remain to be elucidated. Furthermore, whether Tcf4 haplodeficiency similarly impacts the T1 B-cell subset in the Tlr7-tg model is unclear.
In addition, pDCs may mediate emergency myelopoiesis, as indicated by the abated expansion of myeloid cells in Tcf4+/− Tlr7-tg mice (63). A previous study found that pDCs in the bone marrow (BM) of Tlr7-tg mice constitutively express IFN-I, which was hypothesized to drive the proliferation of Sca-1+ granulocyte/macrophage progenitors and subsequent expansion of peripheral myeloid cells (64). Whether the curbed myeloproliferation is mediated by the compromised IFN-I expression from BM pDCs remains to be determined. Of note, the normalization of the myeloid cell compartment was not reported in BXSB mice, which also harbor duplicated Tlr7, after continuous DT treatment (15). The reason for this discrepancy is unclear, but transgenic mouse lines knowingly expressing DTR in cDC lineage reportedly have depletion of additional cell types and elicit neutrophilia and monocytosis upon DT injection (65, 66). Nevertheless, IFN-I+ pDCs may functionally affect hematopoietic cells in the BM of mice with lupus. Indeed, pDCs from adult MRL-Faslpr mouse BM transcribe IFNα and likely hinder the survival of B-cell progenitor cells (67).
Altogether, these recent studies are exceedingly significant and informative. The findings from diverse lupus models converge on the profound effect of pDCs toward humoral autoimmunity in general; while at the same time raise additional questions. Further detailed analysis is needed to determine whether pDCs differentially promote autoimmune B-cell responses in certain genetic and cellular contexts.
Clinical Implications
Recent findings in lupus models about the pivotal function pDCs have in lupus pathogenesis support the tactic of therapeutically targeting pDCs in humans with SLE. TLR activation in pDCs under systemic autoimmune conditions hampers the effect of glucocorticoid treatment (68). In lupus-prone mice, pDCs seem to be more sensitive to Bcl2 antagonism, whose effectiveness could probably be improved with the addition of glucocorticoid therapy (69).
In their search for an effective treatment for SLE, researchers have developed numerous schemes to neutralize IFN-I or interfere with the function of IFNα/β receptor (8, 70, 71). Strategies to selectively deplete pDCs or suppress their IFN-I response should also help alleviate lupus (3). Given the predominant role of the pDC-IFN-I pathway in the early stages of lupus development, IFN-I and pDC blockade should be administrated to patients with relatively low disease scores. More detailed mechanistic studies will provide insight into disease-specific processes and enable innovation in SLE treatments.
Conflict of Interest Statement
Yihong Yao is an employee of Cellular Biomedicine Group Inc. The other co-authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by an Institutional Research Grant from The University of Texas MD Anderson Cancer Center, National Institutes of Health (NIH) grant AI074809, the Natural Science Foundation of China (31470854), and NIH Cancer Center Support Grant CA016672. SD-E was supported by NIH Training Program Grant T32 CA009598. We thank Joseph Munch from Department of Scientific Publications, The University of Texas MD Anderson Cancer Center, for editing the manuscript.
References
1. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med (2003) 197(6):711–23. doi: 10.1084/jem.20021553
2. Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity (2006) 25(3):383–92. doi:10.1016/j.immuni.2006.08.010
3. Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol (2008) 8(8):594–606. doi:10.1038/nri2358
4. Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol (2004) 5(12):1219–26. doi:10.1038/ni1141
5. Reizis B, Colonna M, Trinchieri G, Barrat F, Gilliet M. Plasmacytoid dendritic cells: one-trick ponies or workhorses of the immune system? Nat Rev Immunol (2011) 11(8):558–65. doi:10.1038/nri3027
6. Di Domizio J, Cao W. Fueling autoimmunity: type I interferon in autoimmune diseases. Expert Rev Clin Immunol (2013) 9(3):201–10. doi:10.1586/eci.12.106
7. Ronnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus (2008) 17(5):394–9. doi:10.1177/0961203308090020
8. Crow MK. Advances in understanding the role of type I interferons in systemic lupus erythematosus. Curr Opin Rheumatol (2014) 26(5):467–74. doi:10.1097/BOR.0000000000000087
9. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell (2008) 135(1):37–48. doi:10.1016/j.cell.2008.09.016
10. Schiavoni G. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8[alpha]+ dendritic dells. J Exp Med (2002) 196:1415–25. doi:10.1084/jem.20021263
11. Tsujimura H, Tamura T, Ozato K. Cutting edge: IFN consensus sequence binding protein/IFN regulatory factor 8 drives the development of type I IFN-producing plasmacytoid dendritic cells. J Immunol (2003) 170(3):1131–5. doi:10.4049/jimmunol.170.3.1131
12. Blasius AL, Arnold CN, Georgel P, Rutschmann S, Xia Y, Lin P, et al. Slc15a4, AP-3, and Hermansky-Pudlak syndrome proteins are required for toll-like receptor signaling in plasmacytoid dendritic cells. Proc Natl Acad Sci U S A (2010) 107(46):19973–8. doi:10.1073/pnas.1014051107
13. Baccala R, Gonzalez-Quintial R, Blasius AL, Rimann I, Ozato K, Kono DH, et al. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc Natl Acad Sci U S A (2013) 110(8):2940–5. doi:10.1073/pnas.1222798110
14. Sisirak V, Ganguly D, Lewis KL, Couillault C, Tanaka L, Bolland S, et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med (2014) 211(10):1969–76. doi:10.1084/jem.20132522
15. Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med (2014) 211(10):1977–91. doi:10.1084/jem.20132620
16. Davison LM, Jorgensen TN. Sialic acid-binding immunoglobulin-type lectin H-positive plasmacytoid dendritic cells drive spontaneous lupus-like disease development in B6.Nba2 mice. Arthritis Rheumatol (2015) 67(4):1012–22. doi:10.1002/art.38989
17. Wang H, Morse H III. IRF8 regulates myeloid and B lymphoid lineage diversification. Immunol Res (2009) 43(1–3):109–17. doi:10.1007/s12026-008-8055-8
18. Aliberti J, Schulz O, Pennington DJ, Tsujimura H, Reis e Sousa CR, Ozato K, et al. Essential role for ICSBP in the in vivo development of murine CD8alpha + dendritic cells. Blood (2003) 101(1):305–10. doi:10.1182/blood-2002-04-1088
19. Sasawatari S, Okamura T, Kasumi E, Tanaka-Furuyama K, Yanobu-Takanashi R, Shirasawa S, et al. The solute carrier family 15A4 regulates TLR9 and NOD1 functions in the innate immune system and promotes colitis in mice. Gastroenterology (2011) 140(5):1513–25. doi:10.1053/j.gastro.2011.01.041
20. Kobayashi T, Shimabukuro-Demoto S, Yoshida-Sugitani R, Furuyama-Tanaka K, Karyu H, Sugiura Y, et al. The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity (2014) 41(3):375–88. doi:10.1016/j.immuni.2014.08.011
21. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science (2006) 312(5780):1669–72. doi:10.1126/science.1124978
22. Subramanian S, Tus K, Li Q-Z, Wang A, Tian X-H, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A (2006) 103(26):9970–5. doi:10.1073/pnas.0603912103
23. Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity (2007) 27(5):801–10. doi:10.1016/j.immuni.2007.09.009
24. Morel L. Genetics of SLE: evidence from mouse models. Nat Rev Rheumatol (2010) 6(6):348–57. doi:10.1038/nrrheum.2010.63
25. Bar-On L, Birnberg T, Lewis KL, Edelson BT, Bruder D, Hildner K, et al. CX3CR1+ CD8alpha+ dendritic cells are a steady-state population related to plasmacytoid dendritic cells. Proc Natl Acad Sci U S A (2010) 107(33):14745–50. doi:10.1073/pnas.1001562107
26. Belle I, Zhuang YE. Proteins in lymphocyte development and lymphoid diseases. In: Reshma T, editor. Current Topics in Developmental Biology, (Vol. 110). Waltham, MA: Academic Press (2014). p. 153–87.
27. Forrest MP, Hill MJ, Quantock AJ, Martin-Rendon E, Blake DJ. The emerging roles of TCF4 in disease and development. Trends Mol Med (2014) 20(6):322–31. doi:10.1016/j.molmed.2014.01.010
28. Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity (2010) 33(6):955–66. doi:10.1016/j.immuni.2010.11.020
29. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med (2008) 358(9):929–39. doi:10.1056/NEJMra071297
30. Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol (2010) 6(12):683–92. doi:10.1038/nrrheum.2010.176
31. Cusick MF, Libbey JE, Fujinami RS. Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol (2012) 42(1):102–11. doi:10.1007/s12016-011-8293-8
32. Pollard KM, Kono DH. Requirements for innate immune pathways in environmentally induced autoimmunity. BMC Med (2013) 11(1):100. doi:10.1186/1741-7015-11-100
34. Sideras K, Gertz MA. Amyloidosis. Adv Clin Chem (2009) 47:1–44. doi:10.1016/S0065-2423(09)47001-X
35. Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell (2012) 148(6):1188–203. doi:10.1016/j.cell.2012.02.022
36. Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature (2003) 426(6968):895–9. doi:10.1038/nature02263
37. Schnabel J. Protein folding: the dark side of proteins. Nature (2010) 464(7290):828–9. doi:10.1038/464828a
38. Selkoe DJ. Folding proteins in fatal ways. Nature (2003) 426(6968):900–4. doi:10.1038/nature02264
39. Jiménez JS. Protein-DNA interaction at the origin of neurological diseases: a hypothesis. J Alzheimers Dis (2010) 22(2):375–91. doi:10.3233/JAD-2010-100189
40. Di Domizio J, Zhang R, Stagg LJ, Gagea M, Zhuo M, Ladbury JE, et al. Binding with nucleic acids or glycosaminoglycans converts soluble protein oligomers to amyloid. J Biol Chem (2012) 287(1):736–47. doi:10.1074/jbc.M111.238477
41. Di Domizio J, Dorta-Estremera S, Gagea M, Ganguly D, Meller S, Li P, et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc Natl Acad Sci U S A (2012) 109(36):14550–5. doi:10.1073/pnas.1206923109
42. Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA composites of bacterial biofilms stimulate autoimmunity. Immunity (2015) 42(6):1171–84. doi:10.1016/j.immuni.2015.06.002
43. Munch J, Rucker E, Standker L, Adermann K, Goffinet C, Schindler M, et al. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell (2007) 131(6):1059–71. doi:10.1016/j.cell.2007.10.014
44. Roan NR, Muller JA, Liu H, Chu S, Arnold F, Sturzel CM, et al. Peptides released by physiological cleavage of semen coagulum proteins form amyloids that enhance HIV infection. Cell Host Microbe (2011) 10(6):541–50. doi:10.1016/j.chom.2011.10.010
45. Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol (2006) 60:131–47. doi:10.1146/annurev.micro.60.080805.142106
46. Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, Boersma FG, et al. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev (2003) 17(14):1714–26. doi:10.1101/gad.264303
47. Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science (2009) 325(5938):328–32. doi:10.1126/science.1173155
48. Badtke MP, Hammer ND, Chapman MR. Functional amyloids signal their arrival. Sci Signal (2009) 2(80):e43. doi:10.1126/scisignal.280pe43
49. Baccala R, Gonzalez-Quintial R, Schreiber RD, Lawson BR, Kono DH, Theofilopoulos AN. Anti-IFN-alpha/beta receptor antibody treatment ameliorates disease in lupus-predisposed mice. J Immunol (2012) 189(12):5976–84. doi:10.4049/jimmunol.1201477
50. Cao W. Pivotal functions of plasmacytoid dendritic cells in systemic autoimmune pathogenesis. J Clin Cell Immunol (2014) 5:e212. doi:10.4172/2155-9899.1000212
51. Shlomchik MJ, Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunol Rev (2012) 247(1):52–63. doi:10.1111/j.1600-065X.2012.01124.x
52. Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol (2010) 11(8):681–8. doi:10.1038/ni.1900
53. MacLennan IC. Germinal centers. Annu Rev Immunol (1994) 12:117–39. doi:10.1146/annurev.iy.12.040194.001001
54. Kuchroo VK, Ohashi PS, Sartor RB, Vinuesa CG. Dysregulation of immune homeostasis in autoimmune diseases. Nat Med (2012) 18(1):42–7. doi:10.1038/nm.2621
55. Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol (2012) 8(6):337–47. doi:10.1038/nrrheum.2012.58
56. Soni C, Wong EB, Domeier PP, Khan TN, Satoh T, Akira S, et al. B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers. J Immunol (2014) 193(9):4400–14. doi:10.4049/jimmunol.1401720
57. Giltiay NV, Chappell CP, Sun X, Kolhatkar N, Teal TH, Wiedeman AE, et al. Overexpression of TLR7 promotes cell-intrinsic expansion and autoantibody production by transitional T1 B cells. J Exp Med (2013) 210(12):2773–89. doi:10.1084/jem.20122798
58. Shlomchik MJ. Sites and stages of autoreactive B cell activation and regulation. Immunity (2008) 28(1):18–28. doi:10.1016/j.immuni.2007.12.004
59. Mathian A, Gallegos M, Pascual V, Banchereau J, Koutouzov S. Interferon-alpha induces unabated production of short-lived plasma cells in pre-autoimmune lupus-prone (NZBxNZW)F1 mice but not in BALB/c mice. Eur J Immunol (2011) 41(3):863–72. doi:10.1002/eji.201040649
60. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood (2011) 118(5):1305–15. doi:10.1182/blood-2011-01-331462
61. Rubtsov AV, Rubtsova K, Kappler JW, Marrack P. TLR7 drives accumulation of ABCs and autoantibody production in autoimmune-prone mice. Immunol Res (2013) 55(1–3):210–6. doi:10.1007/s12026-012-8365-8
62. Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood (2011) 118(5):1294–304. doi:10.1182/blood-2011-01-330530
63. Sawai CM, Sisirak V, Ghosh HS, Hou EZ, Ceribelli M, Staudt LM, et al. Transcription factor Runx2 controls the development and migration of plasmacytoid dendritic cells. J Exp Med (2013) 210(11):2151–9. doi:10.1084/jem.20130443
64. Buechler MB, Teal TH, Elkon KB, Hamerman JA. Cutting edge: type I IFN drives emergency myelopoiesis and peripheral myeloid expansion during chronic TLR7 signaling. J Immunol (2013) 190(3):886–91. doi:10.4049/jimmunol.1202739
65. van Blijswijk J, Schraml BU, Reis e Sousa C. Advantages and limitations of mouse models to deplete dendritic cells. Eur J Immunol (2013) 43(1):22–6. doi:10.1002/eji.201243022
66. Roberts LM, Ledvina HE, Tuladhar S, Rana D, Steele SP, Sempowski GD, et al. Depletion of alveolar macrophages in CD11c diphtheria toxin receptor mice produces an inflammatory response. Immun Inflamm Dis (2015) 3(2):71–81. doi:10.1002/iid3.51
67. Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity (2013) 38(3):528–40. doi:10.1016/j.immuni.2012.11.017
68. Guiducci C, Gong M, Xu Z, Gill M, Chaussabel D, Meeker T, et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature (2010) 465(7300):937–41. doi:10.1038/nature09102
69. Zhan Y, Carrington EM, Ko HJ, Vikstrom IB, Oon S, Zhang JG, et al. Bcl-2 antagonists kill plasmacytoid dendritic cells from lupus-prone mice and dampen interferon-alpha production. Arthritis Rheumatol (2015) 67(3):797–808. doi:10.1002/art.38966
70. Lichtman EI, Helfgott SM, Kriegel MA. Emerging therapies for systemic lupus erythematosus – focus on targeting interferon-alpha. Clin Immunol (2012) 143(3):210–21. doi:10.1016/j.clim.2012.03.005
Keywords: plasmacytoid dendritic cells, systemic lupus erythematosus, type I interferon, amyloid, autoantibody, autoimmunity, animal models, B cells
Citation: Huang X, Dorta-Estremera S, Yao Y, Shen N and Cao W (2015) Predominant role of plasmacytoid dendritic cells in stimulating systemic autoimmunity. Front. Immunol. 6:526. doi: 10.3389/fimmu.2015.00526
Received: 27 July 2015; Accepted: 28 September 2015;
Published: 12 October 2015
Edited by:
James Harris, Monash University, AustraliaReviewed by:
Raymond John Steptoe, University of Queensland, AustraliaMeredith O’Keeffe, Monash University, Australia
Copyright: © 2015 Huang, Dorta-Estremera, Yao, Shen and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Cao, wei.cao.phd@gmail.com