 Kok P. M. van Kessel
Kok P. M. van Kessel Jovanka Bestebroer
Jovanka Bestebroer Jos A. G. van Strijp
Jos A. G. van Strijp- Medical Microbiology, University Medical Center Utrecht, Utrecht, Netherlands
Initial elimination of invading Staphylococcus aureus from the body is mediated by professional phagocytes. The neutrophil is the major phagocyte of the innate immunity and plays a key role in the host defense against staphylococcal infections. Opsonization of the bacteria with immunoglobulins and complement factors enables efficient recognition by the neutrophil that subsequently leads to intracellular compartmentalization and killing. Here, we provide a review of the key processes evolved in neutrophil-mediated phagocytosis of S. aureus and briefly describe killing. As S. aureus is not helpless against the professional phagocytes, we will also highlight its immune evasion arsenal related to phagocytosis.
Introduction
Staphylococcus aureus is a human commensal but also a common course of serious infections, ranging from mild skin infections to more serious life-threating wound and bloodstream infections (1). The innate immunity is an important part of the host defense in elimination of infections caused by S. aureus. Notably, many virulence factors of S. aureus are directed toward elements of the innate immune defense including its principal phagocyte, the neutrophil (2–4).
In the human blood, neutrophils are the predominant phagocytic cell type, accounting for 50–60% of all leukocytes. The acknowledgment for professional phagocytes started with the first description of motile cells capable of engulfing other matter by Ilya Ilyich Mechnikov, a Russian biologist, best known for his pioneering research on the immune system. Mechnikov received the Nobel Prize in Medicine, jointly with Paul Ehrlich, in 1908 for his work on phagocytosis, which is defined as the uptake of bacteria, parasites, dead host cells, and foreign debris. In addition to neutrophils, dendritic cells, monocytes, and macrophages are considered professional phagocytes, and all cell types are critical in controlling bacterial infection, all be it through different means.
Neutrophils are ready-to-go cells, show a fast response, and have a generally believed short half-life of <7 h. Recent in vivo labeling studies, however, estimated the lifespan of neutrophils to be much longer, i.e., 5.4 days (5). Neutrophils are rapidly mobilized from the bone marrow into the circulation, and several subtypes are now characterized based on differential surface antigen expression and function in innate immunity (6–8). The last decade, the role of neutrophils in several other aspects of immunity is appreciated as it has become clear that neutrophils also participate in processes associated with the adaptive immunity and tumor immunology. They display cross talk with adaptive immune cells, i.e., dendritic cells, lymphocytes, and natural killer cells, through secretion of cytokines and reactive oxygen species (ROS), and they interact directly with cells of adaptive immunity via cell surface molecules (9), functions that are most likely associated with different subpopulations or activation states (10, 11). As neutrophils are circulating cells, they first need to leave the bloodstream via diapedesis to reach the site of infection through directed migration along an increasing gradient of chemoattractants, which are derived from bacteria, generated via complement activation or secreted by activated cells including leukocytes (12). For efficient phagocytosis, bacteria need to be covered with opsonins provided by specific immunoglobulins (Igs), the complement system, and others. Uptake of bacteria leads to full activation of the anti-microbial arsenal of the neutrophil leading to killing of the ingested bacteria. The neutrophil is equipped with two major pathways for killing, generation of ROS, and degranulation of granules packed with proteases and specific anti-microbial peptides. The active phagocytosis by neutrophils is eventually followed by a more passive form of elimination of the micro-organisms as the lifetime of the cell is consumed through the formation of neutrophil extracellular traps (NETs) that consist of chromatin and granule content (13). It should be noted that neutrophils and its arsenal of anti-microbials employed to fight infection, sometimes turn against the host itself causing inflammation (14).
Opsonization and Recognition of S. aureus
For neutrophils, initiation of phagocytosis requires decoration of bacteria with opsonins that are recognized by specific surface receptors. Igs and complement components are the predominant factor in serum that enables efficient opsonization. They are deposited on the surface of the bacterium and enable recognition by Fc receptors (FcRs) and complement receptors (CRs), respectively, which trigger the phagocytic machinery (Figure 1). Therefore, the interface between the specific opsonins and their receptors dictates phagocytosis and is influenced by both the target (bacterium) and effector (neutrophil) characteristics. Indirectly, the availability and nature of the opsonins are major determinants in the process but are dependent on the target bacterium, and the generation of specific Igs is part of the adaptive immune response and depends on antigen presentation, memory, and affinity maturation.
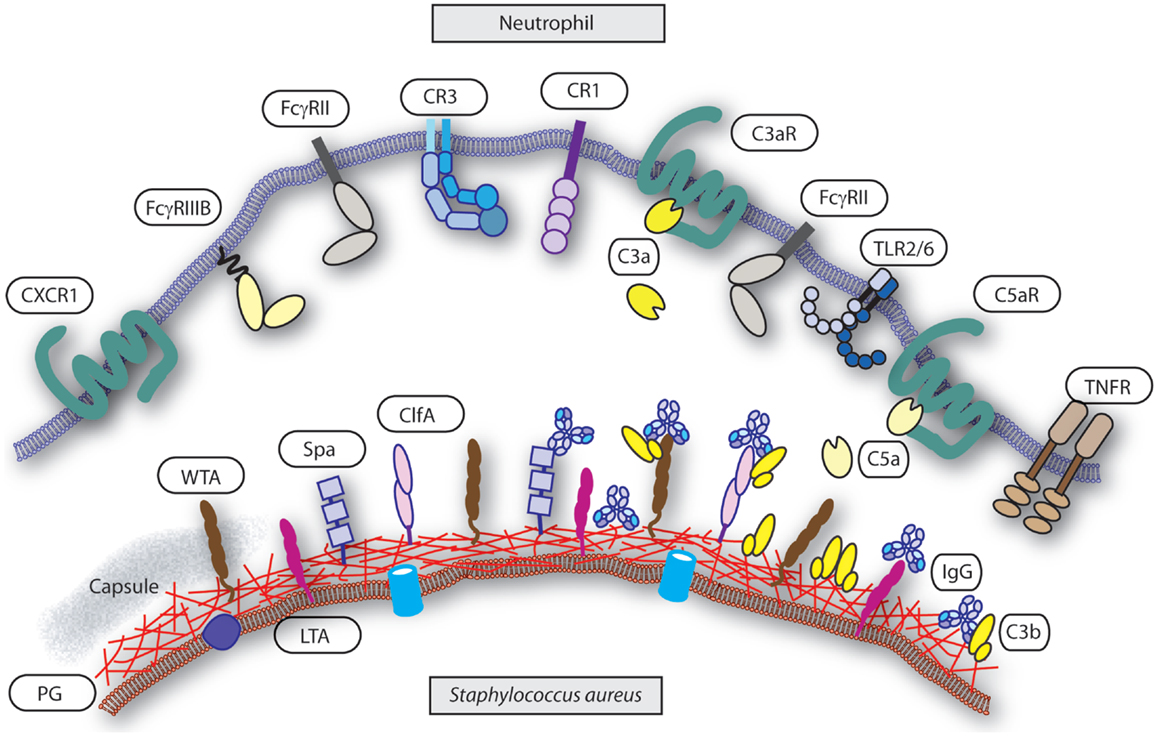
Figure 1. Interface neutrophil and S. aureus. Several groups of receptors mediate neutrophils recognition of S. aureus upon opsonization and others are involved in activation or priming of phagocytosis. Targets on the S. aureus surface are the cell wall components peptidoglycan (PG), wall teichoic acid (WTA), lipoteichoic acid (LTA), capsule (“gray area”), and representative associated proteins clumping factor A (ClfA) and protein-A (Spa). Targets are decorated with serum derived opsonins IgG (binding with their Fab part) and C3b. Note the reverse Fc-dependent association of IgG with Spa. Receptors on the neutrophil surface involved in recognition of the opsonized S. aureus are FcγRII and FcγRIII for IgG, and CR1 and CR3 for C3b (and iC3b). Examples of receptors on the neutrophil involved in priming or activation of phagocytosis are complement receptors C3aR for C3a and C5aR for C5a, CXCR1 for il-8, and TNFR for TNFα. The heterodimer TLR2/TLR6 represents a common pattern recognition receptor for bacterial lipoproteins.
The most logical target candidates are of course surface-exposed proteins and general cell wall components, like peptidoglycan and wall teichoic acid (WTA) (15, 16). Since most humans are exposed to staphylococci already early in life without causing serious diseases, common structures, and/or proteins present on most staphylococci generate adequate Ig levels in normal healthy people. These structures provide sufficient natural occurring IgG that mediate recognition through FcγRs and also initiate classical pathway complement activation and thereby increase the amount of surface-bound opsonins, enabling uptake by neutrophils. Although the individual IgG levels by themselves do not seem to be that high, the power of the host defense is the combination of multiple IgG against several target molecules with complement activation. Lectin and alternative pathway-mediated complement activation results in the deposition of C3b/C3bi on the bacterial surface that is recognized by neutrophil CRs. Although phagocytosis is initiated, this system seems not to be so efficient in promotion of uptake of bacteria. On the other side of the spectrum, antibodies alone, and more specifically IgG, do trigger the FcγRs on the neutrophil to ingest the bacteria. For opsonization, location of the bound antibodies in relation to complement deposition and presentation to the neutrophil FcRs and CRs influences the opsonophagocytic potential. Thus, the efficacy of phagocytosis depends on the presence of specific IgG that activates the complement system via classical pathway resulting in C3b/C3bi deposition and binding to the FcγRs. The combination of these two key opsonins, complement, and IgG, triggers the phagocytic machinery into a high speed mode (17).
The attributed role of opsonins in neutrophil-mediated phagocytosis may, however, be dependent on the methodology, related to the site of infection. A recent example (18) showed that phagocytosis of S. aureus by human neutrophils in suspension depends on opsonization, while adherent neutrophils internalize more bacteria independent of opsonization. Consequently, addition of specific IgG did not enhance uptake by adherent neutrophils but improved the process in suspension. Remarkably, killing of the bacteria was only evident with adherent neutrophils when serum-induced aggregation phenomena were excluded. Under suspension conditions, persistence of free extracellular bacteria contributed substantially to the poor killing.
Immunoglobulins and Their Receptors in Recognition of S. aureus
Immunoglobulin is the second most abundant protein in serum/plasma after albumin and provides life-long protection against infectious agents. Igs are important for proper opsonization of bacteria and subsequent recognition by specific FcR on the surface of phagocytes. Several Ig subtypes exist and they display specific functions; while IgG (IgG1, IgG2, IgG3, and IgG4), IgM and secretory IgA of the mucosal tissues have roles in infection control, IgE does not. Different IgG subclasses bind different FcγRs. Normal peripheral blood neutrophils express FcγRII (CD32) and FcγRIIIB (CD16), and do not express the FcγR1 (CD64) that is found on monocytes. However, during systemic infections and sepsis neutrophils do express FcγRI. Together with other activation-related surface antigens, expression of CD64 is used as specific sign of bacterial infections and marks the activation status of the neutrophil (19, 20). Most phagocytes including neutrophils express specific receptor for IgA (FcαR or CD89). When appropriate specific IgA antibodies are present, the FcαR initiated responses are comparable with those of IgG and FcγRII. In contrast to IgG, IgA does not activate the complement system. The binding characteristics between different Ig classes and the various FcRs are well documented; please refer to some excellent reviews (21–23).
For proper Ig opsonization, generation of target-specific Igs is crucial. In the case of S. aureus, effective Igs are directed against its surface molecules. The outer surface of Gram-positive bacteria, including S. aureus, contains a thick layer of peptidoglycan that is decorated with covalently anchored proteins that can initiate both IgG and complement deposition. The cell wall-associated proteins are often virulence factors and have been shown to influence pathogenesis (24). Naturally occurring antibodies directed against these proteins, like clumping factor A (ClfA) and protein-A, circulate in human serum, and several of these proteins are used as vaccine candidates to elicit protective antibodies. The presence of specific naturally occurring IgG directed against several surface-exposed cell wall components and proteins of S. aureus has been described by several studies that demonstrate that IgG provides the host with a handle to recognize and phagocytose the bacterium.
Much research has been performed on the efficacy of antibodies directed against specific staphylococcal targets. However, several studies also document a larger approach. Using a bead-based Luminex screening against 56 staphylococcal antigens – including many known virulence factors, surface-exposed, and secreted proteins, but also peptidoglycan and WTA – IgG and IgA levels in multiple serum samples of patients with a S. aureus bacteremia and non-infected controls were compared (25). IgG directed against all antigens were detected in healthy controls as well as patients at the time of diagnosis. In the majority of bacteremia patients, the IgG levels showed a temporal increase. Using classical ELISAs on 19 staphylococcal cell surface and secreted proteins, a wide range of antibody levels in both healthy donors and patients was observed (26). Here, the majority of IgG was induced by lipoteichoic acid (LTA) and peptidoglycan. The amount of anti-staphylococcal antibodies accounted for 0.1–3% of total serum antibodies and was already obvious in healthy adolescents aged 13–15 years. The total anti-staphylococcal IgG levels correlated with a functionality opsonophagocytosis assay using fluorescently labeled bacteria and P388.D1 mouse macrophage cells. A comparable ELISA screening study (27) was performed against 11 different purified antigens from S. aureus, including surface antigens (such as teichoic acid and clumping factors A and B) and secreted proteins (such as alpha-toxin, lipase, enterotoxin A, toxic shock syndrome toxin, scalded-skin syndrome toxin, fibrinogen-binding protein, and extracellular adherence protein). Most healthy individuals (15–89 years) had circulating IgG antibodies against these antigens, whereby titers against teichoic acid were the highest. An alternative strategy to demonstrate the antibody repertoire used 2-D immunoblots with sera from patients with S. aureus bacteremia on the extracellular protein profile of the infecting S. aureus strain (28). Pre-existing antibodies were present and the pattern of antibody response was distinct for carriers and non-carriers, but also did contain a common signature.
In addition to the wide studies pertaining to groups of S. aureus surface epitopes, much research has been done on specific staphylococcal antigens, i.e., peptidogylcan, the cell WTA (a glycopolymer covalently linked to peptidoglycan), polymeric-N-acetylglucosamine (PNAG) (29, 30), N-acetylglucosamine (GlcNac) (31), iron-regulated surface protein B (IsdB) (32, 33), and ClfA (34, 35). As an example, interesting studies have been published on the opsonizing capacity of peptidoglycan. Elevated IgG levels to peptidoglycan were observed in patients with deep tissue infection with S. aureus but hardly in patients with superficial staphylococcal infection (36). Peptidoglycan appeared to be the key cell wall component involved in staphylococcal opsonization, both by IgG and activating complement (37). By itself, peptidoglycan and its soluble parts are also potent activators of the host defense leading to pro-inflammatory cytokine release from monocytes (38) and oxidative burst in neutrophils (39), only valid for the polymeric form of peptidoglycan. In addition, small dipeptides or tripeptides derived from polymeric peptidoglycan trigger cytoplasmic nucleotide oligomerization domain (NOD) receptors leading to NF-kB activation (40). For Bacillus anthracis-derived peptidoglycan it has been shown that specific IgG mediates bacterial binding to monocytes and neutrophils. All tested healthy donors contained IgG recognizing peptidoglycan. The authors suggest that IgG facilitates uptake of complex peptidoglycan, where after small derivatives trigger NODs for cytokine release (41). Alternatively, the pentraxin serum amyloid P binds to peptidoglycan on the surface of S. aureus (in the absence of WTA in a dTagO strain) and subsequently to FcγR, which induces complement and IgG-independent phagocytosis (42). Notably, another pentraxin, C-reactive protein, was ineffective under identical conditions, and the presence of WTA seems to prevent this process. Pentraxins are known to bind and activate FcγRs in a structural similar fashion as IgGs do (43). In addition to Igs and pentraxins, mannose binding lectin (MBL) also recognizes peptidoglycan resulting in cytokine release from macrophages (44). WTA is also recognized by MBL and functions as an opsonins but only when anti-WTA IgG are not yet present like in infants (45). On the other hand, affinity purified anti-WGA IgG induces classical complement-mediated opsonophagocytosis of S. aureus (46). Using glycosyltransferase KO/mutant strains, beta-GlcNAc residues of WTA are shown to be required for the induction of the classical complement pathway-dependent opsonophagocytosis of S. aureus. MBL recognized both α- and β-GlcNAc residues of WTA. In normal human serum, the major antibodies to WTA are specific to β-GlcNAc-modified form (47).
Next to IgG, natural occurring IgM antibodies activate complement and thereby also have opsonophagocytic capacity. IgM against S. aureus is present in human serum as was demonstrated by a study using a multiplex Luminex assay 34. The level of S. aureus-specific IgM was relatively low, and IgM was directed mostly to microbial surface components recognizing adhesive matrix molecules (MSCRAMMs). When healthy donors and patients with infections were examined by ELISA, IgM antibodies to peptidoglycan were not detected (36). Dimeric IgA linked via the J chain and monomeric IgA found in serum are good opsonins for bacteria and promote neutrophil oxidative burst and phagocytosis as efficient as IgG (48).
Because IgG is of the highest importance for S. aureus opsonization, many studies focus on the identification of likely antigens for possible therapy. Some studies have used intravenous immunoglobulin (IVIgG) as a source to identify new IgG targets. IVIgG is a pool of IgG isolated from hundreds of healthy blood donors that is commonly used as replacement therapy in immunocompromised patients. The broad antibody spectrum present in such a pool provides protection against otherwise life-threatening infections and autoimmune diseases (49). Subtractive proteome analysis of S. aureus anchorless cell wall proteins was performed by screening for proteins reacting with IVIgG but not with IVIgG depleted of S. aureus-specific opsonizing antibodies. Three of the 40 candidate proteins, i.e., enolase, oxoacyl reductase, and hypothetical protein hp2160, were used as vaccine candidates in mice and provided protection. Affinity isolation of the IgGs from IVIgG resulted in opsonization, phagocytosis, and killing of S. aureus by human neutrophils (50). In addition, on studies using IVIgG, others have applied bioinformatics for antigen prediction selecting for lipoproteins and cell wall-anchored proteins from Streptococcus sanguinis. Eight protein candidates were randomly picked, pooled, and used to immunize rabbits. From the raised antibody pool, affinity chromatography demonstrated opsonic activity for the individual IgGs with human neutrophils and rabbit complement (51). Using DNA bar-coding and deep sequencing of IgG genes from S. aureus infected humans identified protective antibodies that promoted opsonophagocytosis (52).
Complement Activation and Recognition during S. aureus Infection
The complement system is part of the innate immune defense and is a complex regulated system of proteases leading to soluble and surface-bound factors that contribute to efficient phagocytosis (53). Complement activation proceeds via three pathways, all leading to the cleavage of C3 into surface-deposited C3b (that also gets fragmented to iC3b) and released soluble mediator C3a. The “classical” activation of complement is mediated by specific antibodies bound to the bacterial surface that activate C1. Bacterially exposed carbohydrates are recognized by host lectins, mainly MBL, initiating the “lectin pathway.” Spontaneous and/or surface antigen-specific activation of C3 is possible as well as an amplification of the already-deposited C3b through the “alternative pathway.” The surface-bound C3b convertases activate new C3 molecules leading to more deposition of C3b onto the bacterial surface. The binding of C5 to the surface-bound C3b convertases subsequently initiates the release of the soluble mediator C5a and the formation of the membrane attack complex composed of C5b, C6, C7, C8, and multiple C9 molecules. The pores formed by the membrane attack complex may directly induce lysis of susceptible of mainly Gram-negative bacteria. The soluble cleavage products C3a and C5a, also known as anaphylatoxins (54), are chemoattractants and activators for phagocytes that express the C3aR and C5aR. With respect to phagocytosis, the bacterial surface-bound C3b is a key opsonin and is recognized by CRs on neutrophils. Both the complement factors as their receptors play several roles in phagocytosis. As the classical pathway is dependent on IgG, which was discussed in the previous section, more emphasis will be placed on other complement opsonin strategies in this section. Important group of CRs on neutrophils are lectins, those for bacterial surface-bound C3b or iC3b that belong to the adhesion receptor family and those for the soluble anaphylatoxins C3a and C5a that are G protein-coupled receptors (GPCRs).
Receptors that recognize surface-deposited complement factors are CR1 (CD35), CR2 (CD21), CR3 (CD11b/CD18 or MAC-1), CR4 (CD11c/CD18, integrin αXβ2), and CR of the Ig superfamily, CRIg (55). They all recognize C3b and/or iC3b, through distinct binding sites. Neutrophils express CR1 and CR3, whereas, macrophages express CR3 and CR4. CRIg is found on monocyte-derived macrophages and liver Kupfer cells (56). CR1 is a membrane glycoprotein with specificity for the complement products C3b, C4b and, with lower affinity, iC3b. It shares structural similarities with several regulators of complement activation, and the extracellular domain of CR1 consists of an array of 30 or more homologous units (57, 58). CR3 and CR4 are heterodimeric glycoproteins with a shared β-chain (CD18). Both receptors show specificity for the iC3b fragment (59, 60). Finally, CRIg is shown to mediate efficient phagocytosis of complement-opsonized particles and participates in the initial stage of phagosome formation (56).
Three cell-associated proteins have been described that show affinity for complement factor C1q, which is deposited upon recognition of surface-bound IgM. cC1qR, which closely resembles calreticulin, binds the collagen-like tail of C1q. C1qRp is a phagocytosis-promoting receptor that has similar ligand specificity, and gC1qR recognizes the globular head regions of C1q. Receptors for C1q are thought to play a role in both triggering and regulation of complement activation, and neutrophils express all three types of receptors (55, 61).
Pertaining to complement and specific recognition of S. aureus, although all three complement pathways play a role, lectins have thoroughly been investigated. Direct binding of MBL to several micro-organisms, including S. aureus, was demonstrated by flow cytometry, and bound MBL promoted deposition of C4 (62). The contribution of the MBL–MASP pathway relative to other pathways of complement activation on S. aureus and the consequence for C3b deposition and phagocytosis was demonstrated using MBL-deficient sera (63). MBL–MASP added to deficient sera enhanced the generation of C3b and iC3b on the surface of staphylococci and subsequently increased phagocytosis by human neutrophils. It must be noted that others have observed that while yeast species are preferentially opsonized and subsequently phagocytosed via activation of the lectin pathway of complement, the uptake of bacterial strains, including S. aureus, was largely MBL independent (64). Using MBL-deficient sera, it was shown that there was less C3 deposition on zymosan and Candida, while C3 deposition was not different on S. aureus using MBL-sufficient and MBL-deficient serum. Same authors have shown that inhibition of the classical pathway of complement activation during opsonization with either MBL-sufficient or MBL-deficient sera induced a two to threefold reduction in the subsequent phagocytosis of S. aureus. An alternative role for MBL in S. aureus infection is suggested independent of opsonization or complement activation. Binding of MBL to LTA via its carbohydrate recognition domain and subsequent complexing with TLR2 to increase ligand delivery is described to enhance TLR2 responses, as was measured by cytokine release by murine macrophages. This TLR2-mediated responses were only effective when S. aureus was delivered into the phagosome (65).
In addition to MBL, other sugar pattern recognition molecules as ficolins and collectins play a part in the lectin complement pathway and aid in S. aureus recognition. They both contain lectin activities within the C-terminus. Ficolins consists of collagen-like long thin stretches and fibrinogen-like globular domains with lectin activity, usually specific for N-acetylglucosamine (GlcNAc) (66). L-ficolin specifically binds to LTA, and immobilized LTA from S. aureus binds L-ficolin complexes from sera. These complexes are described to initiate lectin pathway-dependent C4 turnover (67). Collectins are a family of proteins that contain both collagen-like regions and lectin domains and play a role in opsonization and activation of neutrophils. In addition to MBL, conglutinin and the surfactant proteins SP-A and SP-D, which participate in the innate immunity of the lung, are other members of this group. The collectins share structural similarities with complement component C1q. SP-A may act directly as an opsonin for micro-organisms by binding via their lectin domains and enhance phagocytosis by alveolar macrophages. In addition, SP-A directly binds to C1q and thereby facilitates uptake of C1q-coated targets (68). SP-A and SP-D enhance bacterial (Escherichia coli, S. pneumoniae, and S. aureus) uptake by human neutrophils through a mechanism that involves both bacterial aggregation and direct actions on neutrophils. Multimerization of SP-D is an important parameter in its ability to increase uptake and differs thereby from the classical opsonins IgG and complement (69). Sp-A enhances the uptake of IgG-coated polystyrene beads by inflammatory neutrophils via direct interaction with both the opsonized beads and the neutrophils. In this case, the rat neutrophils were obtained from a lavage of LPS challenged lungs, and here SP-A did not have a generalized activation effect on neutrophils (70).
Finally, an important group of CRs on neutrophils are those for the soluble anaphylatoxins C3a and C5a. The C3a receptor (C3aR) and C5aR belong to the family of GPCRs. Neutrophils express a large number of GPCRs that participate in the host defense. They can sense bacterial peptides and toxins, e.g., formyl-peptide receptors (FPR1 and FPR2), lipid mediators, e.g., leukotriene B4 receptor and platelet activating factor receptor, and chemokine receptors (71, 72). Although known for their role in migration of neutrophils, most of these ligands, including C3a and C5a, also trigger direct cell activation, like ROS production, or “prime” cells for subsequent activation by other agonists. A crucial role for C5a–C5aR interaction was demonstrated for E. coli-induced phagocytosis in lepirudin anticoagulated whole blood. Generation of the anaphylatoxin C5a was essential and preceded the up-regulation of CR3, which was required for the subsequent oxidative burst and phagocytosis (73). Also for S. aureus in lepirudin treated whole blood, complement was responsible for phagocytosis as well as leukocyte activation (74).
Neutrophil Priming
Neutrophil priming is a process that causes a dramatic increase in the response of the cells and allows for faster and more efficient response, including phagocytosis of invading pathogens (12, 75). Activating agents have direct effects on cell surface receptor expression, but also intracellular in superoxide anion generation, degranulation, and mediator release (70). In the primed state, there is no increase in oxidase activity, yet subsequent stimulation provokes a response that is larger than in non-primed, activated cells. Neutrophil priming for enhanced oxidative burst, phagocytosis, and killing is initiated by several agents, such as soluble complement activators C3a and C5a (74), interferon-γ (IFNγ) (76, 77), interleukin-8 (il-8) (78), tumor necrosis factor-α (TNF-α) (79), and granulocyte/macrophage colony-stimulating factor (G-CSF & GM-CSF) (80). Stimulating receptors for G-CSF, GM-CSF, and IFNγ also delay apoptosis thereby affecting survival of neutrophils (81).
As mentioned, sensing of C5a is important in priming neutrophils by inducing up-regulation of CR3, which is crucial for subsequent phagocytosis in whole blood. Regarding TNF-α, neutrophils express several members of the TNF receptor family with diverse biological functions, including TNFR1 and TNFR2. TNF-α is an important pro-inflammatory cytokine that both directly activates neutrophils and primes the cells for subsequent stimulation including phagocytosis (82). Interleukin-1 plays also a major role in the inflammatory response and S. aureus infections and triggers a modest neutrophil activation (83, 84). They do express the IL-1R1, a member of the IL-1/TLR super family with comparable intracellular domains. The predominant receptor on neutrophils is the decoy receptor IL-1RII lacking intracellular signaling domains. Different classes of chemotactic agents, including C5a and il-8, cause a rapid reduction in the IL-1 binding capacity by human neutrophils (85).
Neutrophils express several innate immune receptors that are involved in recognition of pathogens and danger signals and allow priming of the cells. Those are found on the cell surface as well as in intracellular endocytic compartments and lead to cell activation, priming, and transcriptional changes. Neutrophils express most of the known TLRs that recognize bacterial structures, most importantly TLR4, which recognizes lipopolysaccharide from Gram-negative bacteria, and TLR2, which recognizes lipoproteins like LTA from Gram-positive bacteria. Stimulation of neutrophils with several TLR ligands stimulates phagocytosis of latex beads (86) and enhance the oxidative burst (87). Neutrophils also express members of the C-type lectins, such as Dectin-1, the receptor for fungal β-glucans and the components of the NLRP3 inflammasome that include the cytoplasmic NOD-like receptors sensing bacterial proteoglycan degradation products and danger signals (88). TLRs are not phagocytic receptors per se, but they are also internalized in the process and therefore participate to the link between phagocytosis and inflammatory responses by triggering the production of cytokines.
Phagocytosis
Phagocytosis is defined as the engulfing of other cells, cell fragments, and micro-organisms. Thereby it leads to sequestration and elimination of e.g., dead cells after apoptosis, pathogens like bacteria and uptake or delivery of substances by therapeutic micro- or nanoparticles. Physical parameters, i.e., particle size, shape, deformability, as well as biological parameters determine the effectiveness of phagocytosis.
The actual phagocytosis process can be distinguished in several phases: (1) attachment of the opsonized particle upon recognition by specific receptors, (2) pseudopod extensions around attached particle whereby it is still exposed to the environment (“zippering”), and (3) completion of the engulfment resulting in the formation of a phagosome, which is an outside-in compartment inside the cell (Figure 2). The next steps involve mobilization and fusion of the phagosome with different granule types resulting in the liberation of granule content that is required for killing of the microorganism (89, 90). Concurrently, a strong oxidative burst is initiated in the phagosome by NADPH-dependent oxidases upon triggering of specific cell surface receptors, leading to the generation of highly toxic ROS. Together with the granular content, ROS play an important role in bacterial killing (91).
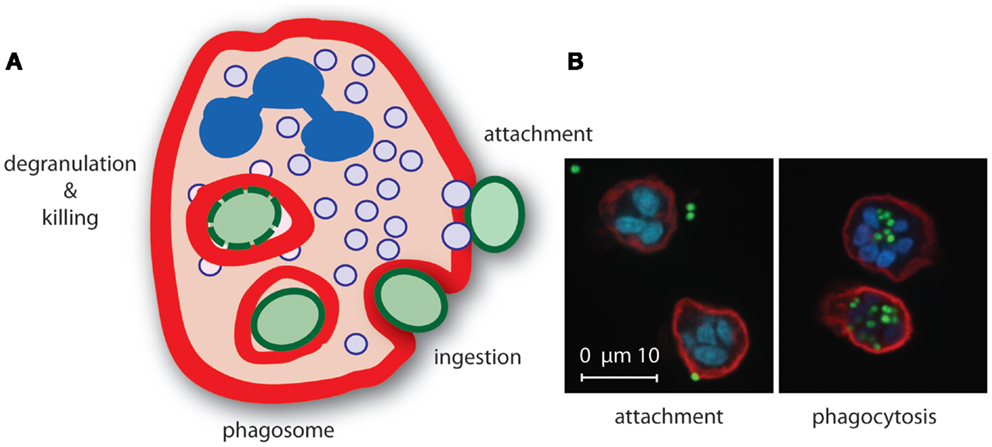
Figure 2. Neutrophil phagocytosis of S. aureus. (A) Upon recognition of opsonized S. aureus, neutrophils internalize the bacterium in a phagosome where secretory granule content is released and ROS are produced that mediate killing of the bacterium. (B) Confocal microscopy image of S. aureus expressing green fluorescence protein (green) attached (left) to and phagocytosed (right) by neutrophils [red, membrane stain wheat germ agglutinin (WGA)-Alexa647, blue, nucleic acid stain Syto82].
Neutrophils are capable of engulfing as many as 50 bacteria whereby a substantial amount of surface area is internalized while maintaining cell size and shape. For macrophages it is shown that the surface membrane internalized during engulfment is replaced from intracellular reservoirs. It is believed that during this replacement of the plasma membrane originating from endoplasmatic vesicles, focal exocytosis is effective leading to cytokine release. To form an autonomous internal compartment, the phagosome must undergo fission from the plasma membrane. The actual proteins involved in this process remain not fully understood, but involve dynamin and myosins. An important notion for any phagocytosis process is that not all phagosomes are created equal (89).
Besides the appropriate opsonins and their ligands, neutrophil versus monocytes/macrophages are of influence on this process, also physical parameters have their influence, like target particle size (>5 μm) and even their shape as shown for S. pneumoniae. Increasing the bacterial chain length, by natural changes in cellular morphology or via antibody-mediated agglutination, promoted complement-dependent killing (92). The density of both the ligand, e.g., IgG bound to the surface of the target particle, and the receptor, e.g., FcγR, also influence phagocytosis. Using RAW 264.7 murine macrophage cell line, IgG density had different effects on uptake depending on the particle size (93). Phagocytosis and IgG-mediated triggering are sometimes misleading if immune complexes are used to stimulate cells. Although these complexes could be considered as very small particles, the nature of these complexes differs from that of e.g., bacteria. Others call opsonized particles immune complexes. By loading sheep red blood cells (ShRBC) with increasing amounts of IgG, the increase in IgG density corresponds with increase in phagocytosis by mouse macrophages, both in number of positive cells and ShRBC per cell (94). For high-throughput assays, automatic imaging methods or flow cytometry are becoming more popular and rely on fluorescent beads and cell lines in microplate formats (95, 96).
Neutrophils are extremely efficient phagocytes and can internalize IgG-opsonized latex beads in <20 s (97). Localized granule secretion is important for phagocytosis and the generation of an anti-microbial phagosome. Phagocytosis of IgG-opsonized zymosan particles was very fast and most phagosomes were sealed within 1–3 min (98). Fusion of azurophilic granules and specific granules with the plasma membrane preceded phagosome sealing, and the granule membrane markers (CD63 and CD66b) were observed at the site of phagocytosis. Whereas, azurophilic granules are mainly secreted toward the forming/formed phagosome, specific granules can fuse with the plasma membrane at any location. A consumption of protons increases the pH, and early studies even indicated alkaline levels in early phagosomes before the pH gradually decreased (90, 99). It must be noted that during phagocytosis several signaling pathways are initiated that direct cellular activities but are beyond the scope of this review (81, 100–103).
Killing Process
The major task for neutrophils in the host defense is the elimination of pathogens by efficient uptake and subsequent killing. Therefore, neutrophils are equipped with an arsenal of anti-bacterial products stored in granules and an efficient massive generation of ROS by the membrane-associated NADPH-oxidase that can kill microorganism upon phagocytosis. In addition, neutrophils can kill bacteria extracellularly by release of chromatin covered with granule contents and selected cytoplasmic constituents providing NETs.
Neutrophils contain several granule subtypes that are subdivided into peroxidase-positive granules [containing myeloperoxidase (MPO)], which are also called primary or azurophil granules, and peroxidase-negative granules termed specific or secondary granules. However, granules are much more heterogeneous with subsets defined by a selection of marker proteins (104). MPO is one of the most abundant proteins in the neutrophil azurophil granules and catalyzes the oxidation of halide ions in the presence of hydrogen peroxide and generates hypochlorous acid that aids in bacterial killing (105). The granules contain a broad-spectrum of bactericidal and degradative proteins, but also growth depriving factors. Lactoferrin binds iron and neutrophil gelatinase-associated lipocalin (NGAL) interferes with siderophore-mediated iron acquisition thereby sequestering an essential microbial growth factor (106). Lysozyme hydrolyzes β (1–4) glycosidic linkages in the peptidoglycan layer, compromising bacterial integrity. Cationic anti-microbial peptides (CAPs) such as defensins and cathelicidin-type peptides (LL37) bind with high affinity into the bacterial membrane causing disruption (107–109). Azurophil granules also contain a family of structurally related serine proteases, cathepsin G, elastase, and proteinase 3, with anti-microbial and regulatory activity (110, 111).
The neutrophil NADPH-oxidase is a multicomponent enzyme complex that transfers electrons from NADPH onto molecular oxygen, thereby generating superoxide anion. Spontaneous dismutation together with the granule-derived MPO and iron-catalyzed reactions leads to the formation of several reactive ROS that include hydrogen peroxide, hydroxyl radicals, hypochlorous acid, and singlet oxygen. The transport of electrons is mediated by the redox center that contains Cytochrome b558, a flavo-hemeprotein composed of two subunits, gp91phox and p22phox. Assembly of the NADPH-oxidase involves translocation of three soluble components, p47phox, p67phox, p40phox, and a GTP-binding protein onto the plasma membrane to form the complete oxidase. This complex system is also carefully regulated in place and time, and it is concurrently assembled with phagosome formation (91). Specific KO mice are used to study the contribution of the NADPH-oxidase complex in bacterial pathogenesis and survival of the host. The oxidase response of these KO neutrophils is severely reduced upon stimulation with particles (IgG latex beads or S. aureus), soluble mediators like fMLF, or is adhesion dependent. Phagocytosis of bacteria by KO neutrophils is normal, but a clear defect is observed in killing of S. aureus in vitro as well as in vivo (112). Both reactive oxygen metabolites and neutrophil serine proteases contribute to the host defense depending on the pathogen studied, as was shown in the case of Aspergillus (111). The importance of an intact oxidative burst in neutrophils is demonstrated by chronic granulomatous disease (CGD; a primary immunodeficiency disorder of phagocytes) patients with a genetic defect in one of the NADPH-oxidase subunits. These patients suffer from recurrent bacterial infections, most commonly S. aureus, Aspergillus spp, and Salmonella spp (113). The role of ROS in killing of bacteria by neutrophils in vitro is evident when cells are treated with diphenyleneiodonium (DPI), which binds strongly to flavoproteins and is thereby a powerful inhibitor of several important enzymes including the NADPH-oxidase. Another defect is found for MPO deficiency; patients are fairly asymptomatic and defects are mainly found in the formation of NETs (9, 114). The oxidative burst pumps electrons into the phagosome that is compensated by a flux of K+ ions across the membrane in a pH dependent matter. This is an important trigger for the release of cationic granule proteins (115, 116).
Oxidative deamination of L-arginine by nitric oxide (NO) synthetase generates NO that together with superoxide anion forms reactive nitrogen intermediates with anti-microbial activity. The production of NO within phagocytes is an important component of the innate immune response to infection, and requires inducible NOS (iNOS). In mice macrophages are the source for NO, but in human leukocytes levels of iNOS are far more regulated (105, 117, 118). The role of iNOS in human neutrophils is limited and requires cytokine activation (119).
For effective bacterial killing, a critical concentration of neutrophils is required in suspension. Mathematical models have been applied to examine this ratio, which was supported by experimental data. By varying both neutrophil and bacterial concentrations (S. epidermidis), it was documented that the killing rate requires a critical neutrophil concentration of 3–4 × 105 per ml, is independent of ratio neutrophils to bacteria, and fitted an exponential function. The mathematical model fits the killing of Gram-positive and Gram-negative bacteria, opsonized by IgG and C3, by neutrophils in suspension (120). Further mathematical modeling and in vitro experimental killing of serum-opsonized S. aureus demonstrated that a critical neutrophil concentration dictates the outcome. Here, the individual maximum bearable bacterial concentration depended on neutrophil concentration, phagocytic activity, and patient barrier integrity and varied by orders of magnitude between patients (121).
Neutrophils can kill bacteria extracellular by release of NETs that trap bacteria covered with anti-microbials. This release and formation of NETs is the last step in an active neutrophil death termed NETosis and requires the formation of ROS. Several different agonists trigger the NET formation, including cytokines, microbial components, and bacteria itself. The extracellular traps are described to be formed in vivo and to contribute to clearing of infections (13, 122). In response to S. aureus, neutrophils employ a very rapid (5–60 min) alternative process of NET formation, independent of ROS, employing budding of vescicles. This response requires live bacteria and was mimicked by Panton–Valentine leukocidin as soluble mediator (123). Another member of this pore-forming leukotoxins, LukGH (also known as LukAB), also promotes the release of NETs (124). IgA-opsonized S. aureus also rapidly initiates the formation of NETs probably due to the robust production of ROS. In these circumstances killed bacteria or beads coated with IgA are also effective and engagement of the FcαR is a prerequisite (125).
Killing of S. aureus by neutrophils is not only effective in suspension but is also observed when bacteria are found in a biofilm. In contrast to mouse macrophages, neutrophils are capable of phagocytizing biofilm-associated S. aureus in vitro (126). Neutrophils migrate into the biofilm and enable clearance of bacteria by phagocytosis, whereby the extent of cleareance is dependent on the maturation state of the biofilm. Young developing biofilms are more sensitive toward the attack by neutrophils compared to more mature biofilms (127).
Opsonization with normal human serum did not change the phagocytosis of bacteria in biofilms, while opsonization of dispersed S. aureus was enhanced. IgG coating of the biofilm induced oxygen radical production that improved clearance of the biofilms due to bacterial killing (128).
Anti-Opsonic/Phagocytic Strategies
Phagocytosis by neutrophils is a very effective mechanism for S. aureus clearance. However, staphylococci are not just mere bystanders, and the bacteria can employ several anti-opsonic and anti-phagocytic means to survive. Escape strategies can be found for every step of phagocytosis, from binding and cleavage of antibodies to escape from phagosomes.
The best way to prevent phagocytosis is to prevent the initial proper opsonization, either directly by taking a sugar coating (capsule) that covers most of the more antigenic/immunogenic surface-exposed proteins, or indirectly by employing decoy molecules and active removal of opsonins. A rather thick polysaccharide capsule is best known for the many pneumococcal serotypes, but also for staphylococci a capsular polysaccharide efficiently hinders phagocytosis by neutrophils. Up to 50% of clinical S. aureus isolates have a capsule or microcapsule divided in up to 11 serotypes, whereby most of them react with antibodies directed to capsule type 5 or 8. These bacteria resist opsonophagocytosis and hence killing when bacteria were grown under conditions for optimal capsule production. These capsule polysaccharides and their components are also used as candidates for vaccine development against staphylococci. Specific antibodies directed against the capsule provide efficient opsonization and thereby uptake by neutrophils (129, 130). Recently an indirect covering of S. aureus with a shield of fibrinogen is shown by the staphylococcal extracellular fibrinogen-binding protein (Efb) that links the surface C3b deposition with fibrinogen. Thereby Efb prevents binding of phagocytic receptors to the opsonins C3b and IgG and subsequent phagocytosis (131).
Another anti-opsonic strategy is the secretion of IgG-binding proteins. The best known IgG-binding protein is staphylococcal protein-A (SpA). This ubiquitously expressed protein is a prominent cell wall-anchored protein, but it is also found in the supernate. Typically, its C-terminal part is linked to the cell wall, while the N-terminus contains five highly homologous extracellular Ig-binding domains in tandem. Each domain binds Igs through the Fc and restricted Fab domain of the human heavy chain VH3 family. The residues involved in binding to Fab are distinct from the residues that mediate binding to Fc part as the Fab fragments through distinct residues (132). Fc-binding to SpA is thought to protect staphylococci from opsonophagocytic killing (133–135). However, the precise mechanism is still not clear. Binding of Ig is essential though for S. aureus escape from host immune surveillance in mice as a protein-A-deficient strain induces a less severe arthritis and septic death, indicating that protein-A is a virulence factor (136). S. aureus also contains another IgG-binding protein, S. aureus binder of IgG (Sbi) that exhibits two Ig-binding domains similar to those of protein-A with comparable specificity (137, 138). Sbi is a multifunctional bacterial protein, which also acts a complement inhibitor and interferes with innate immune recognition (139). In a whole blood assay, Sbi prevents neutrophil-mediated opsonophagocytosis thereby promoting bacterial survival. The Sbi-KO bacteria show increased survival in whole blood and better uptake by neutrophils (140). However, Sbi does not contribute to S. aureus virulence in contrast to the Fcγ and VH3-type Fab binding activities of SpA (141). The impact of “reverse” IgG-binding on the interaction with host defense and invasive infection is shown for streptococcal protein M1 and H. Antibodies bound via Fab facilitate opsonization and killing by neutrophils whereas Fc-binding to protein M and H protected against phagocytosis (142).
Another strategy to escape killing, is through cleavage of IgG and C3, stretegies predominantly described for streptococci. Staphylokinase (SAK) is a secreted protein that can form a complex with human plasminogen resulting in the formation of active plasmin, a broad-spectrum proteolytic enzyme. This facilitates bacterial penetration into the surrounding tissues and is shown to cleave bacterial surface-bound IgG and C3b molecules. Cleavage of these important opsonins results in diminished uptake of the bacteria by neutrophils (143). Both staphylococcal proteins Sbi and Efb bind simultaneously C3/C3b and plasminogen on the surface of the bacterium and thereby enable the recruited plasmin or SAK to cleave bound C3 and C3b as well as soluble C3a (144).
In addition to strategies targeting IgG for anti-phagocytic purposes, staphylococci secrete many complement inhibitors to halt effective phagocytosis. These include, amongst others, staphylococcal complement inhibitor SCIN, extracellular complement-binding protein Ecb, and staphylococcal superantigen-like protein SSL7. They generally affect conversion of complement through binding major complement convertase componants and halt the complement cascade at several stages. As staphylococci have a large arsenal of complement inhibitors, please refer to excellent overviews (3, 145–147).
In addition to anti-opsonic strategies, bacteria, including S. aureus, have developed strategies to evade killing after internalization. Staphylococci can survive within the phagosome and have developed several ways to escape neutrophil killing. Escape is accomplished by employing toxins, like phenol-soluble modulins and leukocidin AB, that lyse the neutrophil from within after phagocytosis (148, 149). Toxin production in this bacterium is mainly regulated by activation of the agr operon (150), and the survival of the bacterium inside the neutrophil could even contribute to spread of the bacteria in the host (151). S. aureus also evades the ROS mediated killing mechanism using scavengers like catalase, superoxide dismutase (152, 153), and the golden carotenoid pigment (154). With regard to NETs, some bacteria can escape entrapment through secretion of endonucleases that can liberate them from the traps. An alternative NET escape mechanism for S. aureus is by converting NETs to deoxyadenosine, which induces macrophage cytotoxicity. Two secreted proteins, nuclease and adenosine synthethase are required for this host cell death (155).
Concluding Remarks
For invading staphylococci, phagocytosis and killing by human neutrophils is the biggest threat. Neutrophils are the only cells that can effectively kill staphylococci by engulfment and subsequent bombardment with proteases, amidases, anti-microbial peptides, and proteins in concert with ROS that are generated during the metabolic burst. Both complement and antibodies are crucial for effective uptake and neutrophil activation. S. aureus is not an innocent bystander in this process. It actively secretes several proteins to impair every single step in this process from receptor modulation, to complement inhibition to neutrophil lysis to protease, anti-microbial peptide inhibition and resistance to ROS. For the design of future novel anti-microbial strategies: therapeutic antibodies, vaccines, and novel antibiotics, all this should be taken into account. Still the best way to treat diseases is to help to enhance the natural defense mechanisms that are already in place.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Jovanka Bestebroer is supported by a ZonMW VENI grant (916.126.018).
References
1. Lowy FD. Staphylococcus aureus infections. N Engl J Med (1998) 339:520–32. doi:10.1056/NEJM199808203390806
2. Rigby KM, DeLeo FR. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin Immunopathol (2012) 34:237–59. doi:10.1007/s00281-011-0295-3
3. Rooijakkers SH, van Strijp JA. Bacterial complement evasion. Mol Immunol (2007) 44:23–32. doi:10.1016/j.molimm.2006.06.011
4. Spaan AN, Surewaard BG, Nijland R, van Strijp JA. Neutrophils versus Staphylococcus aureus : a biological tug of war. Annu Rev Microbiol (2013) 67:629–50. doi:10.1146/annurev-micro-092412-155746
5. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood (2010) 116:625–7. doi:10.1182/blood-2010-01-259028
6. Borregaard N. Neutrophils, from marrow to microbes. Immunity (2010) 33:657–70. doi:10.1016/j.immuni.2010.11.011
7. Tsuda Y, Takahashi H, Kobayashi M, Hanafusa T, Herndon DN, Suzuki F. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity (2004) 21:215–26. doi:10.1016/j.immuni.2004.07.006
8. Beyrau M, Bodkin JV, Nourshargh S. Neutrophil heterogeneity in health and disease: a revitalized avenue in inflammation and immunity. Open Biol (2012) 2:120134. doi:10.1098/rsob.120134
9. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol (2014) 9:181–218. doi:10.1146/annurev-pathol-020712-164023
10. Mocsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med (2013) 210:1283–99. doi:10.1084/jem.20122220
11. McFarlin BK, Williams RR, Venable AS, Dwyer KC, Haviland DL. Image-based cytometry reveals three distinct subsets of activated granulocytes based on phagocytosis and oxidative burst. Cytometry A (2013) 83:745–51. doi:10.1002/cyto.a.22330
12. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi:10.1038/nri3399
13. Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol (2012) 198:773–83. doi:10.1083/jcb.201203170
14. Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The balancing act of neutrophils. Cell Host Microbe (2014) 15:526–36. doi:10.1016/j.chom.2014.04.011
15. Brown S, Santa MJ Jr, Walker S. Wall teichoic acids of gram-positive bacteria. Annu Rev Microbiol (2013) 67:313–36. doi:10.1146/annurev-micro-092412-155620
16. Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol (2010) 2:a000414. doi:10.1101/cshperspect.a000414
18. Lu T, Porter AR, Kennedy AD, Kobayashi SD, DeLeo FR. Phagocytosis and killing of Staphylococcus aureus by Human Neutrophils. J Innate Immun (2014) 6:639–49. doi:10.1159/000360478
19. Fjaertoft G, Hakansson LD, Pauksens K, Sisask G, Venge P. Neutrophil CD64 (FcgammaRI) expression is a specific marker of bacterial infection: a study on the kinetics and the impact of major surgery. Scand J Infect Dis (2007) 39:525–35. doi:10.1080/00365540601113693
20. Venet F, Lepape A, Monneret G. Clinical review: flow cytometry perspectives in theICU from diagnosis of infection to monitoring of injury-induced immune dysfunctions. Crit Care (2011) 15:231. doi:10.1186/cc10333
21. Hogarth PM, Pietersz GA. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov (2012) 11:311–31. doi:10.1038/nrd2909
22. Woof JM, Burton DR. Human antibody-Fc receptor interactions illuminated by crystal structures. Nat Rev Immunol (2004) 4:89–99. doi:10.1038/nri1266
23. Radaev S, Sun P. Recognition of immunoglobulins by Fcgamma receptors. Mol Immunol (2002) 38:1073–83. doi:10.1016/S0161-5890(02)00036-6
24. Foster TJ, Geoghegan JA, Ganesh VK, Hook M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol (2014) 12:49–62. doi:10.1038/nrmicro3161
25. den Reijer PM, Lemmens-den Toom N, Kant S, Snijders SV, Boelens H, Tavakol M, et al. Characterization of the humoral immune response during Staphylococcus aureus bacteremia and global gene expression by Staphylococcus aureus in human blood. PLoS One (2013) 8:e53391. doi:10.1371/journal.pone.0053391
26. Dryla A, Prustomersky S, Gelbmann D, Hanner M, Bettinger E, Kocsis B, et al. Comparison of antibody repertoires against Staphylococcus aureus in healthy individuals and in acutely infected patients. Clin Diagn Lab Immunol (2005) 12:387–98. doi:10.1128/CDLI.12.387-398.2005
27. Colque-Navarro P, Jacobsson G, Andersson R, Flock JI, Mollby R. Levels of antibody against 11 Staphylococcus aureus antigens in a healthy population. Clin Vaccine Immunol (2010) 17:1117–23. doi:10.1128/CVI.00506-09
28. Kolata J, Bode LG, Holtfreter S, Steil L, Kusch H, Holtfreter B, et al. Distinctive patterns in the human antibody response to Staphylococcus aureus bacteremia in carriers and non-carriers. Proteomics (2011) 11:3914–27. doi:10.1002/pmic.201000760
29. Kelly-Quintos C, Cavacini LA, Posner MR, Goldmann D, Pier GB. Characterization of the opsonic and protective activity against Staphylococcus aureus of fully human monoclonal antibodies specific for the bacterial surface polysaccharide poly-N-acetylglucosamine. Infect Immun (2006) 74:2742–50. doi:10.1128/IAI.74.5.2742-2750.2006
30. Cywes-Bentley C, Skurnik D, Zaidi T, Roux D, Deoliveira RB, Garrett WS, et al. Antibody to a conserved antigenic target is protective against diverse prokaryotic and eukaryotic pathogens. Proc Natl Acad Sci U S A (2013) 110:E2209–18. doi:10.1073/pnas.1303573110
31. Hazenbos WL, Kajihara KK, Vandlen R, Morisaki JH, Lehar SM, Kwakkenbos MJ, et al. Novel staphylococcal glycosyltransferases SdgA and SdgB mediate immunogenicity and protection of virulence-associated cell wall proteins. PLoS Pathog (2013) 9:e1003653. doi:10.1371/journal.ppat.1003653
32. Zorman JK, Esser M, Raedler M, Kreiswirth BN, Ala’Aldeen DA, Kartsonis N, et al. Naturally occurring IgG antibody levels to the Staphylococcus aureus protein IsdB in humans. Hum Vaccin Immunother (2013) 9:1857–64. doi:10.4161/hv.25253
33. Fowler VG, Allen KB, Moreira ED, Moustafa M, Isgro F, Boucher HW, et al. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: a randomized trial. JAMA (2013) 309:1368–78. doi:10.1001/jama.2013.3010
34. Verkaik NJ, de Vogel CP, Boelens HA, Grumann D, Hoogenboezem T, Vink C, et al. Anti-staphylococcal humoral immune response in persistent nasal carriers and noncarriers of Staphylococcus aureus. J Infect Dis (2009) 199:625–32. doi:10.1086/596743
35. Hawkins J, Kodali S, Matsuka YV, McNeil LK, Mininni T, Scully IL, et al. A recombinant clumping factor A-containing vaccine induces functional antibodies to Staphylococcus aureus that are not observed after natural exposure. Clin Vaccine Immunol (2012) 19:1641–50. doi:10.1128/CVI.00354-12
36. Verbrugh HA, Peters R, Rozenberg-Arska M, Peterson PK, Verhoef J. Antibodies to cell wall peptidoglycan of Staphylococcus aureus in patients with serious staphylococcal infections. J Infect Dis (1981) 144:1–9. doi:10.1093/infdis/144.1.1
37. Peterson PK, Wilkinson BJ, Kim Y, Schmeling D, Douglas SD, Quie PG, et al. The key role of peptidoglycan in the opsonization of Staphylococcus aureus. J Clin Invest (1978) 61:597–609. doi:10.1172/JCI108971
38. Martinez-Martinez L, Timmerman CP, Fleer A, Verhoef J. Chemiluminescence of human polymorphonuclear leucocytes after stimulation with whole cells and cell-wall components of Staphylococcus epidermidis. J Med Microbiol (1993) 39:196–203. doi:10.1099/00222615-39-3-196
39. Timmerman CP, Mattsson E, Martinez-Martinez L, De Graaf L, Van Strijp JA, Verbrugh HA, et al. Induction of release of tumor necrosis factor from human monocytes by Staphylococci and staphylococcal peptidoglycans. Infect Immun (1993) 61:4167–72.
40. Royet J, Dziarski R. Peptidoglycan recognition proteins: pleiotropic sensors and effectors of antimicrobial defences. Nat Rev Microbiol (2007) 5:264–77. doi:10.1038/nrmicro1620
41. Sun D, Raisley B, Langer M, Iyer JK, Vedham V, Ballard JL, et al. Anti-peptidoglycan antibodies and Fcgamma receptors are the key mediators of inflammation in Gram-positive sepsis. J Immunol (2012) 189:2423–31. doi:10.4049/jimmunol.1201302
42. An JH, Kurokawa K, Jung DJ, Kim MJ, Kim CH, Fujimoto Y, et al. Human SAP is a novel peptidoglycan recognition protein that induces complement-independent phagocytosis of Staphylococcus aureus. J Immunol (2013) 191:3319–27. doi:10.4049/jimmunol.1300940
43. Lu J, Marjon KD, Mold C, Du Clos TW, Sun PD. Pentraxins and Fc receptors. Immunol Rev (2012) 250:230–8. doi:10.1111/j.1600-065X.2012.01162.x
44. Nadesalingam J, Dodds AW, Reid KB, Palaniyar N. Mannose-binding lectin recognizes peptidoglycan via the N-acetyl glucosamine moiety, and inhibits ligand-induced proinflammatory effect and promotes chemokine production by macrophages. J Immunol (2005) 175:1785–94. doi:10.4049/jimmunol.175.3.1785
45. Park KH, Kurokawa K, Zheng L, Jung DJ, Tateishi K, Jin JO, et al. Human serum mannose-binding lectin senses wall teichoic acid glycopolymer of Staphylococcus aureus, which is restricted in infancy. J Biol Chem (2010) 285:27167–75. doi:10.1074/jbc.M110.141309
46. Jung DJ, An JH, Kurokawa K, Jung YC, Kim MJ, Aoyagi Y, et al. Specific serum Ig recognizing staphylococcal wall teichoic acid induces complement-mediated opsonophagocytosis against Staphylococcus aureus. J Immunol (2012) 189:4951–9. doi:10.4049/jimmunol.1201294
47. Kurokawa K, Jung DJ, An JH, Fuchs K, Jeon YJ, Kim NH, et al. Glycoepitopes of staphylococcal wall teichoic acid govern complement-mediated opsonophagocytosis via human serum antibody and mannose-binding lectin. J Biol Chem (2013) 288:30956–68. doi:10.1074/jbc.M113.509893
48. Vidarsson G, van der Pol WL, van den Elsen JM, Vilé H, Jansen M, Duijs J, et al. Activity of human IgG and IgA subclasses in immune defense against Neisseria meningitidis serogroup B. J Immunol (2001) 166:6250–6. doi:10.4049/jimmunol.166.10.6250
49. Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol (2013) 13:176–89. doi:10.1038/nri3401
50. Glowalla E, Tosetti B, Kronke M, Krut O. Proteomics-based identification of anchorless cell wall proteins as vaccine candidates against Staphylococcus aureus. Infect Immun (2009) 77:2719–29. doi:10.1128/IAI.00617-08
51. Ge X, Kitten T, Munro CL, Conrad DH, Xu P. Pooled protein immunization for identification of cell surface antigens in Streptococcus sanguinis. PLoS One (2010) 5:e11666. doi:10.1371/journal.pone.0011666
52. Lu DR, Tan YC, Kongpachith S, Cai X, Stein EA, Lindstrom TM, et al. Identifying functional anti-Staphylococcus aureus antibodies by sequencing antibody repertoires of patient plasmablasts. Clin Immunol (2014) 152:77–89. doi:10.1016/j.clim.2014.02.010
53. Walport MJ. Complement. First of two parts. N Engl J Med (2001) 344:1058–66. doi:10.1056/NEJM200104053441406
54. Haas PJ, van SJ. Anaphylatoxins: their role in bacterial infection and inflammation. Immunol Res (2007) 37:161–75. doi:10.1007/BF02697367
55. Holers VM. Complement and its receptors: new insights into human disease. Annu Rev Immunol (2014) 32:433–59. doi:10.1146/annurev-immunol-032713-120154
56. Helmy KY, Katschke KJ Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell (2006) 124:915–27. doi:10.1016/j.cell.2005.12.039
57. Ahearn JM, Fearon DT. Structure and function of the complement receptors. CR1 (CD35) and CR2 (CD21). Adv Immunol (1989) 46:183–219. doi:10.1016/S0065-2776(08)60654-9
58. Krych-Goldberg M, Atkinson JP. Structure-function relationships of complement receptor type 1. Immunol Rev (2001) 180:112–22. doi:10.1034/j.1600-065X.2001.1800110.x
59. Wright SD, Reddy PA, Jong MT, Erickson BW. C3bi receptor (complement receptor type 3) recognizes a region of complement protein C3 containing the sequence Arg-Gly-Asp. Proc Natl Acad Sci U S A (1987) 84:1965–8. doi:10.1073/pnas.84.7.1965
60. Ehlers MR. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect (2000) 2:289–94. doi:10.1016/S1286-4579(00)00299-9
61. Eggleton P, Tenner AJ, Reid KB. C1q receptors. Clin Exp Immunol (2000) 120:406–12. doi:10.1046/j.1365-2249.2000.01218.x
62. Neth O, Jack DL, Dodds AW, Holzel H, Klein NJ, Turner MW. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect Immun (2000) 68:688–93. doi:10.1128/IAI.68.2.688-693.2000
63. Neth O, Jack DL, Johnson M, Klein NJ, Turner MW. Enhancement of complement activation and opsonophagocytosis by complexes of mannose-binding lectin with mannose-binding lectin-associated serine protease after binding to Staphylococcus aureus. J Immunol (2002) 169:4430–6. doi:10.4049/jimmunol.169.8.4430
64. Brouwer N, Dolman KM, van Houdt M, Sta M, Roos D, Kuijpers TW. Mannose-binding lectin (MBL) facilitates opsonophagocytosis of yeasts but not of bacteria despite MBL binding. J Immunol (2008) 180:4124–32. doi:10.4049/jimmunol.180.6.4124
65. Ip WK, Takahashi K, Moore KJ, Stuart LM, Ezekowitz RA. Mannose-binding lectin enhances toll-like receptors 2 and 6 signaling from the phagosome. J Exp Med (2008) 205:169–81. doi:10.1084/jem.20071164
66. Matsushita M. Ficolins in complement activation. Mol Immunol (2013) 55:22–6. doi:10.1016/j.molimm.2012.08.017
67. Lynch NJ, Roscher S, Hartung T, Morath S, Matsushita M, Maennel DN, et al. L-ficolin specifically binds to lipoteichoic acid, a cell wall constituent of Gram-positive bacteria, and activates the lectin pathway of complement. J Immunol (2004) 172:1198–202. doi:10.4049/jimmunol.172.2.1198
68. Watford WT, Smithers MB, Frank MM, Wright JR. Surfactant protein A enhances the phagocytosis of C1q-coated particles by alveolar macrophages. Am J Physiol Lung Cell Mol Physiol (2002) 283:L1011–22. doi:10.1152/ajplung.00366.2001
69. Hartshorn KL, Crouch E, White MR, Colamussi ML, Kakkanatt A, Tauber B, et al. Pulmonary surfactant proteins A and D enhance neutrophil uptake of bacteria. Am J Physiol (1998) 274:L958–69.
70. Wofford JA, Wright JR. Surfactant protein A regulates IgG-mediated phagocytosis in inflammatory neutrophils. Am J Physiol Lung Cell Mol Physiol (2007) 293:L1437–43. doi:10.1152/ajplung.00239.2007
71. Lattin J, Zidar DA, Schroder K, Kellie S, Hume DA, Sweet MJ. G-protein-coupled receptor expression, function, and signaling in macrophages. J Leukoc Biol (2007) 82:16–32. doi:10.1189/jlb.0107051
72. Sun L, Ye RD. Role of G protein-coupled receptors in inflammation. Acta Pharmacol Sin (2012) 33:342–50. doi:10.1038/aps.2011.200
73. Mollnes TE, Brekke OL, Fung M, Fure H, Christiansen D, Bergseth G, et al. Essential role of the C5a receptor in E. coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood (2002) 100:1869–77.
74. Skjeflo EW, Christiansen D, Espevik T, Nielsen EW, Mollnes TE. Combined inhibition of complement and CD14 efficiently attenuated the inflammatory response induced by Staphylococcus aureus in a human whole blood model. J Immunol (2014) 192:2857–64. doi:10.4049/jimmunol.1300755
75. El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol (2008) 30:279–89. doi:10.1007/s00281-008-0118-3
76. Edwards SW, Say JE, Hughes V. Gamma interferon enhances the killing of Staphylococcus aureus by human neutrophils. J Gen Microbiol (1988) 134:37–42.
77. Ellis TN, Beaman BL. Interferon-gamma activation of polymorphonuclear neutrophil function. Immunology (2004) 112:2–12. doi:10.1111/j.1365-2567.2004.01849.x
78. Mitchell GB, Albright BN, Caswell JL. Effect of interleukin-8 and granulocyte colony-stimulating factor on priming and activation of bovine neutrophils. Infect Immun (2003) 71:1643–9. doi:10.1128/IAI.71.4.1643-1649.2003
79. Rainard P, Riollet C, Poutrel B, Paape MJ. Phagocytosis and killing of Staphylococcus aureus by bovine neutrophils after priming by tumor necrosis factor-alpha and the des-arginine derivative of C5a. Am J Vet Res (2000) 61:951–9. doi:10.2460/ajvr.2000.61.951
80. Khwaja A, Carver JE, Linch DC. Interactions of granulocyte-macrophage colony-stimulating factor (CSF), granulocyte CSF, and tumor necrosis factor alpha in the priming of the neutrophil respiratory burst. Blood (1992) 79:745–53.
81. Futosi K, Fodor S, Mocsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol (2013) 17:638–50. doi:10.1016/j.intimp.2013.11.010
82. Klebanoff SJ, Vadas MA, Harlan JM, Sparks LH, Gamble JR, Agosti JM, et al. Stimulation of neutrophils by tumor necrosis factor. J Immunol (1986) 136:4220–5.
83. Moxey-Mims MM, Simms HH, Frank MM, Lin EY, Gaither TA. The effects of IL-1, IL-2, and tumor necrosis factor on polymorphonuclear leukocyte Fc gamma receptor-mediated phagocytosis. IL-2 down-regulates the effect of tumor necrosis factor. J Immunol (1991) 147:1823–30.
84. Miller LS, Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol (2011) 11:505–18. doi:10.1038/nri3010
85. Colotta F, Orlando S, Fadlon EJ, Sozzani S, Matteucci C, Mantovani A. Chemoattractants induce rapid release of the interleukin 1 type II decoy receptor in human polymorphonuclear cells. J Exp Med (1995) 181:2181–6. doi:10.1084/jem.181.6.2181
86. Hayashi F, Means TK, Luster AD. Toll-like receptors stimulate human neutrophil function. Blood (2003) 102:2660–9. doi:10.1182/blood-2003-04-1078
87. Jann NJ, Schmaler M, Ferracin F, Landmann R. TLR2 enhances NADPH oxidase activity and killing of Staphylococcus aureus by PMN. Immunol Lett (2011) 135:17–23. doi:10.1016/j.imlet.2010.09.007
88. Ekman AK, Cardell LO. The expression and function of nod-like receptors in neutrophils. Immunology (2010) 130:55–63. doi:10.1111/j.1365-2567.2009.03212.x
89. Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol (2012) 7:61–98. doi:10.1146/annurev-pathol-011811-132445
90. Nordenfelt P, Tapper H. Phagosome dynamics during phagocytosis by neutrophils. J Leukoc Biol (2011) 90:271–84. doi:10.1189/jlb.0810457
91. Roos D, van BR, Meischl C. Oxidative killing of microbes by neutrophils. Microbes Infect (2003) 5:1307–15. doi:10.1016/j.micinf.2003.09.009
92. Dalia AB, Weiser JN. Minimization of bacterial size allows for complement evasion and is overcome by the agglutinating effect of antibody. Cell Host Microbe (2011) 10:486–96. doi:10.1016/j.chom.2011.09.009
93. Pacheco P, White D, Sulchek T. Effects of microparticle size and Fc density on macrophage phagocytosis. PLoS One (2013) 8:e60989. doi:10.1371/journal.pone.0060989
94. Gallo P, Goncalves R, Mosser DM. The influence of IgG density and macrophage Fc (gamma) receptor cross-linking on phagocytosis and IL-10 production. Immunol Lett (2010) 133:70–7. doi:10.1016/j.imlet.2010.07.004
95. Ackerman ME, Moldt B, Wyatt RT, Dugast AS, McAndrew E, Tsoukas S, et al. A robust, high-throughput assay to determine the phagocytic activity of clinical antibody samples. J Immunol Methods (2011) 366:8–19. doi:10.1016/j.jim.2010.12.016
96. Yeo JC, Wall AA, Stow JL, Hamilton NA. High-throughput quantification of early stages of phagocytosis. Biotechniques (2013) 55:115–24. doi:10.2144/000114075
97. Segal AW, Dorling J, Coade S. Kinetics of fusion of the cytoplasmic granules with phagocytic vacuoles in human polymorphonuclear leukocytes. Biochemical and morphological studies. J Cell Biol (1980) 85:42–59. doi:10.1083/jcb.85.1.42
98. Tapper H, Grinstein S. Fc receptor-triggered insertion of secretory granules into the plasma membrane of human neutrophils: selective retrieval during phagocytosis. J Immunol (1997) 159:409–18.
99. Segal AW, Geisow M, Garcia R, Harper A, Miller R. The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature (1981) 290:406–9. doi:10.1038/290406a0
100. Araki N. Role of microtubules and myosins in Fc gamma receptor-mediated phagocytosis. Front Biosci (2006) 11:1479–90. doi:10.2741/1897
101. Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr Opin Immunol (2002) 14:136–45. doi:10.1016/S0952-7915(01)00309-0
102. Zhang Y, Hoppe AD, Swanson JA. Coordination of Fc receptor signaling regulates cellular commitment to phagocytosis. Proc Natl Acad Sci U S A (2010) 107:19332–7. doi:10.1073/pnas.1008248107
103. Hawkins PT, Stephens LR, Suire S, Wilson M. PI3K signaling in neutrophils. Curr Top Microbiol Immunol (2010) 346:183–202. doi:10.1007/82_2010_40
104. Borregaard N, Sorensen OE, Theilgaard-Monch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol (2007) 28:340–5. doi:10.1016/j.it.2007.06.002
105. Klebanoff SJ, Kettle AJ, Rosen H, Winterbourn CC, Nauseef WM. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukoc Biol (2013) 93:185–98. doi:10.1189/jlb.0712349
106. Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell (2002) 10:1033–43. doi:10.1016/S1097-2765(02)00708-6
107. Hancock RE, Diamond G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol (2000) 8:402–10. doi:10.1016/S0966-842X(00)01823-0
108. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol (2003) 3:710–20. doi:10.1038/nri1180
109. Durr UH, Sudheendra US, Ramamoorthy A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta (2006) 1758:1408–25. doi:10.1016/j.bbamem.2006.03.030
110. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol (2006) 6:541–50. doi:10.1038/nri1841
111. Vethanayagam RR, Almyroudis NG, Grimm MJ, Lewandowski DC, Pham CT, Blackwell TS, et al. Role of NADPH oxidase versus neutrophil proteases in antimicrobial host defense. PLoS One (2011) 6:e28149. doi:10.1371/journal.pone.0028149
112. Ellson CD, Davidson K, Ferguson GJ, O’Connor R, Stephens LR, Hawkins PT. Neutrophils from p40phox-/- mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J Exp Med (2006) 203:1927–37. doi:10.1084/jem.20052069
113. Andrews T, Sullivan KE. Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev (2003) 16:597–621. doi:10.1128/CMR.16.4.597-621.2003
114. Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood (2011) 117:953–9. doi:10.1182/blood-2010-06-290171
115. Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature (2002) 416:291–7. doi:10.1038/416291a
116. Segal AW. How neutrophils kill microbes. Annu Rev Immunol (2005) 23:197–223. doi:10.1146/annurev.immunol.23.021704.115653
117. Burgner D, Rockett K, Kwiatkowski D. Nitric oxide and infectious diseases. Arch Dis Child (1999) 81:185–8. doi:10.1136/adc.81.2.185
118. Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol (2004) 2:820–32. doi:10.1038/nrmicro1004
119. Evans TJ, Buttery LD, Carpenter A, Springall DR, Polak JM, Cohen J. Cytokine-treated human neutrophils contain inducible nitric oxide synthase that produces nitration of ingested bacteria. Proc Natl Acad Sci U S A (1996) 93:9553–8. doi:10.1073/pnas.93.18.9553
120. Li Y, Karlin A, Loike JD, Silverstein SC. A critical concentration of neutrophils is required for effective bacterial killing in suspension. Proc Natl Acad Sci U S A (2002) 99:8289–94. doi:10.1073/pnas.122244799
121. Malka R, Wolach B, Gavrieli R, Shochat E, Rom-Kedar V. Evidence for bistable bacteria-neutrophil interaction and its clinical implications. J Clin Invest (2012) 122:3002–11. doi:10.1172/JCI59832
122. von Kockritz-Blickwede M, Nizet V. Innate immunity turned inside-out: antimicrobial defense by phagocyte extracellular traps. J Mol Med (Berl) (2009) 87:775–83. doi:10.1007/s00109-009-0481-0
123. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (2010) 185:7413–25. doi:10.4049/jimmunol.1000675
124. Malachowa N, Kobayashi SD, Freedman B, Dorward DW, DeLeo FR. Staphylococcus aureus leukotoxin GH promotes formation of neutrophil extracellular traps. J Immunol (2013) 191:6022–9. doi:10.4049/jimmunol.1301821
125. Aleyd E, van Hout MW, Ganzevles SH, Hoeben KA, Everts V, Bakema JE, et al. IgA enhances NETosis and release of neutrophil extracellular traps by polymorphonuclear cells via Fcalpha receptor I. J Immunol (2014) 192:2374–83. doi:10.4049/jimmunol.1300261
126. Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, et al. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol (2011) 186:6585–96. doi:10.4049/jimmunol.1002794
127. Günther F, Wabnitz GH, Stroh P, Prior B, Obst U, Samstag Y, et al. Host defence against Staphylococcus aureus biofilms infection: phagocytosis of biofilms by polymorphonuclear neutrophils (PMN). Mol Immunol (2009) 46:1805–13. doi:10.1016/j.molimm.2009.01.020
128. Stroh P, Günther F, Meyle E, Prior B, Wagner C, Hänsch GM. Host defence against Staphylococcus aureus biofilms by polymorphonuclear neutrophils: oxygen radical production but not phagocytosis depends on opsonisation with immunoglobulin G. Immunobiology (2011) 216:351–7. doi:10.1016/j.imbio.2010.07.009
129. Verbrugh HA, Peterson PK, Nguyen BY, Sisson SP, Kim Y. Opsonization of encapsulated Staphylococcus aureus: the role of specific antibody and complement. J Immunol (1982) 129:1681–7.
130. O’Riordan K, Lee JC. Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev (2004) 17:218–34. doi:10.1128/CMR.17.1.218-234.2004
131. Ko YP, Kuipers A, Freitag CM, Jongerius I, Medina E, van Rooijen WJ, et al. Phagocytosis escape by a Staphylococcus aureus protein that connects complement and coagulation proteins at the bacterial surface. PLoS Pathog (2013) 9:e1003816. doi:10.1371/journal.ppat.1003816
132. Graille M, Stura EA, Corper AL, Sutton BJ, Taussig MJ, Charbonnier JB, et al. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc Natl Acad Sci U S A (2000) 97:5399–404. doi:10.1073/pnas.97.10.5399
133. Dossett JH, Kronvall G, Williams RC Jr, Quie PG. Antiphagocytic effects of staphylococfcal protein A. J Immunol (1969) 103:1405–10.
134. Forsgren A, Quie PG. Effects of staphylococcal protein A on heat labile opsonins. J Immunol (1974) 112:1177–80.
135. Peterson PK, Verhoef J, Sabath LD, Quie PG. Effect of protein A on staphylococcal opsonization. Infect Immun (1977) 15:760–4.
136. Palmqvist N, Foster T, Tarkowski A, Josefsson E. Protein A is a virulence factor in Staphylococcus aureus arthritis and septic death. Microb Pathog (2002) 33:239–49. doi:10.1006/mpat.2002.0533
137. Zhang L, Jacobsson K, Vasi J, Lindberg M, Frykberg L. A second IgG-binding protein in Staphylococcus aureus. Microbiology (1998) 144(Pt 4):985–91. doi:10.1099/00221287-144-4-985
138. Atkins KL, Burman JD, Chamberlain ES, Cooper JE, Poutrel B, Bagby S, et al. S. aureus IgG-binding proteins SpA and Sbi: host specificity and mechanisms of immune complex formation. Mol Immunol (2008) 45:1600–11. doi:10.1016/j.molimm.2007.10.021
139. Haupt K, Reuter M, van den Elsen J, Burman J, Hälbich S, Richter J, et al. The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement Factor H and C3b. PLoS Pathog (2008) 4:e1000250. doi:10.1371/journal.ppat.1000250
140. Smith EJ, Visai L, Kerrigan SW, Speziale P, Foster TJ. The Sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus. Infect Immun (2011) 79:3801–9. doi:10.1128/IAI.05075-11
141. Falugi F, Kim HK, Missiakas DM, Schneewind O. Role of protein A in the evasion of host adaptive immune responses by Staphylococcus aureus. MBio (2013) 4:e575–513. doi:10.1128/mBio.00575-13
142. Nordenfelt P, Waldemarson S, Linder A, Mörgelin M, Karlsson C, Malmström J, et al. Antibody orientation at bacterial surfaces is related to invasive infection. J Exp Med (2012) 209:2367–81. doi:10.1084/jem.20120325
143. Rooijakkers SH, van Wamel WJ, Ruyken M, van Kessel KP, van Strijp JA. Anti-opsonic properties of staphylokinase. Microbes Infect (2005) 7:476–84. doi:10.1016/j.micinf.2004.12.014
144. Koch TK, Reuter M, Barthel D, Böhm S, van den Elsen J, Kraiczy P, et al. Staphylococcus aureus proteins Sbi and Efb recruit human plasmin to degrade complement C3 and C3b. PLoS One (2012) 7:e47638. doi:10.1371/journal.pone.0047638
145. Laarman A, Milder F, van SJ, Rooijakkers S. Complement inhibition by gram-positive pathogens: molecular mechanisms and therapeutic implications. J Mol Med (Berl) (2010) 88:115–20. doi:10.1007/s00109-009-0572-y
146. Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol (2008) 6:132–42. doi:10.1038/nrmicro1824
147. Geisbrecht BV. Staphylococcal complement inhibitors: biological functions, recognition of complement components, and potential therapeutic implications. Adv Exp Med Biol (2008) 632:221–36. doi:10.1007/978-0-387-78952-1_16
148. DuMont AL, Yoong P, Surewaard BG, Benson MA, Nijland R, van Strijp JA, et al. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun (2013) 81:1830–41. doi:10.1128/IAI.00095-13
149. Surewaard BG, de Haas CJ, Vervoort F, Rigby KM, DeLeo FR, Otto M, et al. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell Microbiol (2013) 15:1427–37. doi:10.1111/cmi.12130
150. Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM. agr-Dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J Innate Immun (2010) 2:546–59. doi:10.1159/000319855
151. Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol (2000) 164:3713–22. doi:10.4049/jimmunol.164.7.3713
152. Mandell GL. Catalase, superoxide dismutase, and virulence of Staphylococcus aureus. In vitro and in vivo studies with emphasis on staphylococcal – leukocyte interaction. J Clin Invest (1975) 55:561–6. doi:10.1172/JCI107963
153. Karavolos MH, Horsburgh MJ, Ingham E, Foster SJ. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology (2003) 149:2749–58. doi:10.1099/mic.0.26353-0
154. Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, et al. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med (2005) 202:209–15. doi:10.1084/jem.20050846
Keywords: neutrophil, phagocytosis, Staphylococcus aureus
Citation: van Kessel KPM, Bestebroer J and van Strijp JAG (2014) Neutrophil-mediated phagocytosis of Staphylococcus aureus. Front. Immunol. 5:467. doi: 10.3389/fimmu.2014.00467
Received: 23 July 2014; Accepted: 12 September 2014;
Published online: 26 September 2014.
Edited by:
Fabio Bagnoli, Novartis Vaccines, ItalyReviewed by:
Sukanya Narasimhan, Yale University School of Medicine, USAOlivier Dussurget, University Paris Diderot, France
Copyright: © 2014 van Kessel, Bestebroer and van Strijp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jos A. G. van Strijp, Medical Microbiology, University Medical Center Utrecht, Heidelberglaan 100, Utrecht 3584 CX, Netherlands e-mail: j.vanstrijp@umcutrecht.nl