 Elena Panzeri
Elena Panzeri Andrea Citterio
Andrea Citterio Andrea Martinuzzi
Andrea Martinuzzi Vera Ancona2
Vera Ancona2 Eleonora Martini
Eleonora Martini Maria Teresa Bassi
Maria Teresa Bassi- 1Laboratory of Molecular Biology, Scientific Institute IRCCS E. Medea, Bosisio Parini, Italy
- 2Department of Neurorehabilitation, Scientific Institute IRCCS E. Medea, Conegliano, Italy
Defects in FARS2 are associated with either epileptic phenotypes or a spastic paraplegia subtype known as SPG77. Here, we describe an 8-year-old patient with severe and complicated spastic paraplegia, carrying a missense variant (p.Pro361Leu) and a novel intragenic deletion in FARS2. Of note, the disease is unexpectedly progressing rapidly and in a biphasic way differently from the previously reported cases. Our study provides the first detailed molecular characterization of a FARS2 deletion and its underlying molecular mechanism, and demonstrates the need for combining different tools to improve the diagnostic rate.
Introduction
The human FARS2 gene encodes mitochondrial phenylalanyl-tRNA synthetase, which contributes to the accuracy of the translation of mitochondrial proteins, avoiding ATP deficiency in the cells (Roy et al., 2005). To date, 41 pathogenic FARS2 variants, inherited in an autosomal recessive pattern, have been reported (Figure 1). The variants are related to three main phenotypic manifestations: infantile-onset epileptic mitochondrial encephalopathy, later-onset spastic paraplegia (SPG77), and juvenile-onset refractory epilepsy. Usually, the epileptic groups have a poorer prognosis, while SPG77 is associated with a less severe disease and prolonged survival (Shamseldin et al., 2012; Yang et al., 2016; Hotait et al., 2020). Here, we describe a patient affected by a complicated and rapidly progressive form of hereditary spastic paraplegia (HSP) with upper limb involvement, bilateral single palmar creases, clinodactyly, mild intellectual disability, and dysmorphic features, carrying compound heterozygous variants in FARS2, the known missense variant p.Pro361Leu, and a novel intragenic deletion involving the coding exons 2, 3, and 4. Herein, we provide the first detailed molecular characterization of the intragenic deletion, identifying the molecular mechanism underlying the genomic rearrangement.
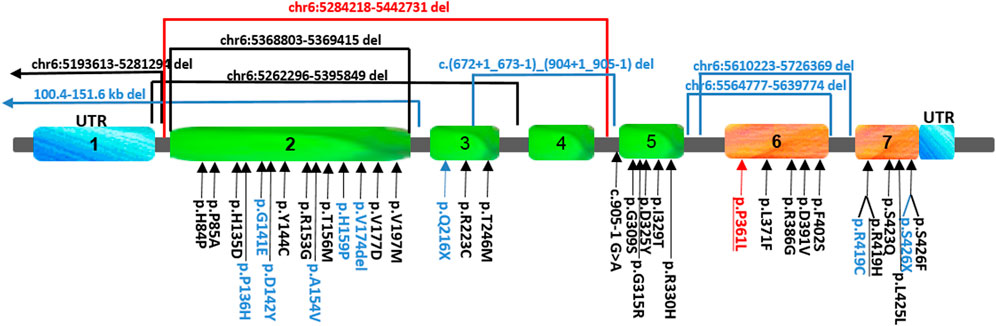
FIGURE 1. Schematic view and location of the FARS2 variants reported so far. The variants found in our patient are indicated in red, those found either in epileptic or spastic patients are underlined, and those found only in epileptic patients are indicated in black, while the variants associated only with spastic paraplegia are indicated in blue. To date, 41 pathogenic FARS2 variants have been found to be spread throughout the gene, including missense, non-sense, and splice-site variants, and deletions.
Materials and methods
Case presentation
The patient was referred to our clinic at the age of 8 years for cerebral palsy and developmental delay. The patient has a healthy brother and sister aged 18 and 15 years, respectively. The child was born at term, the pregnancy was uneventful, and the delivery and postnatal period had been uncomplicated. The child began walking with support at 26 months of age. The subject was admitted to an outpatient rehabilitation program since 2 years of age. At 6 years of age, the patient showed transient signs of ideomotor slowdown and linguistic regression, associated with nocturnal awakenings that spontaneously resolved over 2 weeks. The brain magnetic resonance imaging (MRI) and electroencephalogram (EEG) results were negative. The patient was diagnosed with a complicated and rapidly progressive form of HSP with upper limb involvement, dyspraxia, irregular action tremor, pronounced pyramidal signs in the lower limbs (deep tendon hyperreflexia, ankle clonus, and Babinski sign), bradykinesia, dorsal kyphoscoliosis, pronated valgus feet, and postural imbalance. Gait analysis at 8 years of age detected spasticity of hamstring/gastrocnemius muscles and a weakness of the dorsal flexors of the feet. In spite of this, autonomous gait was still possible; the spastic paraplegia rating scale (SPRS) score was 20/52, and the gross motor function measure (GMFM) score was 210/264. The child also presented bilateral single palmar creases on their hands, clinodactyly, mild intellectual disability, and dysmorphic features (macrocephaly, micrognathia, ogival palate, and macrodontia with severe caries). The subject’s social skills were fairly expressed, but verbal understanding was limited; speech was markedly dysarthric and not always intelligible. Creatine kinase, thyroid hormones, and sensory and motor nerve conduction velocities (NCVs) of the lower limbs were normal. Needle electromyography did not show any sign of active denervation. The child had exophoria and myopic astigmatism, but the fundus oculi results were negative. There was no sensory deficit. At the last clinical evaluation at the age of 12 years, a significant progression of the disability was evident, with a complete loss of ambulation. The SPRS score was 40/52, and the GMFM score was 145/264 (timeline in Figure 2). A unique aspect of this case is the modality of disease progression, apparently in a biphasic pattern, with decreasing development in the first 8 years of life, when an autonomous gait was still possible, followed by a very rapid worsening of the motor function with a complete loss of ambulation by the age of 12 years.
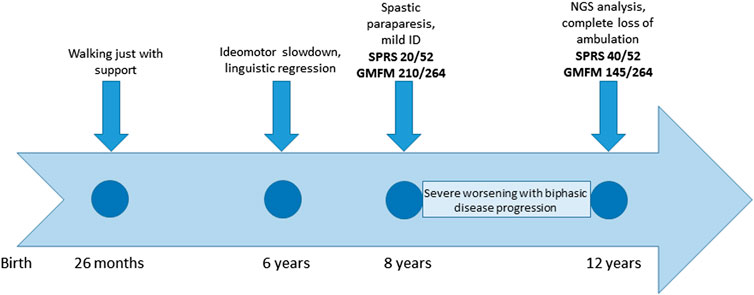
FIGURE 2. Timeline with relevant episodes in the case report presented.
Genetic analysis
Genomic DNA (gDNA) was extracted from the patient’s peripheral blood and screened using a targeted next-generation sequencing (NGS) approach with a custom gene panel from Agilent Technologies (Santa Clara, CA, United States), including 231 genes related to HSP, ataxias, and neuropathies (Supplementary Material). The targeted regions were run on a NextSeq platform (Illumina, San Diego, CA, United States), and the variants located in the coding regions, including the splice site, that exhibited a MAF <1% or were not present in variant databases were then analyzed with different tools to predict a possible pathogenicity. Sanger sequencing was used to confirm the variant in the patient and to verify the segregation in the patient’s family. The data were then further analyzed with ExomeDepth, a powerful bioinformatic tool, which is able to detect even small copy number variants (CNVs) using a robust beta-binomial model (Plagnol et al., 2012). Every CNV called with a Bayes factor (BF) higher than 20 was tested by qPCR. In view of the complicated features of the patient (developmental delay, dysmorphic features, intellectual disability, and skeletal abnormalities) and of the early onset of the disease, we also performed array CGH and whole-exome sequencing (WES) to exclude a possible coexistence of other genetic abnormalities (see Supplementary Material for a detailed description of the materials and methods used in this study).
Real-time PCR
To validate the deletion found with ExomeDepth, we performed a quantitative PCR (qPCR) on three regions downstream of exon 1 and three regions downstream of exon 4 within the putative deleted region. The primer pairs for qPCR analysis were selected within the non-repeated portions of the chromosome by Primer Express software (Applied Biosystems, Foster City, CA, United States) (See Supplementary Material for a detailed description).
Breakpoint characterization
A long-range PCR was performed with Herculase II fusion DNA polymerase (Agilent Technologies, Santa Clara, CA, United States), following the protocol for a fragment larger than 10 kb (Supplementary Material). Amplification was then tested by electrophoresis with 0.8% agarose gel, and the smaller size DNA fragment, found only in the proband, was sequenced with a BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Waltham, MA, United States) and run on an ABI 3500xL Genetic Analyzer. In silico analysis carried out with the RepeatMasker program (http://www.repeatmasker.org) and CENSOR utility (http://ebi.acuk/Tools/censor) were used to find possible elements of homology in the breakpoint regions. BLAST2 software (http://www.ncbi.nml.nih.gov/BLAST/) was used to align sequences close to the two breakpoints.
RNA extraction and cDNA sequencing
RNA was obtained from patient’s skin fibroblasts and from the PAXgene blood samples from their parents and extracted using either the Direct-zol RNA MiniPrep Kit (Zymo Research, Irvine, CA, United States) or the QIAGEN PAXgene Blood RNA Kit (PreAnalytiX, QIAGEN, Hombrechtikon, CH). A volume of 1 μg of RNA/sample was reverse-transcribed into cDNA using the SuperScript First-Strand Synthesis System for the reverse transcription (RT)-PCR kit (Thermo Fisher Scientific, Waltham, MA) and used for the PCR (Supplementary Material).
Results
A single heterozygous pathogenic variant was found in FARS2, c.1082C>T (p.Pro361Leu) (Figure 3A); the subsequent search for CNVs using the ExomeDepth tool led to the identification of an intragenic deletion spanning the region from intron 1 to 4 of FARS2. Amplification, subcloning, and sequencing of the junction fragment at the breakpoint revealed a deletion spanning 158,513 bp at the genomic level (g.5284218-5442731del). The absence of any repeats or homology regions suggested that a non-homologous recombination event had likely mediated the rearrangement (Figure 3B). At the cDNA level, the deletion involved 925 nucleotides (c.21_904del), eliminating the first three coding exons of the gene (exon 2 containing the ATG, and exons 3 and 4), corresponding to the first 319 amino acid residues of FARS2 protein. An in-frame ATG, 56 nucleotides downstream of the breakpoint, is still maintained, and a putative polypeptide containing the last 131 amino acids of FARS2 might still have been translated. This putative protein product (if any) is likely inactive because it lacks the catalytic functional domain (p.Met1_Ala320del), while it retains the FARS2 linker region and the anticodon-binding domain (Figure 3C). Segregation analysis showed that the missense variant was maternally inherited and transmitted to the proband’s unaffected sister, while the deletion was paternally inherited and segregated in the proband’s unaffected brother (Figure 3A). We confirmed these data by performing WES and array CGH on the trio. No additional pathogenic variants or CNVs were identified.

FIGURE 3. (A) On the left, the family pedigree with the proband is indicated by an arrow and a black square. The non-affected individuals are indicated by unfilled symbols. On the right, electropherogram of the FARS2 p.Pro361Leu maternal variant found in the patient was compared to the wild-type sequence (wt). (B) Schematic view of FARS2 at the genomic level with an indication of breakpoint locations, deleted exons in light green, and hypotheses of the underlying genomic event (non-homologous end joining, NHEJ). gDNA electrophoresis of the junction fragment and the schematic view of the gDNA resulting from the deletion are shown here. In the following figure, we see electropherogram of the sequence generated from the deletion, which spans from intron 1 to 4 of FARS2. (C) On the left, we observe the cDNA electrophoresis of the wild-type sequence and the fragment generated by the paternal intragenic deletion (del). On the right, we observe the schematic view of FARS2 at the cDNA level with the indication of the deleted exons in light green and representation of the cDNA resulting from the deletion. In the following figure, we see electropherograms of the 925 nucleotide deletion that lead to the loss of exons 2–3–4, including the canonical starting codon in exon 2. An ATG, located 56 nucleotides downstream of the breakpoint, might generate a putative protein with 131 residues instead of the canonical 451. This putative protein product (if any) is likely inactive because it lacks the catalytic functional domain, while it retains the FARS2 linker region and the anticodon-binding domain as shown here.
Discussion
Several FARS2 patients have been described with clinical phenotypes ranging from different types of epilepsy to pure and complicated forms of HSP SPG77. To date, 41 pathogenic FARS2 variants were found, including missense, nonsense, splice-site variants, and deletions. Among all the variants, seven are deletions and four of them are associated with SPG77 (Vernon et al., 2015; Forman et al., 2019; Jou et al., 2019; Meszarosova et al., 2020) (Figure 1). Although the deletions were not molecularly characterized with breakpoint subcloning and sequencing, in one case (Meszarosova et al., 2020), the analysis of genomic sequences surrounding the deletion had revealed the presence of highly homologous ALU repeats in FARS2 intron 2 and in LYRM4 intron 1, likely mediating the rearrangement. Here, we describe a child affected by a complicated form of SPG77, carrying compound heterozygous variants in FARS2, the known p.Pro361Leu pathogenic variant, and a novel intragenic deletion of 158513 bp, involving the first three coding exons of the gene, including the start codon. Unlike the case reported by Meszarosova and colleagues, the breakpoint sequence analysis in our patient revealed that the rearrangement was not mediated by any homologous region, thereby indicating a non-homologous end joining (NHEJ) recombination event had likely occurred. The NHEJ pathway is well known to be the election pathway for repairing double-strand breaks because their misrepair can cause severe changes, such as deletions or chromosomal translocations (Shibata, 2017). Based on the putative effects of FARS2 variants, we hypothesize a dramatic decrease of the FARS2 protein function in our patient. Indeed, the maternal p.Pro361Leu allele may likely retain a residual activity since this variant was already reported as affecting the stability of the anticodon-binding domain, thereby leading to a decreased protein interaction with tRNAPhe (Vantroys et al., 2017), while the paternally deleted allele may generate a likely putative inactive protein (if any) without the functional catalytic domain. This genotype is associated with clinical features partially overlapping those of a family reported by Vernon and colleagues (Table 1), with an early-onset severe spastic paraplegia, global developmental delay, and several dysmorphisms. In addition, our patient showed mild intellectual disability and additional dysmorphic features, such as bilateral single palmar creases on the hands, and clinodactily that were not observed in patients reported by Vernon and colleagues. Furthermore, a unique aspect of this case is the disease that seems to progress in a biphasic pattern, with decreasing development in the first 8 years of life, when an autonomous gait was still possible, followed by a very rapid worsening of the motor function with a complete loss of ambulation by the age of 12 years. Considering the clinical features of all SPG77 patients described so far, including the novel variant reported here, genotype–phenotype correlations are difficult to be established, given that the variants, such as p.Arg419His, are associated with both HSP and epileptic encephalopathy (Jou et al., 2019; Barcia et al., 2021). Analogously, even when considering only the HSP-related variants, there is no clear correlation between the genetic defect and disease severity, with the p.Pro361Leu variant (also carried by the patient described here) found in both pure and complicated forms (Table 1). Furthermore, neither the presence of a deletion nor its size (large ones or single amino acid deletions) could be correlated with a specific phenotype. Indeed, the loss of a single residue (p.Val174del) is associated with a complicated HSP form with a very early onset and severe presentation (Vantroys et al., 2017), while a deletion of about 151.6 kb was found in a patient with a pure form of HSP (Meszarosova et al., 2020). Thus, the phenotypic differences observed may be due to different combinations of FARS2 variants, as previously postulated (Barcia et al., 2021). Overall, the clinical and genetic data presented here further confirm the lack of any genotype–phenotype correlations in FARS2 patients. This represents a major drawback in genetic counseling and clinical prognoses in these patients. Therefore, a better understanding of the molecular mechanism(s) of FARS2 variants should allow a better risk assessment for patients and their families. In addition, this may also lead to the development of novel therapies. To this aim, the Drosophila dFARS2 mutants, mimicking many disease features, might represent a useful tool to better define the pathogenic mechanisms underlying the variable degree of FARS2-related disease severity (Fan et al., 2021).
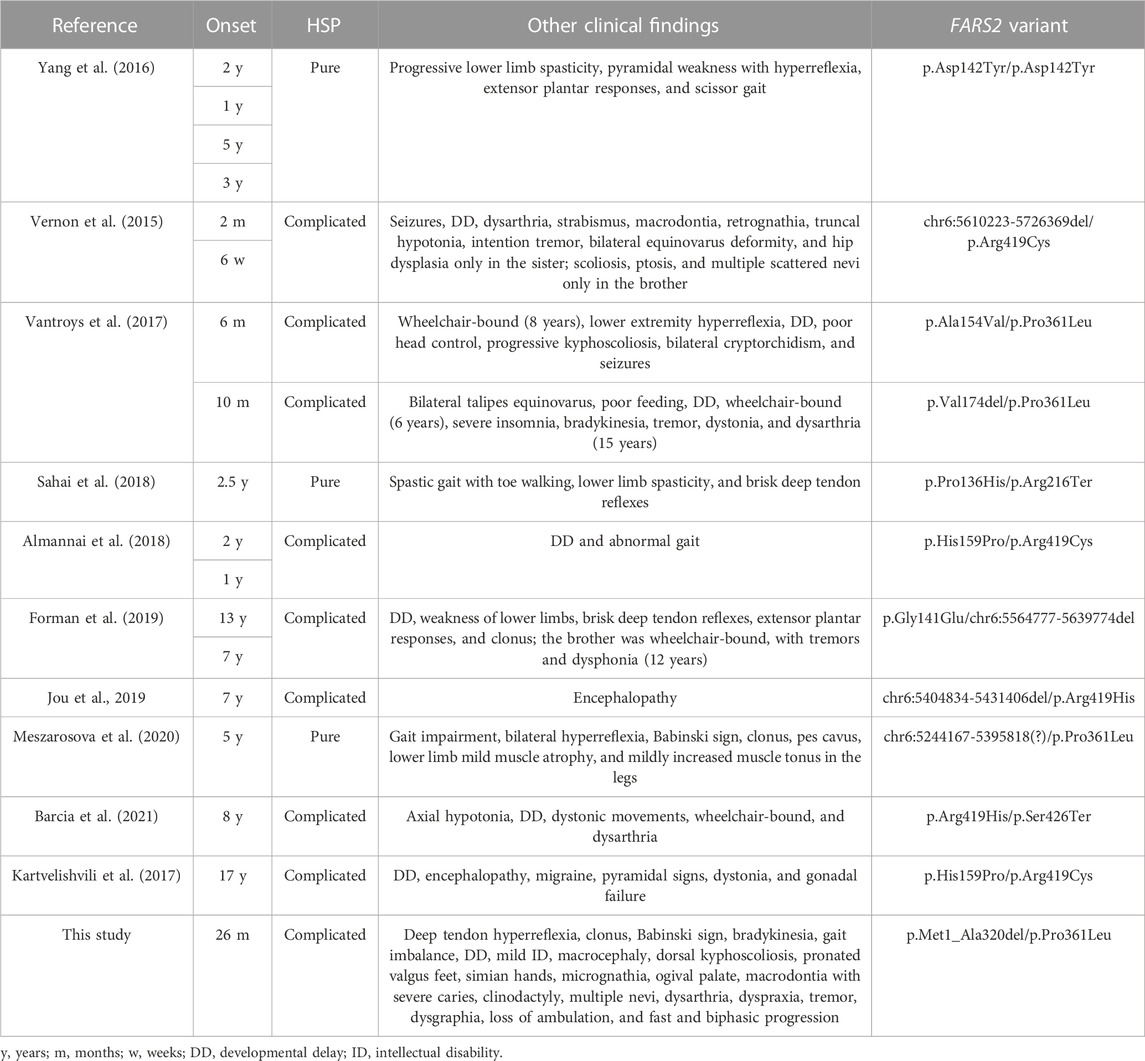
TABLE 1. Overview of the FARS2 variations related to SPG77 to date.
Conclusion
Our study widens the phenotypic spectrum and heterogeneity associated with FARS2 variants. Furthermore, we report the first molecular characterization of a FARS2 deletion and stress on the importance of combining different tools (i.e., sequencing and deletion/duplication studies) for variant screening in HSP patients, especially when the gene panels are inconclusive.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of IRCCS Eugenio Medea. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
EP performed the molecular study and wrote the manuscript. AC performed the molecular study and data analysis. VA and EM were involved in the patient’s evaluation. AM was responsible for the patient’s evaluation, data analysis, and funding acquisition, and was involved in critically revising the manuscript. MTB was involved in supervising the study, critically revising the manuscript, and in funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the funds from the Italian Ministry of Health (Grants #RC2021 and RC2022 to MTB and AM) and by Fondazione Regionale per la Ricerca Biomedica (Regione Lombardia), project number CP2_20/2018 to MTB.
Acknowledgments
The authors wish to thank the patient and his family for participating in the study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1130687/full#supplementary-material
References
Almannai, M., Wang, J., Dai, H., El-Hattab, A. W., Faqeih, E. A., Saleh, M. A., et al. (2018). FARS2 deficiency; new cases, review of clinical, biochemical, and molecular spectra, and variants interpretation based on structural, functional, and evolutionary significance. Mol. Genet. Metab. 125, 281–291. doi:10.1016/j.ymgme.2018.07.014
Barcia, G., Rio, M., Assouline, Z., Zangarelli, C., Roux, C-J., de Lonlay, P., et al. (2021). Novel FARS2 variants in patients with early onset encephalopathy with or without epilepsy associated with long survival. Eur. J. Hum. Genet. 29, 533–538. doi:10.1038/s41431-020-00757-x
Fan, W., Jin, X., Xu, M., Xi, Y., Lu, W., Yang, X., et al. (2021). FARS2 deficiency in Drosophila reveals the developmental delay and seizure manifested by aberrant mitochondrial tRNA metabolism. Nucleic Acids Res. 49, 13108–13121. doi:10.1093/nar/gkab1187
Forman, E. B., Gorman, K. M., Ennis, S., and King, M. D. (2019). FARS2 causing complex hereditary spastic paraplegia with dysphonia: Expanding the disease spectrum. J. Child. Neurol. 34, 621. doi:10.1177/0883073819846805
Hotait, M., Nasreddine, W., El-Khoury, R., Dirani, M., Nawfal, O., and Beydoun, A. (2020). FARS2 mutations: More than two phenotypes? A case report. Front. Genet. 11, 787. doi:10.3389/fgene.2020.00787
Jou, C., Ortigoza-Escobar, J. D., O'Callaghan, M. M., Nascimento, A., Darling, A., Pias-Peleteiro, L., et al. (2019). Muscle involvement in a large cohort of pediatric patients with genetic diagnosis of mitochondrial disease. J. Clin. Med. 8, 68. doi:10.3390/jcm8010068
Kartvelishvili, E., Tworowski, D., Vernon, H., Moor, N., Wang, J., Wong, L-J., et al. (2017). Kinetic and structural changes in HsmtPheRS, induced by pathogenic mutations in human FARS2. Protein Sci. 26, 1505–1516. doi:10.1002/pro.3176
Meszarosova, A. U., Seeman, P., Jencik, J., Drabova, J., Cibochova, R., Stellmachova, J., et al. (2020). Two types of recessive hereditary spastic paraplegia in Roma patients in compound heterozygous state; no ethnically prevalent variant found. Neurosci. Lett. 721, 134800. doi:10.1016/j.neulet.2020.134800
Plagnol, V., Curtis, J., Epstein, M., Mok, K. Y., Stebbings, E., Grigoriadou, S., et al. (2012). A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 28, 2747–2754. doi:10.1093/bioinformatics/bts526
Roy, H., Ling, J., Alfonzo, J., and Ibba, M. (2005). Loss of editing activity during the evolution of mitochondrial phenylalanyl-tRNA synthetase. J. Biol. Chem. 280, 38186–38192. doi:10.1074/jbc.M508281200
Sahai, S. K., Steiner, R. E., Au, M. G., Graham, J. M., Salamon, N., Ibba, M., et al. (2018). FARS2 mutations presenting with pure spastic paraplegia and lesions of the dentate nuclei. Ann. Clin. Transl. Neurol. 5, 1128–1133. doi:10.1002/acn3.598
Shamseldin, H. E., Alshammari, M., Al-Sheddi, T., Salih, M. A., Alkhalidi, H., Kentab, A., et al. (2012). Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 49, 234–241. doi:10.1136/jmedgenet-2012-100836
Shibata, A. (2017). Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. 803-805, 51–55. doi:10.1016/j.mrfmmm.2017.07.011
Vantroys, E., Larson, E., Friederich, M., Knight, K., Swanson, M. A., Powell, C. A., et al. (2017). New insights into the phenotype of FARS2 deficiency. Mol. Genet. Metab. 122, 172–181. doi:10.1016/j.ymgme.2017.10.004
Vernon, H. J., McClellan, R., Batista, D. A. S., and Naidu, S. (2015). Mutations in FARS2 and non-fatal mitochondrial dysfunction in two siblings. Am. J. Med. Genet. A 167A, 1147–1151. doi:10.1002/ajmg.a.36993
Keywords: complicated spastic paraplegia, FARS2, deletion, SPG77, severe
Citation: Panzeri E, Citterio A, Martinuzzi A, Ancona V, Martini E and Bassi MT (2023) Case report: A novel FARS2 deletion and a missense variant in a child with complicated, rapidly progressive spastic paraplegia. Front. Genet. 14:1130687. doi: 10.3389/fgene.2023.1130687
Received: 23 December 2022; Accepted: 23 March 2023;
Published: 19 April 2023.
Edited by:
Acary Oliveira, Federal University of São Paulo, BrazilReviewed by:
Min-Yu Lan, Kaohsiung Chang Gung Memorial Hospital, TaiwanAlice Barbara Schindler, National Institute of Neurological Disorders and Stroke, United States
Copyright © 2023 Panzeri, Citterio, Martinuzzi, Ancona, Martini and Bassi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Panzeri, elena.panzeri@lanostrafamiglia.it