 April L. Darling
April L. Darling Vladimir N. Uversky
Vladimir N. Uversky- 1Department of Molecular Medicine, USF Health Byrd Alzheimer’s Institute, Morsani College of Medicine, University of South Florida, Tampa, FL, United States
- 2Laboratory of New Methods in Biology, Institute for Biological Instrumentation, Russian Academy of Sciences, Pushchino, Russia
Intrinsically disordered proteins (IDPs) and intrinsically disordered protein regions (IDPRs) are functional proteins and domains that devoid stable secondary and/or tertiary structure. IDPs/IDPRs are abundantly present in various proteomes, where they are involved in regulation, signaling, and control, thereby serving as crucial regulators of various cellular processes. Various mechanisms are utilized to tightly regulate and modulate biological functions, structural properties, cellular levels, and localization of these important controllers. Among these regulatory mechanisms are precisely controlled degradation and different posttranslational modifications (PTMs). Many normal cellular processes are associated with the presence of the right amounts of precisely activated IDPs at right places and in right time. However, wrecked regulation of IDPs/IDPRs might be associated with various human maladies, ranging from cancer and neurodegeneration to cardiovascular disease and diabetes. Pathogenic transformations of IDPs/IDPRs are often triggered by altered PTMs. This review considers some of the aspects of IDPs/IDPRs and their normal and aberrant regulation by PTMs.
Introduction
Protein posttranslational modifications (PTMs) represent an important means for the natural increase in the proteome complexity and serves as one of the factors (in addition to the allelic variations and alternative splicing, together with some other pre-translational mechanisms, e.g., mRNA editing, allowing production of numerous mRNA variants), defining the ability of one gene to efficiently encode a set of distinct protein molecules, known as proteoforms (Smith et al., 2013). Since in eukaryotic organisms, the number of functionally different proteins dramatically exceeds the number of protein-encoding genes, one can reasonably argue that the complexity of a biological system is mostly determined by its proteome size and not by the size of genome (Schluter et al., 2009). Although the number of human protein-coding genes is approaching 20,700 (ENCODE Project Consortium, 2012), the actual human proteome includes a few hundred thousand, if not millions, of functionally different proteins (Uhlen et al., 2005; Farrah et al., 2013, 2014; Kim et al., 2014; Reddy et al., 2015). Because PTMs can affect protein activity, folding, interactions, localization, stability, and turnover, they play an important role in defining the protein structure-function continuum, according to which a protein has multiple structurally and functionally different states, proteoforms, generated by various mechanisms (including PTMs) (Uversky, 2015, 2016a,b,c). It was also pointed out that combination of alternative splicing, PTMs, and intrinsic disorder represents an important means for promotion of the alternative, context-dependent states of gene regulatory networks, thereby serving as a critical tool for controlling of a broad range of cellular responses, including cell fate specification (Niklas et al., 2015).
Intrinsic disorder and protein conformational dynamics represent another means used by nature to generate proteoforms (Uversky, 2015, 2016a,b,c). In fact, it was pointed out that even without alternative splicing, PTMs, or mutations, any protein, being a dynamic conformational ensemble, represents a set of basic (or intrinsic, or conformational) proteoforms; i.e., molecules with identical amino acid sequences but with different structures and, potentially, with different functions. Obviously, any mutated, modified, or alternatively spliced form of a protein [i.e., any member of the inducible (or modified) proteoforms] also exists as a structural ensemble and thereby represents a set of conformational proteoforms (Uversky, 2015, 2016a,b,c). Protein functions and structural ensembles of basic and induced proteoforms can be significantly altered by placing proteins in crowded cellular environment. This environment originates from high concentrations of various biological macromolecules inside the cells (Zimmerman and Trach, 1991; van den Berg et al., 1999; Rivas et al., 2004) that limit available volume (Ellis and Minton, 2003) and restrict amounts of free water (Fulton, 1982; Zimmerman and Trach, 1991; Zimmerman and Minton, 1993; Minton, 1997; Minton, 2000; Ellis, 2001). Finally, functionality per se can be considered as a factor generating functioning proteoforms (Uversky, 2015, 2016a,b,c). In other words, because of all these factors, any given protein exists as a set of basic, induced and functioning proteoforms. As a result, the actual relationships between a gene and a protein function follow the ‘one-gene – many-proteins – many-functions’ concept (Uversky, 2015, 2016a,b,c). This is in a striking contrast to the classical ‘one-gene – one-protein’ view, where each gene produces a single enzyme for controlling a single step of a metabolic pathway (Beadle and Ephrussi, 1936; Beadle and Tatum, 1941; Horowitz et al., 1945; Horowitz, 1995).
Posttranslational modifications are included into the ‘dark matter of biology’ concept that is attributed to “important, yet invisible species of molecules and proteins that interact weakly but couple together to have huge and important effects in many biological processes… [and that] remains mostly hidden, because our tools were developed to investigate strongly interacting species and folded proteins” (Ross, 2016). Curiously, many PTMs are intimately linked to another component of the ‘dark matter of biology,’ namely intrinsically disordered proteins (IDPs) and IDP regions (IDPRs), making such disorder-centered PTMs the darker side of the biological dark matter. This review is dedicated to the analysis of an intriguing connection between intrinsic disorder and PTMs, thereby representing an attempt to shed some light to the darker corner of the dark matter of biology.
What Is Intrinsic Disorder?
Intrinsically Disordered Proteins, Their Abundance and Biological Functions
The protein functionality is not always associated with the presence of unique 3D structure in a protein molecule. Instead, functional states of many proteins constitute dynamic conformational ensembles of interconverting structures, where a whole protein or its noticeable regions lack stable tertiary and/or secondary structure (Wright and Dyson, 1999; Uversky et al., 2000; Dunker et al., 2001; Tompa, 2002; Uversky and Dunker, 2013). These floppy but biologically active proteins and regions are currently known as IDPs and IDPRs. In relation to the subject of this special issue, IDPRs frequently contain sites of proteolytic attack and include various PTM sites (Dunker et al., 2001; Iakoucheva et al., 2004; Radivojac et al., 2010; Pejaver et al., 2014).
Figure 1 shows that IDPs and IDPRs are abundantly present in all proteomes analyzed to date, with the proteome disorder levels typically increasing with the increase of the organism complexity (Romero et al., 1998; Dunker et al., 2000, 2001; Ward et al., 2004; Uversky, 2010; Xue et al., 2012; Peng et al., 2014). Furthermore, many IDPs/IDPRs are evolutionary conserved, indicating that at least some protein functionality is determined by intrinsic disorder. In fact, order and disorder are both needed for protein functionality, and functional repertoire ascribed to IDPs/IDPRs complement catalytic and transport functions of ordered proteins and domains (Radivojac et al., 2007). Therefore, the unbiased consideration of protein functionality should include both ordered proteins/domains and IDPs/IDPRs (Dunker et al., 2001, 2002a). This idea is illustrated by Figure 2 showing that protein structure-function interrelationship includes two pathways, where the ‘sequence-to-structure-to-function’ paradigm can be utilized for the description of functionality of transport proteins and enzymes, whereas the ‘sequence-to-dynamic conformational ensemble-to-function’ paradigm represents a key for understanding the functions of IDPs/IDPRs preferentially involved in control, recognition, regulation, and signaling (Dunker et al., 2001, 2002a,b; Uversky, 2002a,b). Furthermore, the ‘protein trinity’ (Dunker and Obradovic, 2001) or the ‘protein quartet’ (Uversky, 2002a) models were used for the conceptualization of protein functionality. In these models, function of a protein can be associated with ordered, molten globule-like (collapsed-disordered), pre-molten globule-like (partially collapsed-disordered) or coil-like (extended-disordered) conformations and/or from the transitions between all these conformations (Dunker and Obradovic, 2001; Uversky, 2002a).
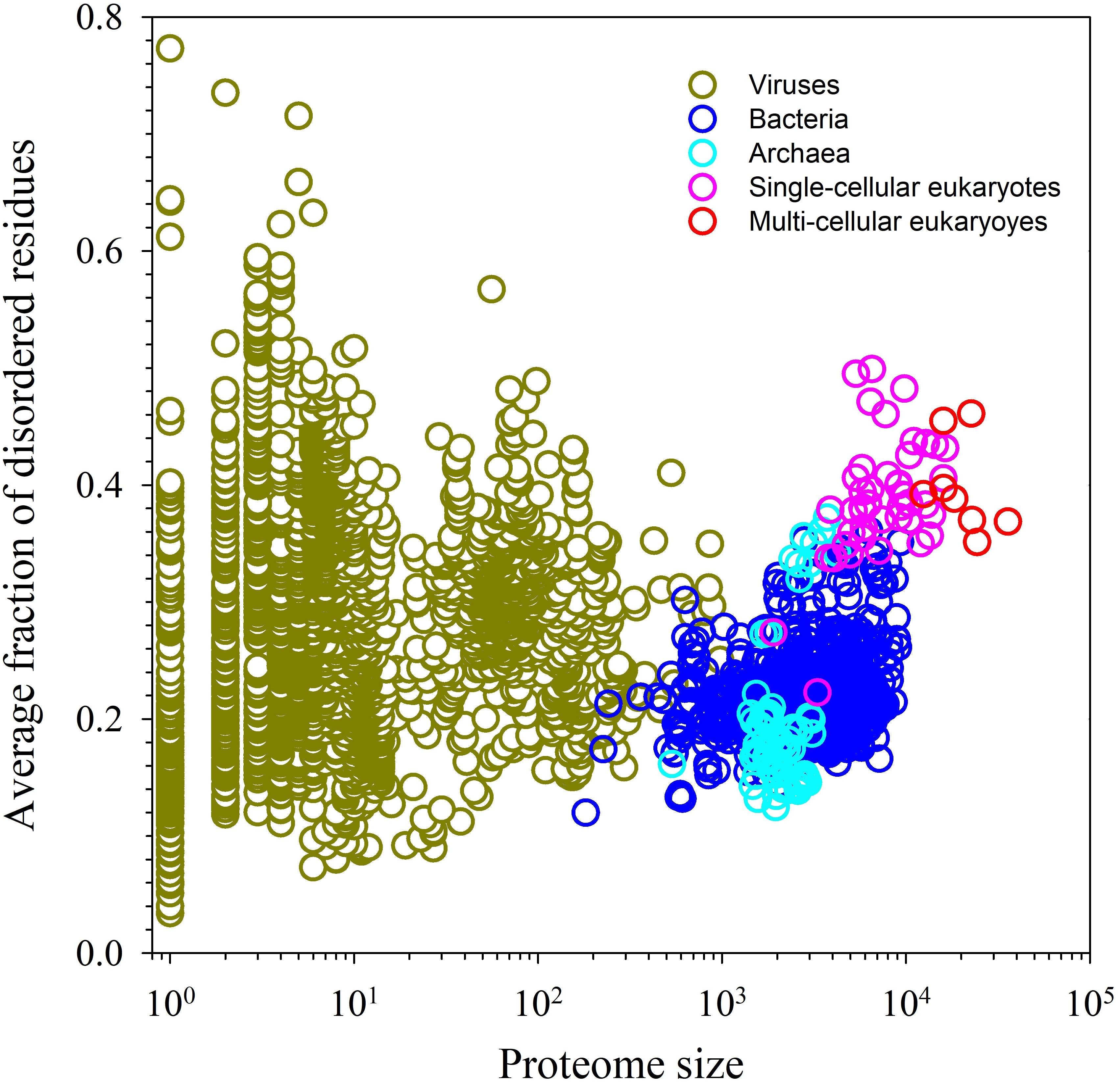
FIGURE 1. Abundance of intrinsic disorder in various proteomes.
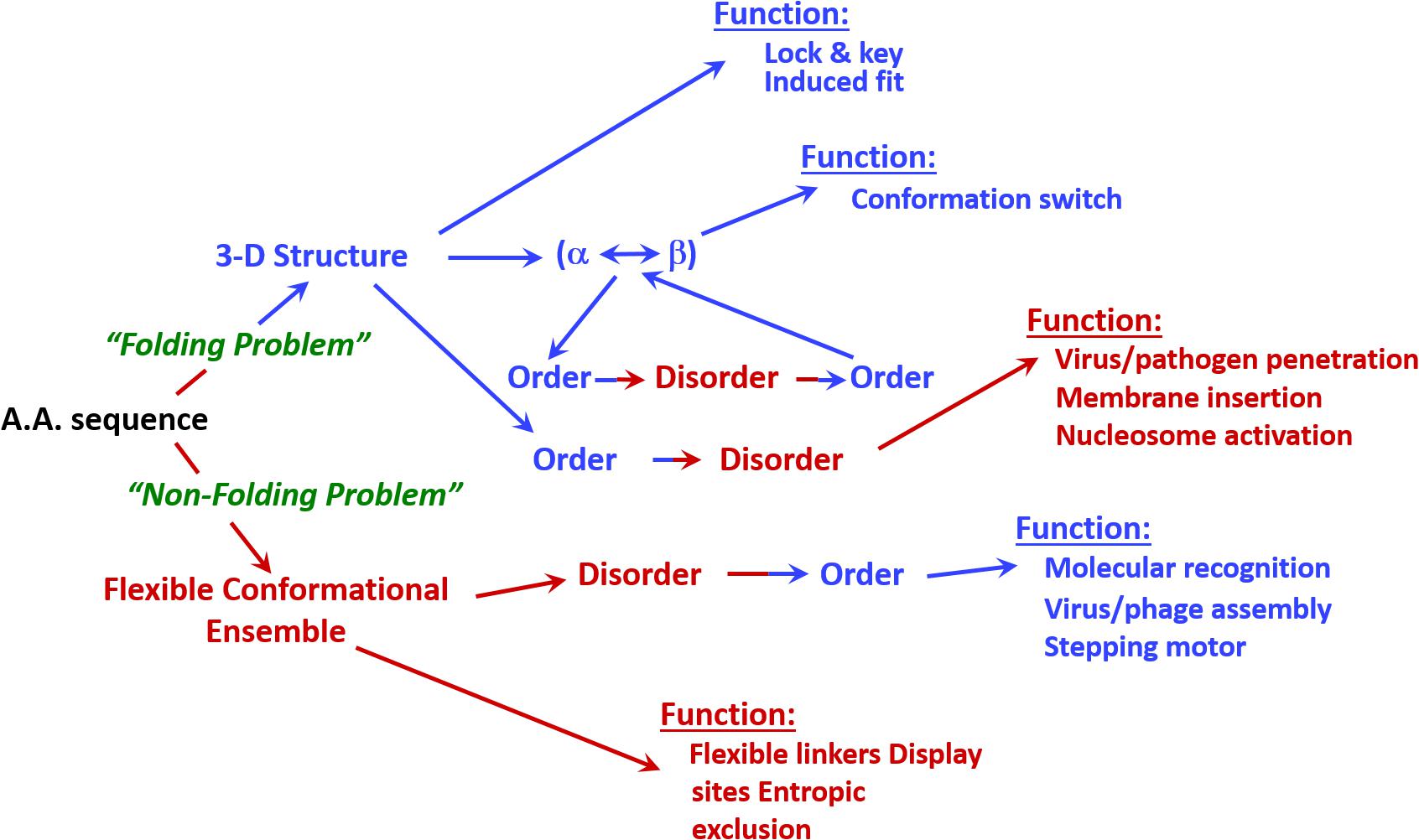
FIGURE 2. Involvement of intrinsic disorder in protein function. Note that the classical structure-function paradigm cannot describe many of the function proteins perform.
Some illustrative biological activities of IDPs/IDPRs include protein and RNA chaperone action, control of transcription and translation, storage of small molecules, location of PTM sites, regulation of the cell cycle and division, control of development, regulation and modulation of the self-assembly of large multi-protein complexes, and moderation of signal transduction, to name a few (Wright and Dyson, 1999; Uversky et al., 2000, 2005; Dunker and Obradovic, 2001; Dunker et al., 2002a,b, 2005, 2008a,b; Iakoucheva et al., 2002; Tompa, 2002, 2005; Uversky, 2002a,b, 2010; Dyson and Wright, 2005; Radivojac et al., 2007; Vucetic et al., 2007; Xie et al., 2007a,b; Cortese et al., 2008; Dunker and Uversky, 2008). The existence of at least 28 separate disorder-based functions was proposed based on the careful analysis of literature on >150 completely disordered proteins or proteins containing functional IDPRs (Dunker et al., 2002a). Globally, functional IDPs/IDPRs have very different modes of action serving as entropic chains, display sites, assemblers, effectors, chaperones, and scavengers (Tompa, 2002; Tompa and Csermely, 2004).
Because of their flexible nature, IDPs/IDPRs have some remarkable functional advantages over their ordered counterparts (Schulz, 1979; Pontius, 1993; Plaxco and Gross, 1997; Wright and Dyson, 1999; Dunker et al., 2001, 2002a, 2005; Dyson and Wright, 2002; Iakoucheva et al., 2002; Dyson and Wright, 2005; Uversky et al., 2005), including their ability to be promiscuous binders engaged in efficient interactions with different and often unrelated targets (Wright and Dyson, 1999; Uversky et al., 2000; Dunker et al., 2001; Uversky and Dunker, 2010). One of the said functional advantages of IDPs/IDPRs is their ability to (partially) fold (or undergo disorder-to-order transitions) upon interaction with specific partners (Schulz, 1979; Pontius, 1993; Spolar and Record, 1994; Plaxco and Gross, 1997; Wright and Dyson, 1999; Dunker et al., 2001, 2002a, 2005; Dyson and Wright, 2002, 2005; Iakoucheva et al., 2002; Oldfield et al., 2005; Uversky et al., 2005; Mohan et al., 2006; Cheng et al., 2007; Vacic et al., 2007; Tompa et al., 2009). In relation to the reversible signaling interactions, such binding-induced disorder-to-order transitions allow IDPs/IDPRs to participate in highly specific but weak interactions (Schulz, 1979; Dunker et al., 2001). Furthermore, IDPs/IDPRs can fold on a template-dependent manner. This provides them with an ability to attain very different structures at interaction with different binding partners (Kriwacki et al., 1996; Dunker et al., 2001; Oldfield et al., 2008; Hsu et al., 2012, 2013).
Although promiscuous interactivity and ability to fold at binding to specific partners represent a characteristic signature of disorder-based functionality, one should remember that IDPs/IDPRs have numerous ‘entropic chain activities.’ These activities rely on the flexibility, plasticity, and pliability of the protein backbone and therefore that do not require coupled binding and folding. The voltage gated ion channel can serve as an illustrative example of such entropic chain activities. Activity of this channel involves cyclic transitions between closed (sensitive to voltage), open, and inactive (insensitive to voltage) states (Armstrong and Bezanilla, 1977; Antz et al., 1997). In structure of such channels, a highly flexible ‘chain’ connects a small folded domain, ‘ball,’ to the channel opening. Channels utilize an entropic clock mechanism for inactivation, where the random motions of the ‘ball-and-chain’ unit provide a means for the ‘ball’ to eventually plug into the open channel and inactivate it (Armstrong and Bezanilla, 1977; Antz et al., 1997). It was pointed out that timing of such channels relies on flexibility and length of their disordered ‘chains.’ In fact, since the time of closure of such channels is inversely proportional to the square of the length of their ‘chains’ (Liebovitch et al., 1992), inactivation is faster when the ‘chain’ is shorter (Hoshi et al., 1990).
Entropic bristle domain (EBD); i.e., the time-averaged three-dimensional area swept out by the thermally driven motion of a disordered polypeptide, represents another example of entropic activity. Obviously, EBD that occupies a defined area but does not have a fixed structure is different from a traditional protein domain, which is a conserved part of a protein structure that can evolve, function, and exist independently of the rest of the polypeptide chain. Illustrative carriers of functional EBDs are given by the neurofilament proteins found in the major cytoskeletal components of the axonal cell, neurofilaments, that among other functions, are needed to maintain the bore of the axon (Brown and Hoh, 1997). The needed spacing between the neurofilaments is kept by the action of the entropic brush formed by EBDs that project from the NF-M and NF-H neurofilament proteins (Brown and Hoh, 1997).
Overall, multiple functional advantages were ascribed to IDPs/IDPRs to explain their prevalence in various proteomes and their broad use in various biological processes (Dunker et al., 1997, 1998, 2001; Wright and Dyson, 1999; Romero et al., 2001; Brown et al., 2002; Cortese et al., 2008). Natural abundance of IDPs/IDPRs is clearly related to their multifunctionality, which, in its turn, is linked to their specific structural features. Sections below consider peculiarities of structural organization and conformational behavior of IDPs/IDPRs to better understand the uniqueness of these intriguing members of protein universe.
Some Basic Structural Properties of IDPs/IDPRs
Structural description of IDPs/IDPRs relies on the dynamic conformational ensemble representation, where the members of such conformational ensembles interconvert on a number of timescales. By analogy with the conformational states of typical globular proteins that, depending on the environmental conditions, may exist in at least four different conformations: folded (ordered), molten globule, pre-molten globule, and coil-like (Uversky and Ptitsyn, 1994, 1996; Fink, 1995; Ptitsyn, 1995; Ptitsyn et al., 1995; Uversky, 2003; Turoverov et al., 2010), IDPs, being considered at the whole protein level, can be classified as native molten globules containing collapsed form of disorder, native pre-molten globules possessing semi-collapsed form disorder, and native coils with extended form of (Dunker et al., 2001; Uversky, 2003). This is illustrated by Figure 3 schematically representing these three types of disorder at the whole protein level.
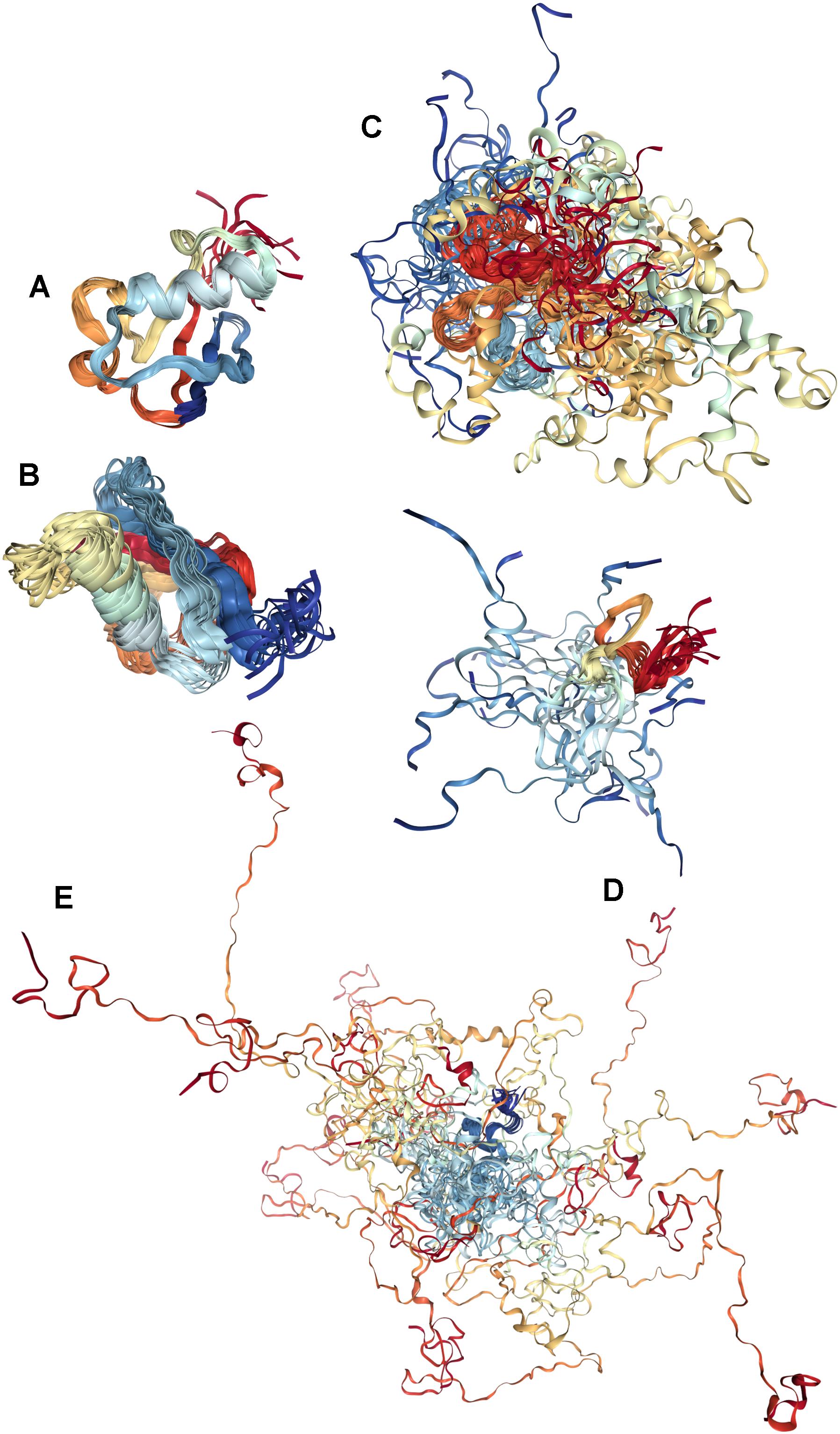
FIGURE 3. Illustrative examples of proteins with different levels of global disorder. (A) Ordered protein: NMR solution structure of bovine ubiquitin (PDB ID: 1V81) (Kitahara et al., 2005). (B) Protein with the collapsed (molten globule-like) disorder: NMR solution structure of Ubiquitin-like domain of NFATC2IP (PDB ID: 2JXX). (C) Protein with semi-extended (pre-molten globule-like) disorder: NMR solution structure of the domain II from the blood-stage malarial protein, apical membrane antigen 1 (PDB ID: 1YXE) (Feng et al., 2005). (D) Small protein with extended (coil-like) disorder: sunflower trypsin inhibitor 1 (SFTI-1) (PDB ID: 2AB9) (Mulvenna et al., 2005). (E) Large protein with extended (coil-like) disorder: NMR solution structure of Air2p protein, which is the key component of the Trf4/5p-Air1/2p-Mtr4p polyadenylation complex (TRAMP) (PDB ID: 2LLI) (Holub et al., 2012).
One should keep in mind, however, this classification represents a clear oversimplification, since order and disorder are not homogeneously distributed within a protein molecule. In fact, analysis of crystal structures of various proteins deposited in PDB revealed that they are not only characterized by the presence of regions with high B-factor that reflects the high uncertainty in atom positions in the model, but systematically contain long regions of missing electron density that often correspond to IDPRs (Radivojac et al., 2004) [in fact, only about 7% of PDB entries devoid intrinsic disorder (Le Gall et al., 2007)], and also contain ambiguous regions, where different structures of the same protein disagree in terms of the presence or absence of missing residues (Le Gall et al., 2007; DeForte and Uversky, 2016). It is likely that similar levels of disorderedness (collapsed, semi-collapsed, and extended forms of disorder) discussed for the whole proteins can also be found in protein regions, indicating that both IDPs and IDPRs can be differently disordered. These observations suggest that proteins, in general, are characterized by non-homogeneous distribution of foldability and, as a consequence, by a high spatiotemporal heterogeneity, where different parts of a protein molecule are ordered (or disordered) to a different degree (Uversky, 2013a,c). In other words, globally, structural space of functional proteins represents a continuous spectrum of conformations with different degree and depth of disorder. This structural spectrum spreads from fully ordered to completely disordered proteins and includes a myriad of differently (dis)ordered species between these two extremes (Uversky, 2013a,c). Therefore, protein structural space represents a structure-disorder continuum with no clear boundary between ordered proteins and IDPs (Uversky, 2013c). It was also pointed out that because of the presence of different levels and depths of intrinsic disorder, proteins are characterized by the mosaic structure and typically contain foldons (i.e., independently foldable regions), inducible foldons (disordered regions that can fold at interaction with binding partner), semi-foldons (IDPRs that are always in the semi-folded state), non-foldons (IDPRs with entropic chain activities), and unfoldons (or conditionally disordered protein regions, which, in order to become functional or to make a protein active, have to undergo order-to-disorder transition) (Uversky, 2013c).
Peculiarities of Conformational Behavior of IDPs/IDPRs
The free energy landscape of an extended IDP/IDPR is dramatically different from that of ordered globular protein or domain. In fact, if the energy landscape of an ordered protein has a funnel-like shape with a well-defined global energy minimum corresponding to unique protein structure (Radford, 2000; Jahn and Radford, 2005), the free energy landscape of an IDP can be described as a ‘hilly plateau,’ with hills on the plateau corresponding to the forbidden conformations (Uversky et al., 2008; Turoverov et al., 2010; Fisher and Stultz, 2011) and with numerous local energy minima. Such free energy landscape reflects the existence of numerous conformations that constitutes the dynamic conformational ensemble of an IDP. It also shows that such a protein that does not have stable conformation, possessing instead a highly frustrated nature (Uversky, 2013c). The shape of such energy landscape can be easily changed even by minimal changes in the local environment of an IDP. This contrasts high conformational stability of ordered proteins originated from their relatively robust funnel-like energy landscapes. Therefore, the ‘hilly plateau’-like energy landscape determines conformational plasticity of an IDP/IDPR and its ability to fold differently depending on the environmental conditions, where any (even rather subtle) changes in the IDP/IDPR environment might have very strong effects on the protein/region structure leading to the formation of very different structures (Uversky, 2013c).
Because of the IDPs/IDPRs are characterized by highly biased amino acid compositions, their conformational behavior is noticeably different from that of ordered proteins and domains. In fact, proteins with extended disorder (which are typically depleted in hydrophobic residues, but enriched in charged and polar residues) were shown to partially fold at increase in temperature, which is in striking opposition to globular proteins and domains that typically unfold at heating. This structure-forming effect of elevated temperature was attributed to the temperature-driven increase in the strength of the hydrophobic interactions that leads to a stronger hydrophobic attraction at higher temperatures (Uversky, 2002b). Similarly, a decrease (or increase) in pH was shown to induces partial folding of many extendedly disordered proteins characterized by high net charges at neutral pH. This is because at extreme pH, the net charge of such proteins decreases, causing the decrease in their charge-charge intramolecular repulsion. This permits formation of a partially folded conformation due to the hydrophobicity-driven collapse of polypeptide chain (Uversky, 2002b).
Intrinsically disordered proteins/IDPRs are characterized by highly dynamic and heterogeneous structures that are extremely sensitive to changes in the environment and that determine unique functional repertoire of disordered proteins. Since PTMs do alter local physical and chemical properties of proteins, changing their charge, flexibility, and hydrophobicity, such modifications serve as crucial regulators of IDP/IDPR functions and structures (Uversky, 2013b). Therefore, sections below, in addition to the introduction of the natural PTM variability, provide an outline of the roles that PTMs play in the regulation of IDPs/IDPRs.
Posttranslational Modifications
As it follows from their definitions, PTMs are chemical changes affecting proteins after their biosynthesis, with almost every protein being potentially able to undergo PTMs. Changes induced by PTMs to protein structure are many. Various chemical groups, carbohydrates, lipids, and even entire proteins or nucleic acids can be covalently added to amino acid side chains. Proteins can also undergo enzymatic cleavage of peptide bonds or removal of various chemical groups. Since different PTMs can differently affect physicochemical properties of a protein (Mann and Jensen, 2003), different modifications can graft different functions to the same protein (Jungblut et al., 2008). Although natural variability of PTMs is very broad, these modifications are typically very specific. Many PTMs are catalyzed by special enzymes that recognize particular motifs in target sequences of specific proteins. Some PTMs (e.g., phosphorylation, acetylation, glycosylation, lipidation, methylation, and nitration) are readily reversible due to the concert action of modifying and demodifying enzymes. Such and interplay between the conjugating and deconjugating enzymes represents an economical and rapid way of the controlling the protein function. Furthermore, although mutations (which represent another means of changing the chemical properties of a polypeptide chain) can only occur once per position, different forms of PTMs may happen in tandem (Khoury et al., 2011).
Diversity of PTMs and Their Classifications
Posttranslational modifications represent an important means for the increase in the variability and diversity of protein structures and functions via extension of the range of structures and physico-chemical properties of amino acids (Walsh et al., 2005). It was pointed out that although there are 20 primary amino acids typically utilized in protein biosynthesis, in reality, because of various PTMs, proteins might contain more than 140 chemically different residues. To this end, large portion of the genomes of higher eukaryotes (as much as 5%) represents genes that encode PTM-related enzymes. Altogether, as many as 300 PTMs are known to occur physiologically in proteins (Witze et al., 2007).
Posttranslational modifications are abundantly present in nature. For example, between at least one-fifth (Khoury et al., 2011) and a half of proteins are expected to be glycosylated (Apweiler et al., 1999). The most common PTMs are covalent removal or addition of various groups, formation of disulfide bonds, and specific cleavage of protein precursors (Baumann and Meri, 2004). According to the PTM Curator, most often proteins undergo phosphorylation, acetylation, N-linked glycosylation, amidation, hydroxylation, methylation, O-linked glycosylation, ubiquitylation, attachment of pyrrolidone carboxylic acid or gamma-carboxyglutamic acid, sulfation, sumoylation, palmitoylation, C-linked glycosylation, ADP-ribosylation, myristoylation, farnesylation, nitration, formylation, geranyl-geranylation, deamidation, S-nitrosylation, citrullination, S-diacylglycerol cysteine modification, GPI anchoring, bromination, and FAD addition (Khoury et al., 2011). Diversity of PTMs found in proteins is illustrated by Table 1, which not only shows that due to the various PTMs, side chains of all natural amino acids can be chemically diversified, but also emphasizes that each amino acid residue can be affected by many different PTMs. However, although all amino acid residues can be modified, most commonly PTMs affect side chains that can act as either strong (C, D, E, H, K, M, R, S, T, and Y) or weak (N and Q) nucleophiles (Walsh et al., 2005).

TABLE 1. Variability of posttranslational modifications of side chains in proteins.
All sides of protein life in a cell are affected by PTMs, which can regulate protein folding, target proteins to specific subcellular compartments, control interaction of proteins with their partners, modulate catalytic activity, or affect signaling and recognition functions (Mann and Jensen, 2003; Deribe et al., 2010). Functions of many proteins rely on multiple different PTMs, and modified sites in such multi-PTMed proteins are utilized individually for mediation of specific protein activities or used together for controlling molecular interactions and for modulation of the overall protein activity and stability (Yang, 2005). An illustrative example of such multi-PTMed proteins is given by histones, different stages of action of which require acetylation, ADP-ribosyation, methylation, phosphorylation, SUMOylation and ubiquitylation (Peng et al., 2012). The N-terminal tails of core histones protruding from the nucleosome core and needed to mediate chromatin compaction (Arya and Schlick, 2009) are known to contain an astonishing number of PTM sites (Shliaha et al., 2017). Furthermore, over 30 histone modifications have been also identified in the core domains of these proteins (Mersfelder and Parthun, 2006).
Because of their variability, PTMs can be grouped and classified on multiple ways. For example, depending on the stages of the protein life at which they were introduced, PTMs can be ‘early’ or ‘late’ and can give very different outputs. In fact, some proteins are known to be modified shortly after completion of their biosynthesis and before the final steps of their folding. The corresponding ‘early’ PTMs can influence the efficiency of protein folding, or affect protein conformational stability, or direct the nascent protein to distinct cellular compartments thereby defining its cellular fate (Tokmakov et al., 2012). The ‘late’ PTMs occur after the completion of protein folding and localization. They can activate, deactivate, or modulate the biological activity of target proteins (Ngounou Wetie et al., 2014). PTMs can also be classified based on the new modification-enabled functionality, such as gain of the catalytic function (e.g., resulting from phosphopantetheinylation, biotinylation, or lipoylation), membrane localization (e.g., caused by glypiation, geranylgeranylation, myristoylation, palmitoylation, prenylation, etc.), and proteolytic destruction by proteasomes (via ubiquitylation).
Another classification of PTMs is based on the underlying molecular mechanisms, such as functional group addition (e.g., phosphorylation, acylation, glycosylation, etc.), covalent conjugation of peptides and small proteins to the main polypeptide chain (e.g., ISGylation, neddylation, SUMOylation, ubiquitination, etc.), change in the physico-chemical properties of amino acids (citrullination, deamidation, deimidation, oxidation, etc.), and proteolytic cleavage (Xie et al., 2007a). PTMs can also be classified based on the description of the fragment of coenzyme or cosubstrate attached to the modified protein and the chemical nature of the protein modification, e.g., ATP-dependent phosphorylation, acetyl CoA dependent acetylation, NAD-dependent ADP ribosylation, CoASH-dependent phosphopantetheinylation, phosphoadenosinephosphosulfate (PAPS)-dependent sulfurylation, and S-adenosylmethionine (SAM)-dependent methylation (Walsh et al., 2005).
Posttranslational Modifications and Intrinsic Disorder
Already in early IDP-related studies, it has been pointed out that many PTM sites are frequently associated with IDPRs (Dunker et al., 2002a). Therefore, in addition to the aforementioned classifications of PTMs based on the modulation molecular mechanisms or modulation-enabled functions, PTMs can also be grouped according to the conformational requirements of the potential modification sites. This classification generates two major PTM groups, where modifications are associated either with structured regions or with IDPRs (Xie et al., 2007a). Tables 2, 3 list some of the PTMs associated with IDPs/IDPRs and ordered proteins and domains, respectively (Xie et al., 2007a). Curiously, PTMs targeting ordered regions, such as covalent attachment of quinones and organic radicals, formylation, oxidation, and protein splicing introduce new chemical moieties needed for novel catalytic functions, or changing of the existing enzymatic activity, or modulation of the protein conformational stability. On the other hand, PTMs preferentially targeting IDPRs, such as phosphorylation, acetylation, and methylation, are typically used for modifications of the regulatory and signaling regions in target proteins that are engaged in specific but weak interactions with their partners. For example, p53- and p14-ARF-binding regions of the Mdm2 contain the majority of the phosphorylation sites of this protein. Similarly, the phosphorylation of PEST motifs of many proteins controls their ubiquitin-mediated degradation. The biological activities of various proteins involved in signal transduction are modulated by phosphorylation. Furthermore, this PTM can alter gene expression by modulating the binding affinity of transcription factors to their coactivators and DNA, and also can affect cell growth and differentiation (Zor et al., 2002).
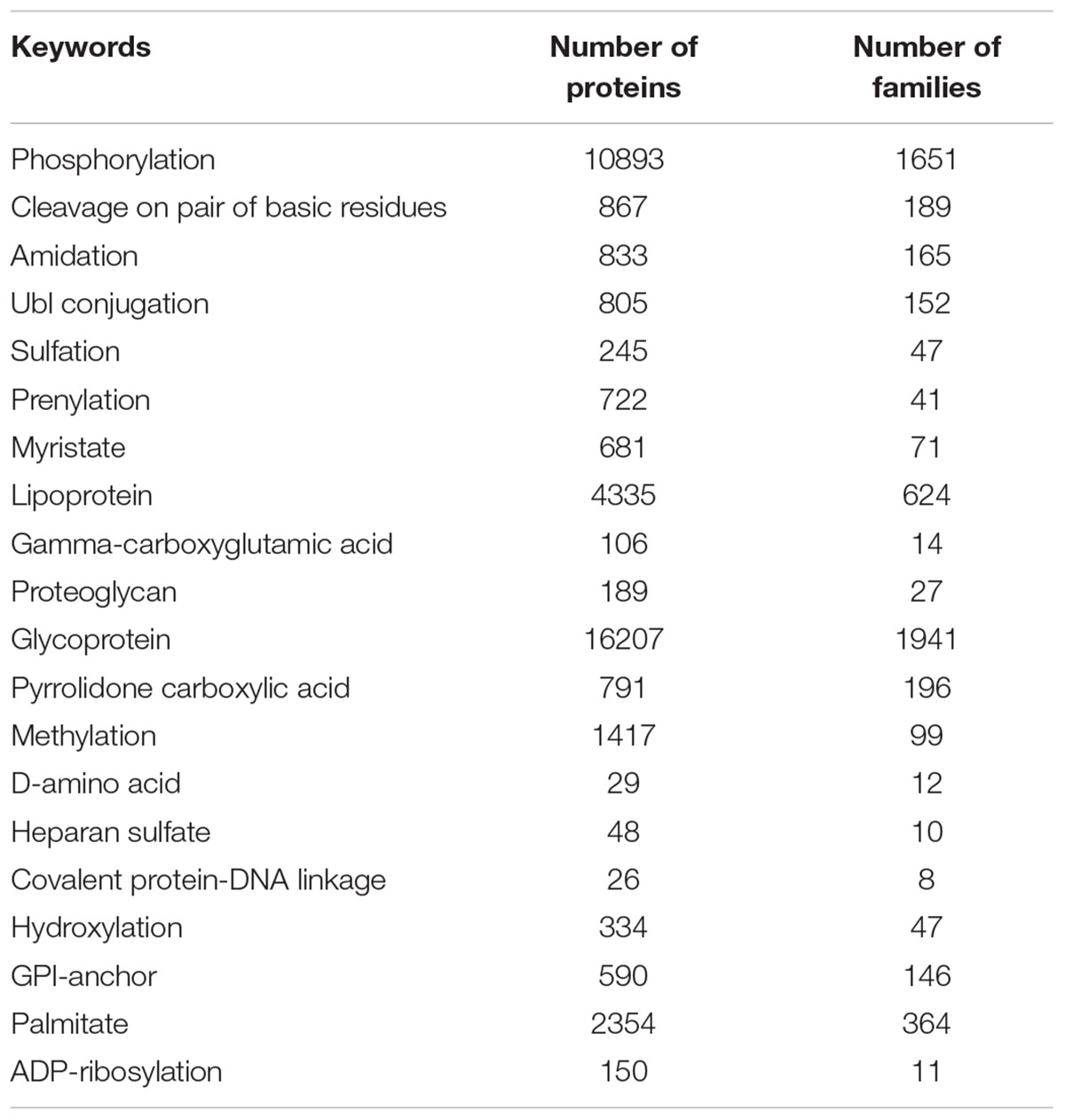
TABLE 2. Top 20 of the PTM-related keywords strongly correlated with predicted disorder.
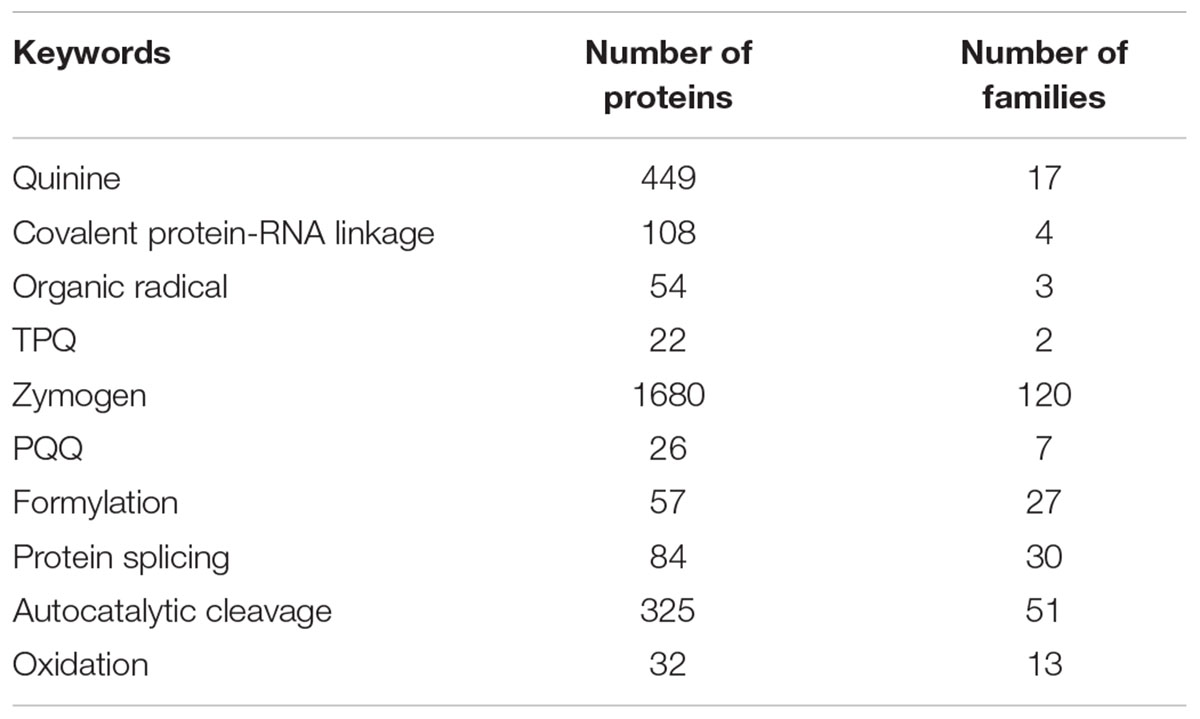
TABLE 3. All PTM-related keywords strongly correlated with predicted order.
Some PTMs Are Preferentially Found in IDPRs, Why?
Mechanistically, the prevalence of intrinsic disorder in the display sites of proteins targeted for certain PTMs is defined by the fact that structural pliability associated with high conformational dynamics of potential modification sites is crucial for the efficient action of a modifying enzyme on a multitude of different target proteins. In other words, such association between PTMs and IDPRs represents a solution for the apparent conundrum, where the modifying enzymes have to act following the ‘one-lock-many-keys’ scenario instead of the classical ‘lock-and-key’ mode of the enzyme action. The scale of the problem with too many ‘keys’ for a given ‘lock’ is given by kinases and phosphatases utilized in protein phosphorylation-dephosphorylation cycles. Protein kinases and phosphatases are among the largest gene families in eukaryotes [e.g., yeast and mouse genomes encode 119 and 540 kinases, respectively, and there are ∼520 (1019) kinase- and 150 (300) phosphatase-coding genes in human (or Arabidopsis thaliana) kinome]. However, in any given proteome, the number of proteins undergoing reversible phosphorylation is noticeably larger than the number of kinases and phosphatases. In fact, phosphorylation is expected to affect functions of at least one-third of eukaryotic proteins (Marks, 1996). In humans, more than two-thirds of the 21,000 proteins were shown to be phosphorylated, and it is likely that more than 90% are actually subjected to this type of PTM (Ardito et al., 2017). Based on the very conservative estimate of the penetrance of phosphorylation in human proteome (66.7%), each human kinase act on 27 target proteins, whereas each human phosphatase can dephosphorylate 93 clients. These numbers increase to 36 and 126 clients for each human kinase and phosphatase, respectively, if one would use the less conservative estimate of the prevalence of phosphorylation in human proteome (90%). However, the reality is even more complex, since many proteins are known to have multiple phosphorylation sites. Similar situation is observed for glycosylation (the covalent addition of carbohydrate moieties to specific amino acids), since almost half of all proteins typically expressed in a cell undergo this modification. Similarly, although the vast majority of human proteins end their lives via proteasomal degradation and although functionality and cellular localization of many proteins are regulated by ubiquitination–deubiquitination sites, the human genome codes for ∼600 E3 ubiquitinating ligases and 80 deubiquitinases (Komander et al., 2009).
Therefore, for each ‘lock’-containing enzyme (kinase, phosphatase, or any other modifying enzyme that targets multiple unrelated proteins) there are numerous ‘keys’ (modification sites of protein targets). This observation raises an important question on what allows these modifying enzymes to be engaged in the ‘one-lock-many-keys’ activity. An obvious solution is in relaxing classical ‘lock-and-key’ model by allowing structural flexibility. In fact, the problem is easily solved if instead of a ‘rigid key’ a modifiable protein is using a ‘flexible lock pick.’ In line with these consideration is a well-known fact that substrates are typically bound to the kinase rather weakly, despite the high specificity of the phosphorylation process. Such combination of low affinity and high specificity is a characteristic feature of signaling interactions (Bossemeyer et al., 1993; McDonald and Thornton, 1994; Hubbard, 1997; Lowe et al., 1997; Narayana et al., 1997; ter Haar et al., 2001), which are commonly based on intrinsic disorder (Dunker et al., 2001; Dunker et al., 2002a, 2005; Uversky et al., 2005). Often, such low affinity – high specificity protein-protein interactions rely on the coupled binding and folding of at least on of the partners. The corresponding disorder-to-order transition is characterized by a positive free energy change that reduces the magnitude of the negative free energy change associated with the interactions and defines the low binding affinity (Schulz, 1979).
Figure 4 further illustrates the mostly disordered nature of regions containing various PTM sites. Crystal structures of complexes between several modifying enzymes, such as kinase, phosphatase, glycosyltransferase, deacetylase, and methyltransferase, and peptides derived from their corresponding target proteins are shown (see Figures 4A–E, respectively). In all these cases, despite the obvious differences between proteins and peptides, the mechanism of interaction, where an extended peptide is bound to the grove of an enzyme, is remarkably similar. Figure 4A shows a crystal structure a 20-amino acid peptide derived from the heat stable protein kinase inhibitor (PKIα) bound to the catalytic subunit of cyclic AMP-dependent protein kinase (cAPK) (PDB ID: 2CPK). In its bound from, the PKIα peptide is characterized by a highly extended conformation. Although this extended bound form is stabilized by a well-developed network of 36 H-bonds, only two of these bonds are intramolecular, with 16 H-bonds being formed with cAPK and remaining 18 H-bonds being formed with water. A very similar situation is observed for complexes between the protein Ser/Thr phosphatase-1 and a 23 amino acid cell-permeable peptide (PDB ID: 4G9J, Figure 4B), or the polypeptide N-acetylgalactosaminyltransferase 2 and the 13 amino acid peptide EA2 (PDB ID: 2FFU, Figure 4C), or human mitochondrial NAD-dependent deacetylase sirtuin-3 and a 12 amino acid peptide derived from the mitochondrial acetyl-coenzyme A synthetase 2-like (PDB ID: 3GLT, Figure 4D), or the protein arginine N-methyltransferase 1 and a 19 amino acid substrate peptide (PDB ID: 1OR8, Figure 4E).
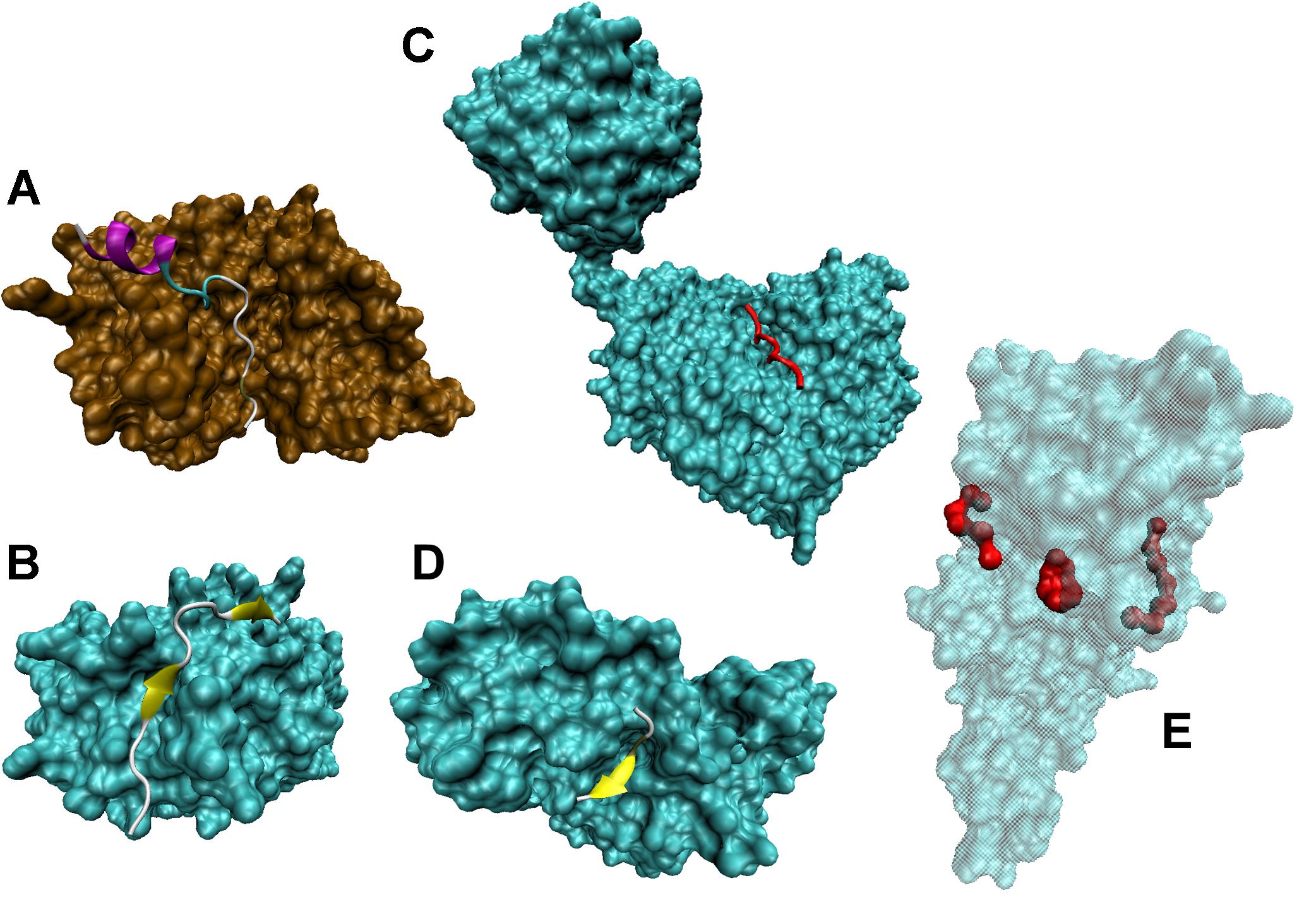
FIGURE 4. Disordered nature of the PTM sites in target proteins. (A) Crystal structure of a complex between a 20-amino acid peptide derived from the heat stable protein kinase inhibitor (PKIα) and the catalytic subunit of cyclic AMP-dependent protein kinase (cAPK) (PDB ID: 2CPK) (Knighton et al., 1991). (B) Crystal structure of a complex between a 23 amino acid cell-permeable peptide and a protein Ser/Thr phosphatase-1 (PDB ID: 4G9J) (Chatterjee J. et al., 2012). (C) Crystal structure of a complex between polypeptide N-acetylgalactosaminyltransferase 2 and a 13 amino acid peptide EA2 (PDB ID: 2FFU) (Abraham and Podell, 1981). (D) Crystal structure of a complex between human mitochondrial NAD-dependent deacetylase sirtuin-3 and a 12 amino acid peptide derived from mitochondrial acetyl-coenzyme A synthetase 2-like (PDB ID: 3GLT) (Jin et al., 2009). (E) Crystal structure of a complex between a protein arginine N-methyltransferase 1 and a 19 amino acid substrate peptide (PDB ID: 1OR8) (Zhang and Cheng, 2003).
Furthermore, systematic bioinformatics analyses of the peculiarities of the IDP/IDPR-located display sites targeted for PTMs and their adjacent regions showed that their sequence attributes (such as sequence complexity, charge, hydrophobicity, amino acid compositions, etc.) are very similar to those of IDPRs. For the first time, this observation was made for protein phosphorylation (Iakoucheva et al., 2004) and later similar trends were found for sites targeted for methylation (Daily et al., 2005), ubiquitination (Radivojac et al., 2010), S-palmitoylation (Reddy et al., 2017), as well as in protein regions that undergo multiple homologous or heterologous PTM events (Pejaver et al., 2014). This last observation is especially interesting, since it clearly indicates the importance of intrinsic disorder for the PTM-based regulation of proteins that occur not only through the individual effect of a given PTM at a single residue, but also through combined effects over multiple sites undergoing the same or different PTMs (Pejaver et al., 2014). These proteins affected by more than one PTM are commonly involved in transcriptional, posttranscriptional, and developmental processes contain multi-PTM or shared-PTM display sites that are characterized by preferences toward IDPRs exceeding those of the single-PTM sites (Pejaver et al., 2014). Furthermore, it was indicated that MoRFs possess significant preferences for PTM sites, particularly shared PTM sites, suggesting that PTMs play crucial roles in the modulation of this specific type of macromolecular recognition (Pejaver et al., 2014).
Also, the facts that >50% of all proteins are glycosylated (Apweiler et al., 1999; Ben-Dor et al., 2004), but only ∼5% of all PDB entries have attached glycan chains (Lutteke et al., 2004) indicate that glycosylated proteins are often disordered. In agreement with this hypothesis, an analysis of the complete proteomes of eight typical monocotyledonous and dicotyledonous plant species revealed that phosphorylation, acetylation, and O-glycosylation sites were preferentially located within the IDPRs of plant proteins (Kurotani et al., 2014). Similarly, a computational analysis of 20 algae proteomes revealed that phosphorylation, O-glycosylation, and ubiquitination sites, as well as PEST motifs [i.e., regions rich in proline, glutamic acid, serine, and threonine that serve as a signal for protein degradation (Rechsteiner and Rogers, 1996)] preferentially occurred in IDPRs (Kurotani and Sakurai, 2015).
Concluding, all these findings strongly suggest that many protein PTM sites are very commonly positioned within the IDPRs. Likely, this is because of the need of the corresponding modifying enzymes to work with a multitude of target sites in a wide variety of rather different protein targets (e.g., to utilize the ‘one-lock-many-keys’ mechanism). Disorder in flanking regions of such PTM sites provides a mean for a single modifying enzyme to bind and modify a wide variety of protein targets via a ‘flexible-lock-pick’ approach (Uversky, 2013b; Sirota et al., 2015).
Structural and Functional Consequences of PTMs in IDPs/IDPRs
Because PTMs are associated with the addition of various chemical groups to target protein, they clearly represent one of the means of altering of the energy landscape of a protein, thereby leading to conformational changes. Curiously, there is no uniform response of a protein structure to PTMs. It was pointed out that the outputs of PTMs are very diverse, ranging from local stabilization or destabilization of transient secondary structure to global disorder-to-order transitions (Bah and Forman-Kay, 2016). PTMs can also drive global changes in the protein phase states, for example, driving transitions between intrinsically disordered and ordered states of a protein molecule or between the dispersed monomeric and phase-separated states (Bah and Forman-Kay, 2016).
Based on the analysis of the effects of phosphorylation on structural properties of 17 proteins with available structural information for their phosphorylated and non-phosphorylated forms it has been concluded that the types and extent of structural changes could be highly diverse, ranging from local to long-range structural changes, leading to both association and disassociation of protein complexes, and causing both order-to-disorder and disorder-to-order transitions (Johnson and Lewis, 2001). Extension of this analysis to all proteins for which structures corresponding to their modified and unmodified forms are known revealed that PTMs typically induce small but statistically significant conformational changes at both local and global levels (Xin and Radivojac, 2012). A few illustrative examples of how PTMs might affect structure and functionality of individual IDPs/IDPRs are given below.
A systematic structural analysis revealed that different PTMs (phosphorylation and acetylation) have a profound effect on the conformational preferences of the intrinsically disordered negative regulatory domain (NRD) of the p53 tumor suppressor, thereby regulating activity of this important protein (McDowell et al., 2013). Phosphorylation induces structural changes in the p65 subunit of NF-κ B, allowing subsequent p65 ubiquitination and interaction with transcriptional cofactors (Milanovic et al., 2014). Phosphorylation of the ETS domain transcription factor Elk-1 by extracellular signal-regulated kinase (ERK) modulates the interaction of Elk-1 with Mediator and histone acetyltransferases (Galbraith et al., 2013). Phosphorylation of the Drosophila transcription factor Hox at multiple sites regulates interactions of this protein with DNA and other proteins, and plays a role in transcription activation (Tan et al., 2002; Bondos et al., 2006; Liu et al., 2008, 2009). Comparison of the solution structures of the phosphorylated and non-phosphorylated forms of the E7 protein from HPV-16 revealed that phosphorylation has significant local effect, changing structural and dynamic properties of the 26–29 region located in the close proximity to the retinoblastoma tumor suppressor (pRb) binding LXCXE motif (residues 22–26), thereby affecting the interactability of this protein (Nogueira et al., 2017). Phosphorylation of a member of the group-3 LEA protein family, PM18 protein, did not affect global intrinsically disordered status of this protein, but had profound effects on the salt-tolerance-related functions of this soybean protein (Liu et al., 2017). Combined experimental and computational analysis of the effect of phosphorylation on the conformational preferences of the synaptotagmin 1 IDPR revealed that phosphorylation of Thr112 resulted in the disruption of a local disorder-to-order transition likely due to the induction of salt bridges unsuitable for helix formation (Fealey et al., 2018).
Progressive acetylation has a cumulative effect on the intrinsically disordered tail of the H4 core histone, leading to the decrease in its conformational heterogeneity, combined with the increase in helical propensity and hydrogen bond occupancies, as well as with the formation of spatially clustered lysines that could serve as recognition patches for interaction with proteins engaged in chromatin regulation (Winogradoff et al., 2015). The aforementioned cumulative effects of acetylation were shown to be associated with the reduction in the protein net charge and the increase in hydrophobicity caused by the addition of the acetyl groups (Winogradoff et al., 2015). Multiple combinatorial PTMs of the C-terminal domain of RNA Polymerase II was shown to coordinate transcription with mRNA processing as well as regulate multiple stages of transcription initiation (Yogesha et al., 2014). Activity of dehydrin/response ABA protein is likely to be regulated by multiple PTMs, such as acetylation, amidation, glycosylation, methylation, myristoylation, nitrosylation, O-linked β-N-acetylglucosamination, palmitoylation, phosphorylation, sumoylation, sulfation, and ubiquitination (Kalemba and Litkowiec, 2015). It was pointed out that human proteins with multiple PTM sites (Mtp-proteins) contain more IDPRs than proteins carrying no known PTM sites (Huang et al., 2014). These Mtp-proteins were shown to be significantly enriched in protein complexes, have more protein partners, and prefer to act as hubs/superhubs in protein–protein interaction (PPI) networks than the proteins carrying no known PTM sites (Huang et al., 2014).
Intrinsic Disorder, PTMs, and Human Diseases
Various PTMs can control, modulate, and regulated functions of IDPs and IDPRs. Therefore, human diseases can be caused by aberrant PTMs. In agreement with this hypothesis, all major PTMs, such as acetylation, glycosylation, methylation, palmitoylation, phosphorylation, proteolytic degradation, and ubiquitination, can be altered in various human maladies, including cancer (Markiv et al., 2012), cardiovascular diseases, diabetes, and neurodegenerative diseases (Uversky, 2014). Systematic computational analysis revealed that ∼5% of the disease-associated mutations in human proteins may affect known PTM sites, with most of the 15 PTM types being found to be disrupted at levels higher than expected by chance (Li et al., 2010). Furthermore, the aforementioned Mtp-proteins were shown to be more prone to be involved in various human diseases than proteins carrying no known PTM sites (Huang et al., 2014).
Many malignancies [e.g., colorectal cancer (CRC) (Park and Lee, 2013)] are characterized by the abnormal glycosylation, which is commonly associated with the oncogenesis and cancer progression (Tuccillo et al., 2014). Many biomarkers used for diagnosis, prediction, and prognosis of various cancers are characterized by the aberrant N-linked glycosylation (Drake et al., 2006; Tian and Zhang, 2013; Hauselmann and Borsig, 2014). It was also shown that both gain and loss of phosphorylation target sites caused by the somatic mutations may play an active role in cancer pathogenesis (Radivojac et al., 2008). The distortion in the tightly controlled multiple modifications of the important nuclear IDPs, histones (Peng et al., 2012), is commonly found in malignancies (Campbell and Turner, 2013). For example, CRC is characterized by the abnormal acetylation and methylation of specific histone residues (Gargalionis et al., 2012), indicating the usefulness of the histone modification analysis for the diagnosis and prognosis in the CRC patients (Gezer and Holdenrieder, 2014). Alterations of the PTMs of lysine residues (such as methylation, acetylation, sumoylation, and ubiquitination) of proteins involved in DNA repair are frequently associated with genomic instability, which is the major cause of different diseases, especially cancers (Chatterjee S. et al., 2012). Normal and pathological activities of one of the high-mobility group transcription factors, the Sry-containing protein Sox2, are controlled by normal and pathological phosphorylation, acetylation, ubiquitination, methylation, and SUMOylation (Liu et al., 2013). Levels of a master transcriptional repressor REST/NRSF protein (RE-1 silencing transcription factor or neuron-restrictive silencer factor) that can serve as a tumor suppressor or oncogene are controlled by ubiquitination-deubiquitination cycles, with the abnormal upregulation of this protein being detected in glioblastoma, medulloblastoma, and neuroblastoma (Huang and Bao, 2012). Ectodomain shedding of syndecans, which is the proteolytic processing of cell-surface proteoglycans (PGs), is associated with the facilitation of cancer development. It also promotes cancer cell motility and invasion, thereby stimulating aggressiveness of various tumors (Theocharis et al., 2010).
In neurodegeneration, Huntington’s disease (HD) is characterized by aberrant acetylation, methylation, phosphorylation, polyamination, and ubiquitination of histones (Moumne et al., 2013). Furthermore, significant alterations in acetylation, palmitoylation, phosphorylation, proteolytic cleavage, sumoylation, and ubiquitination are reported for the HD causative protein, Huntingtin (Htt) (Ehrnhoefer et al., 2011). In Alzheimer’s disease (AD), levels and aggregation of the causative amyloid-β (Aβ) are affected by aberrant proteolytic cleavage of the amyloid precursor protein (APP). Pathogenesis of AD and other tauopathies is associated with altered sumoylation (Lee et al., 2013), abnormal hyperphosphorylation (Hernandez and Avila, 2007; Wang et al., 2013), and abnormal truncations of the microtubule-associated protein tau (Kovacech and Novak, 2010). The pathogenesis of frontotemporal lobar degeneration (FTLD) is associated with the aberrant phosphorylation of a RNA/DNA binding protein TDP-43 (TAR DNA binding protein 43) (Buratti and Baralle, 2008). In the transmissible spongiform encephalopathy (TSE) and other prion diseases, the infectious properties of the prion protein can be altered by changes in the glycosylation status of this protein (Cancellotti et al., 2013).
Acquired cardiac disorders, such as arrhythmias and heart failure, are associated with the aberrant functions of the voltage-gated sodium channel isoform 1.5 (NaV1.5) caused by its altered PTMs (Herren et al., 2013). The myofilament dysfunction in dilated cardiomyopathy (DCM) is associated with the aberrant phosphorylation of troponins I and T and myosin light chain, as well as with altered oxidation and glycation of sarcomeric proteins (LeWinter, 2005). Deregulated phosphorylation and glutathionylation of nitric oxide synthases NOS1 and NOS3 represent an important contributing factor in the pathogenesis of cardiac hypertrophy and failure, myocardial infarction, myocardial ischemia, reperfusion injury, and vascular disease (Carnicer et al., 2013).
In diabetes and hyperhomocysteinemia, the aberrant glycation of fibrinogen (Hammer et al., 1989; Henschen-Edman, 2001) is associated with the increased atherothrombotic risk (Hoffman, 2008). Also, prolonged increase in the O-GlcNAcylation and sustained increase in the O-GlcNAc (O-linked N-acetylglucosamine) levels are associated with the insulin resistance and glucose toxicity (McLarty et al., 2013). This is due to the distortions in the complex interplay between phosphorylation and O-GlcNAcylation (McLarty et al., 2013), since the rise in the GlcNAcylation levels can efficiently modulate the phosphate stoichiometry at most of the sites undergoing phosphorylation-dephosphorylation (Wang et al., 2008). Curiously, 381 proteins affected by these diabetes-distorted dual modifications (phosphorylation and O-GlcNAcylation) were shown to have a multitude of biological functions, acting as metabolic enzymes, cytoskeleton regulatory proteins, chaperones, kinases, RNA processing proteins, or transcription factors (Wang et al., 2008).
Concluding Remarks
Intrinsic disorder represents a common property found in proteins engaged in recognition, regulation, and control of various signaling events and pathways. Interactions with biological binding partners cause partial or complete folding of many IDPs/IDPRs. Due to the multiple binding specificities and mechanisms, these proteins can be involved in one-to-many and many-to-one interactions. Normally, functions and abundance of IDPs/IDPRs are tightly controlled and modulated by various means, including a wide spectrum of PTMs. Alteration of the mechanisms controlling IDP/IDPR functionality, localization, and cellular levels can be detrimental, causing various maladies. Often, protein-based pathogenicity originates from aberrant PTMs and altered degradation of IDPs/IDPRs. It seems that PTMs represent very important cellular mechanisms that affect the abundance, cellular distribution, foldability, or functionality of IDPs/IDPRs, with altered PTMs being commonly associated with pathological transformations of these important cellular players.
Author Contributions
VU conceived the idea. VU and AD collected and analyzed the literature data, and wrote and edited the manuscript.
Funding
This work was supported in part by National Institutes of Health (RF1AG055088 to VU).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abraham, G. N., and Podell, D. N. (1981). Pyroglutamic acid. Non-metabolic formation, function in proteins and peptides, and characteristics of the enzymes effecting its removal. Mol. Cell. Biochem. 38, 181–190. doi: 10.1007/BF00235695
Antz, C., Geyer, M., Fakler, B., Schott, M. K., Guy, H. R., Frank, R., et al. (1997). NMR structure of inactivation gates from mammalian voltage-dependent potassium channels. Nature 385, 272–275. doi: 10.1038/385272a0
Apweiler, R., Hermjakob, H., and Sharon, N. (1999). On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8. doi: 10.1016/S0304-4165(99)00165-8
Ardito, F., Giuliani, M., Perrone, D., Troiano, G., and Lo Muzio, L. (2017). The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 40, 271–280. doi: 10.3892/ijmm.2017.3036
Armstrong, C. M., and Bezanilla, F. (1977). Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 70, 567–590. doi: 10.1085/jgp.70.5.567
Arya, G., and Schlick, T. (2009). A tale of tails: how histone tails mediate chromatin compaction in different salt and linker histone environments. J. Phys. Chem. A 113, 4045–4059. doi: 10.1021/jp810375d
Bah, A., and Forman-Kay, J. D. (2016). Modulation of intrinsically disordered protein function by post-translational modifications. J. Biol. Chem. 291, 6696–6705. doi: 10.1074/jbc.R115.695056
Baumann, M., and Meri, S. (2004). Techniques for studying protein heterogeneity and post-translational modifications. Expert Rev. Proteomics 1, 207–217. doi: 10.1586/14789450.1.2.207
Beadle, G. W., and Ephrussi, B. (1936). The differentiation of eye pigments in Drosophila as studied by transplantation. Genetics 21, 225–247.
Beadle, G. W., and Tatum, E. L. (1941). Genetic control of biochemical reactions in Neurospora. Proc. Natl. Acad. Sci. U.S.A. 27, 499–506. doi: 10.1073/pnas.27.11.499
Ben-Dor, S., Esterman, N., Rubin, E., and Sharon, N. (2004). Biases and complex patterns in the residues flanking protein N-glycosylation sites. Glycobiology 14, 95–101. doi: 10.1093/glycob/cwh004
Bondos, S. E., Tan, X. X., and Matthews, K. S. (2006). Physical and genetic interactions link hox function with diverse transcription factors and cell signaling proteins. Mol. Cell. Proteomics 5, 824–834. doi: 10.1074/mcp.M500256-MCP200
Bossemeyer, D., Engh, R. A., Kinzel, V., Ponstingl, H., and Huber, R. (1993). Phosphotransferase and substrate binding mechanism of the cAMP-dependent protein kinase catalytic subunit from porcine heart as deduced from the 2.0 A structure of the complex with Mn2+ adenylyl imidodiphosphate and inhibitor peptide PKI(5-24). EMBO J. 12, 849–859.
Brown, C. J., Takayama, S., Campen, A. M., Vise, P., Marshall, T. W., Oldfield, C. J., et al. (2002). Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 55, 104–110. doi: 10.1007/s00239-001-2309-6
Brown, H. G., and Hoh, J. H. (1997). Entropic exclusion by neurofilament sidearms: a mechanism for maintaining interfilament spacing. Biochemistry 36, 15035–15040. doi: 10.1021/bi9721748
Buratti, E., and Baralle, F. E. (2008). Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 13, 867–878. doi: 10.2741/2727
Campbell, M. J., and Turner, B. M. (2013). Altered histone modifications in cancer. Adv. Exp. Med. Biol. 754, 81–107. doi: 10.1007/978-1-4419-9967-2_4
Cancellotti, E., Mahal, S. P., Somerville, R., Diack, A., Brown, D., Piccardo, P., et al. (2013). Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J. 32, 756–769. doi: 10.1038/emboj.2013.6
Carnicer, R., Crabtree, M. J., Sivakumaran, V., Casadei, B., and Kass, D. A. (2013). Nitric oxide synthases in heart failure. Antioxid. Redox Signal. 18, 1078–1099. doi: 10.1089/ars.2012.4824
Chatterjee, J., Beullens, M., Sukackaite, R., Qian, J., Lesage, B., Hart, D. J., et al. (2012). Development of a peptide that selectively activates protein phosphatase-1 in living cells. Angew. Chem. Int. Ed. Engl. 51, 10054–10059. doi: 10.1002/anie.201204308
Chatterjee, S., Senapati, P., and Kundu, T. K. (2012). Post-translational modifications of lysine in DNA-damage repair. Essays Biochem. 52, 93–111. doi: 10.1042/bse0520093
Cheng, Y., Oldfield, C. J., Meng, J., Romero, P., Uversky, V. N., and Dunker, A. K. (2007). Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 46, 13468–13477. doi: 10.1021/bi7012273
Cortese, M. S., Uversky, V. N., and Dunker, A. K. (2008). Intrinsic disorder in scaffold proteins: getting more from less. Prog. Biophys. Mol. Biol. 98, 85–106. doi: 10.1016/j.pbiomolbio.2008.05.007
Daily, K. M., Radivojac, P., and Dunker, A. K. (2005). “Intrinsic disorder and protein modifications: building an SVM predictor for methylation,” in Proceeding of the IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), San Diego, CA, 475–481.
DeForte, S., and Uversky, V. N. (2016). Resolving the ambiguity: making sense of intrinsic disorder when PDB structures disagree. Protein Sci. 25, 676–688. doi: 10.1002/pro.2864
Deribe, Y. L., Pawson, T., and Dikic, I. (2010). Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672. doi: 10.1038/nsmb.1842
Drake, R. R., Schwegler, E. E., Malik, G., Diaz, J., Block, T., Mehta, A., et al. (2006). Lectin capture strategies combined with mass spectrometry for the discovery of serum glycoprotein biomarkers. Mol. Cell. Proteomics 5, 1957–1967. doi: 10.1074/mcp.M600176-MCP200
Dunker, A. K., Brown, C. J., Lawson, J. D., Iakoucheva, L. M., and Obradovic, Z. (2002a). Intrinsic disorder and protein function. Biochemistry 41, 6573–6582. doi: 10.1021/bi012159+
Dunker, A. K., Brown, C. J., and Obradovic, Z. (2002b). Identification and functions of usefully disordered proteins. Adv. Protein Chem. 62, 25–49. doi: 10.1016/S0065-3233(02)62004-2
Dunker, A. K., Cortese, M. S., Romero, P., Iakoucheva, L. M., and Uversky, V. N. (2005). Flexible nets: the roles of intrinsic disorder in protein interaction networks. FEBS J. 272, 5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x
Dunker, A. K., Garner, E., Guilliot, S., Romero, P., Albrecht, K., Hart, J., et al. (1998). Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac. Symp. Biocomput. 473–484.
Dunker, A. K., Lawson, J. D., Brown, C. J., Williams, R. M., Romero, P., Oh, J. S., et al. (2001). Intrinsically disordered protein. J. Mol. Graph. Model. 19, 26–59. doi: 10.1016/S1093-3263(00)00138-8
Dunker, A. K., and Obradovic, Z. (2001). The protein trinity–linking function and disorder. Nat. Biotechnol. 19, 805–806. doi: 10.1038/nbt0901-805
Dunker, A. K., Obradovic, Z., Romero, P., Garner, E. C., and Brown, C. J. (2000). Intrinsic protein disorder in complete genomes. Genome Inform. Ser. Workshop Genome Inform. 11, 161–171.
Dunker, A. K., Obradovic, Z., Romero, P., Kissinger, C., and Villafranca, E. (1997). On the importance of being disordered. PDB Newsl. 81, 3–5.
Dunker, A. K., Oldfield, C. J., Meng, J., Romero, P., Yang, J. Y., Chen, J. W., et al. (2008a). The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics 9(Suppl. 2):S1. doi: 10.1186/1471-2164-9-S2-S1
Dunker, A. K., Silman, I., Uversky, V. N., and Sussman, J. L. (2008b). Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 18, 756–764. doi: 10.1016/j.sbi.2008.10.002
Dunker, A. K., and Uversky, V. N. (2008). Signal transduction via unstructured protein conduits. Nat. Chem. Biol. 4, 229–230. doi: 10.1038/nchembio0408-229
Dyson, H. J., and Wright, P. E. (2002). Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 12, 54–60. doi: 10.1016/S0959-440X(02)00289-0
Dyson, H. J., and Wright, P. E. (2005). Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208. doi: 10.1038/nrm1589
Ehrnhoefer, D. E., Sutton, L., and Hayden, M. R. (2011). Small changes, big impact: posttranslational modifications and function of huntingtin in Huntington disease. Neuroscientist 17, 475–492. doi: 10.1177/1073858410390378
Ellis, R. J. (2001). Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26, 597–604. doi: 10.1016/S0968-0004(01)01938-7
Ellis, R. J., and Minton, A. P. (2003). Cell biology: join the crowd. Nature 425, 27–28. doi: 10.1038/425027a
ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Farrah, T., Deutsch, E. W., Hoopmann, M. R., Hallows, J. L., Sun, Z., Huang, C. Y., et al. (2013). The state of the human proteome in 2012 as viewed through PeptideAtlas. J. Proteome Res. 12, 162–171. doi: 10.1021/pr301012j
Farrah, T., Deutsch, E. W., Omenn, G. S., Sun, Z., Watts, J. D., Yamamoto, T., et al. (2014). State of the human proteome in 2013 as viewed through PeptideAtlas: comparing the kidney, urine, and plasma proteomes for the biology- and disease-driven human proteome project. J. Proteome Res. 13, 60–75. doi: 10.1021/pr4010037
Fealey, M. E., Binder, B. P., Uversky, V. N., Hinderliter, A., and Thomas, D. D. (2018). Structural impact of phosphorylation and dielectric constant variation on synaptotagmin’s IDR. Biophys. J. 114, 550–561. doi: 10.1016/j.bpj.2017.12.013
Feng, Z. P., Keizer, D. W., Stevenson, R. A., Yao, S., Babon, J. J., Murphy, V. J., et al. (2005). Structure and inter-domain interactions of domain II from the blood-stage malarial protein, apical membrane antigen 1. J. Mol. Biol. 350, 641–656. doi: 10.1016/j.jmb.2005.05.011
Fink, A. L. (1995). Compact intermediate states in protein folding. Annu. Rev. Biophys. Biomol. Struct. 24, 495–522. doi: 10.1146/annurev.bb.24.060195.002431
Fisher, C. K., and Stultz, C. M. (2011). Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 21, 426–431. doi: 10.1016/j.sbi.2011.04.001
Fulton, A. B. (1982). How crowded is the cytoplasm? Cell 30, 345–347. doi: 10.1016/0092-8674(82)90231-8
Galbraith, M. D., Saxton, J., Li, L., Shelton, S. J., Zhang, H., Espinosa, J. M., et al. (2013). ERK phosphorylation of MED14 in promoter complexes during mitogen-induced gene activation by Elk-1. Nucleic Acids Res. 41, 10241–10253. doi: 10.1093/nar/gkt837
Gargalionis, A. N., Piperi, C., Adamopoulos, C., and Papavassiliou, A. G. (2012). Histone modifications as a pathogenic mechanism of colorectal tumorigenesis. Int. J. Biochem. Cell Biol. 44, 1276–1289. doi: 10.1016/j.biocel.2012.05.002
Gezer, U., and Holdenrieder, S. (2014). Post-translational histone modifications in circulating nucleosomes as new biomarkers in colorectal cancer. In Vivo 28, 287–292.
Hammer, M. R., John, P. N., Flynn, M. D., Bellingham, A. J., and Leslie, R. D. (1989). Glycated fibrinogen: a new index of short-term diabetic control. Ann. Clin. Biochem. 26(Pt 1), 58–62. doi: 10.1177/000456328902600108
Hauselmann, I., and Borsig, L. (2014). Altered tumor-cell glycosylation promotes metastasis. Front. Oncol. 4:28. doi: 10.3389/fonc.2014.00028
Henschen-Edman, A. H. (2001). Fibrinogen non-inherited heterogeneity and its relationship to function in health and disease. Ann. N. Y. Acad. Sci. 936, 580–593. doi: 10.1111/j.1749-6632.2001.tb03546.x
Hernandez, F., and Avila, J. (2007). Tauopathies. Cell. Mol. Life Sci. 64, 2219–2233. doi: 10.1007/s00018-007-7220-x
Herren, A. W., Bers, D. M., and Grandi, E. (2013). Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 305, H431–H445. doi: 10.1152/ajpheart.00306.2013
Hoffman, M. (2008). Alterations of fibrinogen structure in human disease. Cardiovasc. Hematol. Agents Med. Chem. 6, 206–211. doi: 10.2174/187152508784871981
Holub, P., Lalakova, J., Cerna, H., Pasulka, J., Sarazova, M., Hrazdilova, K., et al. (2012). Air2p is critical for the assembly and RNA-binding of the TRAMP complex and the KOW domain of Mtr4p is crucial for exosome activation. Nucleic Acids Res. 40, 5679–5693. doi: 10.1093/nar/gks223
Horowitz, N. H. (1995). One-gene-one-enzyme: remembering biochemical genetics. Protein Sci. 4, 1017–1019. doi: 10.1002/pro.5560040524
Horowitz, N. H., Bonner, D., Mitchell, H. K., Tatum, E. L., and Beadle, G. W. (1945). Genic control of biochemical reactions in Neurospora. Am. Nat. 79, 304–317. doi: 10.1086/281267
Hoshi, T., Zagotta, W. N., and Aldrich, R. W. (1990). Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250, 533–538. doi: 10.1126/science.2122519
Hsu, W. L., Oldfield, C., Meng, J., Huang, F., Xue, B., Uversky, V. N., et al. (2012). Intrinsic protein disorder and protein-protein interactions. Pac. Symp. Biocomput. 116–127.
Hsu, W. L., Oldfield, C. J., Xue, B., Meng, J., Huang, F., Romero, P., et al. (2013). Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 22, 258–273. doi: 10.1002/pro.2207
Huang, Q., Chang, J., Cheung, M. K., Nong, W., Li, L., Lee, M. T., et al. (2014). Human proteins with target sites of multiple post-translational modification types are more prone to be involved in disease. J. Proteome Res. 13, 2735–2748. doi: 10.1021/pr401019d
Huang, Z., and Bao, S. (2012). Ubiquitination and deubiquitination of REST and its roles in cancers. FEBS Lett. 586, 1602–1605. doi: 10.1016/j.febslet.2012.04.052
Hubbard, S. R. (1997). Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581. doi: 10.1093/emboj/16.18.5572
Iakoucheva, L. M., Brown, C. J., Lawson, J. D., Obradovic, Z., and Dunker, A. K. (2002). Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323, 573–584. doi: 10.1016/S0022-2836(02)00969-5
Iakoucheva, L. M., Radivojac, P., Brown, C. J., O’connor, T. R., Sikes, J. G., Obradovic, Z., et al. (2004). The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049. doi: 10.1093/nar/gkh253
Jahn, T. R., and Radford, S. E. (2005). The Yin and Yang of protein folding. FEBS J. 272, 5962–5970. doi: 10.1111/j.1742-4658.2005.05021.x
Jin, L., Wei, W., Jiang, Y., Peng, H., Cai, J., Mao, C., et al. (2009). Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 284, 24394–24405. doi: 10.1074/jbc.M109.014928
Johnson, L. N., and Lewis, R. J. (2001). Structural basis for control by phosphorylation. Chem. Rev. 101, 2209–2242. doi: 10.1021/cr000225s
Jungblut, P. R., Holzhutter, H. G., Apweiler, R., and Schluter, H. (2008). The speciation of the proteome. Chem. Cent. J. 2:16. doi: 10.1186/1752-153X-2-16
Kalemba, E. M., and Litkowiec, M. (2015). Functional characterization of a dehydrin protein from Fagus sylvatica seeds using experimental and in silico approaches. Plant Physiol. Biochem. 97, 246–254. doi: 10.1016/j.plaphy.2015.10.011
Khoury, G. A., Baliban, R. C., and Floudas, C. A. (2011). Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci. Rep. 1:90. doi: 10.1038/srep00090
Kim, M. S., Pinto, S. M., Getnet, D., Nirujogi, R. S., Manda, S. S., Chaerkady, R., et al. (2014). A draft map of the human proteome. Nature 509, 575–581. doi: 10.1038/nature13302
Kitahara, R., Yokoyama, S., and Akasaka, K. (2005). NMR snapshots of a fluctuating protein structure: ubiquitin at 30 bar-3 kbar. J. Mol. Biol. 347, 277–285. doi: 10.1016/j.jmb.2005.01.052
Knighton, D. R., Zheng, J. H., Ten Eyck, L. F., Ashford, V. A., Xuong, N. H., Taylor, S. S., et al. (1991). Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 407–414. doi: 10.1126/science.1862342
Komander, D., Clague, M. J., and Urbe, S. (2009). Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563. doi: 10.1038/nrm2731
Kovacech, B., and Novak, M. (2010). Tau truncation is a productive posttranslational modification of neurofibrillary degeneration in Alzheimer’s disease. Curr. Alzheimer Res. 7, 708–716. doi: 10.2174/156720510793611556
Kriwacki, R. W., Hengst, L., Tennant, L., Reed, S. I., and Wright, P. E. (1996). Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. U.S.A. 93, 11504–11509. doi: 10.1073/pnas.93.21.11504
Kurotani, A., and Sakurai, T. (2015). In silico analysis of correlations between protein disorder and post-translational modifications in algae. Int. J. Mol. Sci. 16, 19812–19835. doi: 10.3390/ijms160819812
Kurotani, A., Tokmakov, A. A., Kuroda, Y., Fukami, Y., Shinozaki, K., and Sakurai, T. (2014). Correlations between predicted protein disorder and post-translational modifications in plants. Bioinformatics 30, 1095–1103. doi: 10.1093/bioinformatics/btt762
Le Gall, T., Romero, P. R., Cortese, M. S., Uversky, V. N., and Dunker, A. K. (2007). Intrinsic disorder in the Protein Data Bank. J. Biomol. Struct. Dyn. 24, 325–342. doi: 10.1080/07391102.2007.10507123
Lee, L., Sakurai, M., Matsuzaki, S., Arancio, O., and Fraser, P. (2013). SUMO and Alzheimer’s disease. Neuromol. Med. 15, 720–736. doi: 10.1007/s12017-013-8257-7
LeWinter, M. M. (2005). Functional consequences of sarcomeric protein abnormalities in failing myocardium. Heart Fail. Rev. 10, 249–257. doi: 10.1007/s10741-005-5254-4
Li, S., Iakoucheva, L. M., Mooney, S. D., and Radivojac, P. (2010). Loss of post-translational modification sites in disease. Pac. Symp. Biocomput. 337–347.
Liebovitch, L. S., Selector, L. Y., and Kline, R. P. (1992). Statistical properties predicted by the ball and chain model of channel inactivation. Biophys. J. 63, 1579–1585. doi: 10.1016/S0006-3495(92)81732-0
Liu, K., Lin, B., Zhao, M., Yang, X., Chen, M., Gao, A., et al. (2013). The multiple roles for Sox2 in stem cell maintenance and tumorigenesis. Cell. Signal. 25, 1264–1271. doi: 10.1016/j.cellsig.2013.02.013
Liu, Y., Matthews, K. S., and Bondos, S. E. (2008). Multiple intrinsically disordered sequences alter DNA binding by the homeodomain of the Drosophila hox protein ultrabithorax. J. Biol. Chem. 283, 20874–20887. doi: 10.1074/jbc.M800375200
Liu, Y., Matthews, K. S., and Bondos, S. E. (2009). Internal regulatory interactions determine DNA binding specificity by a Hox transcription factor. J. Mol. Biol. 390, 760–774. doi: 10.1016/j.jmb.2009.05.059
Liu, Y., Yang, M., Cheng, H., Sun, N., Liu, S., Li, S., et al. (2017). The effect of phosphorylation on the salt-tolerance-related functions of the soybean protein PM18, a member of the group-3 LEA protein family. Biochim. Biophys. Acta 1865, 1291–1303. doi: 10.1016/j.bbapap.2017.08.020
Lowe, E. D., Noble, M. E., Skamnaki, V. T., Oikonomakos, N. G., Owen, D. J., and Johnson, L. N. (1997). The crystal structure of a phosphorylase kinase peptide substrate complex: kinase substrate recognition. EMBO J. 16, 6646–6658. doi: 10.1093/emboj/16.22.6646
Lutteke, T., Frank, M., and von der Lieth, C. W. (2004). Data mining the protein data bank: automatic detection and assignment of carbohydrate structures. Carbohydr. Res. 339, 1015–1020. doi: 10.1016/j.carres.2003.09.038
Mann, M., and Jensen, O. N. (2003). Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261. doi: 10.1038/nbt0303-255
Markiv, A., Rambaruth, N. D., and Dwek, M. V. (2012). Beyond the genome and proteome: targeting protein modifications in cancer. Curr. Opin. Pharmacol. 12, 408–413. doi: 10.1016/j.coph.2012.04.003
McDonald, I. K., and Thornton, J. M. (1994). Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238, 777–793. doi: 10.1006/jmbi.1994.1334
McDowell, C., Chen, J., and Chen, J. (2013). Potential conformational heterogeneity of p53 bound to S100B(betabeta). J. Mol. Biol. 425, 999–1010. doi: 10.1016/j.jmb.2013.01.001
McLarty, J. L., Marsh, S. A., and Chatham, J. C. (2013). Post-translational protein modification by O-linked N-acetyl-glucosamine: its role in mediating the adverse effects of diabetes on the heart. Life Sci. 92, 621–627. doi: 10.1016/j.lfs.2012.08.006
Mersfelder, E. L., and Parthun, M. R. (2006). The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res. 34, 2653–2662. doi: 10.1093/nar/gkl338
Milanovic, M., Kracht, M., and Schmitz, M. L. (2014). The cytokine-induced conformational switch of nuclear factor kappaB p65 is mediated by p65 phosphorylation. Biochem. J. 457, 401–413. doi: 10.1042/BJ20130780
Minton, A. P. (1997). Influence of excluded volume upon macromolecular structure and associations in ‘crowded’ media. Curr. Opin. Biotechnol. 8, 65–69. doi: 10.1016/S0958-1669(97)80159-0
Minton, A. P. (2000). Protein folding: thickening the broth. Curr. Biol. 10, R97—-99. doi: 10.1016/S0960-9822(00)00301-8
Mohan, A., Oldfield, C. J., Radivojac, P., Vacic, V., Cortese, M. S., Dunker, A. K., et al. (2006). Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 362, 1043–1059. doi: 10.1016/j.jmb.2006.07.087
Moumne, L., Betuing, S., and Caboche, J. (2013). Multiple aspects of gene dysregulation in Huntington’s disease. Front. Neurol. 4:127. doi: 10.3389/fneur.2013.00127
Mulvenna, J. P., Foley, F. M., and Craik, D. J. (2005). Discovery, structural determination, and putative processing of the precursor protein that produces the cyclic trypsin inhibitor sunflower trypsin inhibitor 1. J. Biol. Chem. 280, 32245–32253. doi: 10.1074/jbc.M506060200
Narayana, N., Cox, S., Shaltiel, S., Taylor, S. S., and Xuong, N. (1997). Crystal structure of a polyhistidine-tagged recombinant catalytic subunit of cAMP-dependent protein kinase complexed with the peptide inhibitor PKI(5-24) and adenosine. Biochemistry 36, 4438–4448. doi: 10.1021/bi961947+
Ngounou Wetie, A. G., Woods, A. G., and Darie, C. C. (2014). Mass spectrometric analysis of post-translational modifications (PTMs) and protein-protein interactions (PPIs). Adv. Exp. Med. Biol. 806, 205–235. doi: 10.1007/978-3-319-06068-2_9
Niklas, K. J., Bondos, S. E., Dunker, A. K., and Newman, S. A. (2015). Rethinking gene regulatory networks in light of alternative splicing, intrinsically disordered protein domains, and post-translational modifications. Front. Cell Dev. Biol. 3:8. doi: 10.3389/fcell.2015.00008
Nogueira, M. O., Hosek, T., Calcada, E. O., Castiglia, F., Massimi, P., Banks, L., et al. (2017). Monitoring HPV-16 E7 phosphorylation events. Virology 503, 70–75. doi: 10.1016/j.virol.2016.12.030
Oldfield, C. J., Cheng, Y., Cortese, M. S., Romero, P., Uversky, V. N., and Dunker, A. K. (2005). Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 44, 12454–12470. doi: 10.1021/bi050736e
Oldfield, C. J., Meng, J., Yang, J. Y., Yang, M. Q., Uversky, V. N., and Dunker, A. K. (2008). Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 9(Suppl. 1):S1. doi: 10.1186/1471-2164-9-S1-S1
Park, J. J., and Lee, M. (2013). Increasing the alpha 2, 6 sialylation of glycoproteins may contribute to metastatic spread and therapeutic resistance in colorectal cancer. Gut Liver 7, 629–641. doi: 10.5009/gnl.2013.7.6.629
Pejaver, V., Hsu, W. L., Xin, F., Dunker, A. K., Uversky, V. N., and Radivojac, P. (2014). The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 23, 1077–1093. doi: 10.1002/pro.2494
Peng, Z., Mizianty, M. J., Xue, B., Kurgan, L., and Uversky, V. N. (2012). More than just tails: intrinsic disorder in histone proteins. Mol. Biosyst. 8, 1886–1901. doi: 10.1039/c2mb25102g
Peng, Z., Yan, J., Fan, X., Mizianty, M. J., Xue, B., Wang, K., et al. (2014). Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 72, 137–151. doi: 10.1007/s00018-014-1661-9
Plaxco, K. W., and Gross, M. (1997). Cell biology. The importance of being unfolded. Nature 386, 657–659. doi: 10.1038/386657a0
Pontius, B. W. (1993). Close encounters: why unstructured, polymeric domains can increase rates of specific macromolecular association. Trends Biochem. Sci. 18, 181–186. doi: 10.1016/0968-0004(93)90111-Y
Ptitsyn, O. B. (1995). Molten globule and protein folding. Adv. Protein Chem. 47, 83–229. doi: 10.1016/S0065-3233(08)60546-X
Ptitsyn, O. B., Bychkova, V. E., and Uversky, V. N. (1995). Kinetic and equilibrium folding intermediates. Philos. Trans. R. Soc. Lond. B Biol. Sci. 348, 35–41. doi: 10.1098/rstb.1995.0043
Radford, S. E. (2000). Protein folding: progress made and promises ahead. Trends Biochem. Sci. 25, 611–618. doi: 10.1016/S0968-0004(00)01707-2
Radivojac, P., Baenziger, P. H., Kann, M. G., Mort, M. E., Hahn, M. W., and Mooney, S. D. (2008). Gain and loss of phosphorylation sites in human cancer. Bioinformatics 24, i241–i247. doi: 10.1093/bioinformatics/btn267
Radivojac, P., Iakoucheva, L. M., Oldfield, C. J., Obradovic, Z., Uversky, V. N., and Dunker, A. K. (2007). Intrinsic disorder and functional proteomics. Biophys. J. 92, 1439–1456. doi: 10.1529/biophysj.106.094045
Radivojac, P., Obradovic, Z., Smith, D. K., Zhu, G., Vucetic, S., Brown, C. J., et al. (2004). Protein flexibility and intrinsic disorder. Protein Sci. 13, 71–80. doi: 10.1110/ps.03128904
Radivojac, P., Vacic, V., Haynes, C., Cocklin, R. R., Mohan, A., Heyen, J. W., et al. (2010). Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78, 365–380. doi: 10.1002/prot.22555
Rechsteiner, M., and Rogers, S. W. (1996). PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 21, 267–271. doi: 10.1016/S0968-0004(96)10031-1
Reddy, K. D., Malipeddi, J., Deforte, S., Pejaver, V., Radivojac, P., Uversky, V. N., et al. (2017). Physicochemical sequence characteristics that influence S-palmitoylation propensity. J. Biomol. Struct. Dyn. 35, 2337–2350. doi: 10.1080/07391102.2016.1217275
Reddy, P. J., Ray, S., and Srivastava, S. (2015). The quest of the human proteome and the missing proteins: digging deeper. OMICS 19, 276–282. doi: 10.1089/omi.2015.0035
Rivas, G., Ferrone, F., and Herzfeld, J. (2004). Life in a crowded world. EMBO Rep. 5, 23–27. doi: 10.1038/sj.embor.7400056
Romero, P., Obradovic, Z., Kissinger, C. R., Villafranca, J. E., Garner, E., Guilliot, S., et al. (1998). Thousands of proteins likely to have long disordered regions. Pac. Symp. Biocomput. 437–448.
Romero, P., Obradovic, Z., Li, X., Garner, E. C., Brown, C. J., and Dunker, A. K. (2001). Sequence complexity of disordered protein. Proteins 42, 38–49. doi: 10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3
Ross, J. L. (2016). The dark matter of biology. Biophys. J. 111, 909–916. doi: 10.1016/j.bpj.2016.07.037
Schluter, H., Apweiler, R., Holzhutter, H. G., and Jungblut, P. R. (2009). Finding one’s way in proteomics: a protein species nomenclature. Chem. Cent. J. 3:11. doi: 10.1186/1752-153X-3-11
Schulz, G. E. (1979). “Nucleotide binding proteins,” in Molecular Mechanism of Biological Recognition, ed. M. Balaban (New York, NY: Elsevier), 79–94.
Shliaha, P. V., Baird, M. A., Nielsen, M. M., Gorshkov, V., Bowman, A. P., Kaszycki, J. L., et al. (2017). Characterization of complete histone tail proteoforms using differential ion mobility spectrometry. Anal. Chem. 89, 5461–5466. doi: 10.1021/acs.analchem.7b00379
Sirota, F. L., Maurer-Stroh, S., Eisenhaber, B., and Eisenhaber, F. (2015). Single-residue posttranslational modification sites at the N-terminus, C-terminus or in-between: to be or not to be exposed for enzyme access. Proteomics 15, 2525–2546. doi: 10.1002/pmic.201400633
Smith, L. M., Kelleher, N. L., and Consortium for Top Down Proteomics (2013). Proteoform: a single term describing protein complexity. Nat. Methods 10, 186–187. doi: 10.1038/nmeth.2369
Spolar, R. S., and Record, M. T. Jr. (1994). Coupling of local folding to site-specific binding of proteins to DNA. Science 263, 777–784. doi: 10.1126/science.8303294
Tan, X. X., Bondos, S., Li, L., and Matthews, K. S. (2002). Transcription activation by ultrabithorax Ib protein requires a predicted alpha-helical region. Biochemistry 41, 2774–2785. doi: 10.1021/bi011967y
ter Haar, E., Coll, J. T., Austen, D. A., Hsiao, H. M., Swenson, L., and Jain, J. (2001). Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 8, 593–596. doi: 10.1038/89624
Theocharis, A. D., Skandalis, S. S., Tzanakakis, G. N., and Karamanos, N. K. (2010). Proteoglycans in health and disease: novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 277, 3904–3923. doi: 10.1111/j.1742-4658.2010.07800.x
Tian, Y., and Zhang, H. (2013). Characterization of disease-associated N-linked glycoproteins. Proteomics 13, 504–511. doi: 10.1002/pmic.201200333
Tokmakov, A. A., Kurotani, A., Takagi, T., Toyama, M., Shirouzu, M., Fukami, Y., et al. (2012). Multiple post-translational modifications affect heterologous protein synthesis. J. Biol. Chem. 287, 27106–27116. doi: 10.1074/jbc.M112.366351
Tompa, P. (2002). Intrinsically unstructured proteins. Trends Biochem. Sci. 27, 527–533. doi: 10.1016/S0968-0004(02)02169-2
Tompa, P. (2005). The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 579, 3346–3354. doi: 10.1016/j.febslet.2005.03.072
Tompa, P., and Csermely, P. (2004). The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 18, 1169–1175. doi: 10.1096/fj.04-1584rev
Tompa, P., Fuxreiter, M., Oldfield, C. J., Simon, I., Dunker, A. K., and Uversky, V. N. (2009). Close encounters of the third kind: disordered domains and the interactions of proteins. Bioessays 31, 328–335. doi: 10.1002/bies.200800151
Tuccillo, F. M., de Laurentiis, A., Palmieri, C., Fiume, G., Bonelli, P., Borrelli, A., et al. (2014). Aberrant glycosylation as biomarker for cancer: focus on CD43. Biomed Res. Int. 2014:742831. doi: 10.1155/2014/742831
Turoverov, K. K., Kuznetsova, I. M., and Uversky, V. N. (2010). The protein kingdom extended: ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog. Biophys. Mol. Biol. 102, 73–84. doi: 10.1016/j.pbiomolbio.2010.01.003
Uhlen, M., Bjorling, E., Agaton, C., Szigyarto, C. A., Amini, B., Andersen, E., et al. (2005). A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 4, 1920–1932. doi: 10.1074/mcp.M500279-MCP200
Uversky, V. N. (2002a). Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11, 739–756.
Uversky, V. N. (2002b). What does it mean to be natively unfolded? Eur. J. Biochem. 269, 2–12. doi: 10.1046/j.0014-2956.2001.02649.x
Uversky, V. N. (2003). Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell. Mol. Life Sci. 60, 1852–1871. doi: 10.1007/s00018-003-3096-6
Uversky, V. N. (2010). The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010:568068. doi: 10.1155/2010/568068
Uversky, V. N. (2013a). A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 22, 693–724. doi: 10.1002/pro.2261
Uversky, V. N. (2013b). Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 19, 4191–4213.
Uversky, V. N. (2013c). Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 1834, 932–951. doi: 10.1016/j.bbapap.2012.12.008
Uversky, V. N. (2014). Wrecked regulation of intrinsically disordered proteins in diseases: pathogenicity of deregulated regulators. Front. Mol. Biosci. 1:6. doi: 10.3389/fmolb.2014.00006
Uversky, V. N. (2015). Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 282, 1182–1189. doi: 10.1111/febs.13202
Uversky, V. N. (2016a). Dancing protein clouds: the strange biology and chaotic physics of intrinsically disordered proteins. J. Biol. Chem. 291, 6681–6688. doi: 10.1074/jbc.R115.685859
Uversky, V. N. (2016b). (Intrinsically disordered) splice variants in the proteome: implications for novel drug discovery. Genes Genomics 38, 577–594. doi: 10.1007/s13258-015-0384-0
Uversky, V. N. (2016c). p53 proteoforms and intrinsic disorder: an illustration of the protein structure-function continuum concept. Int. J. Mol. Sci. 17:E1874.
Uversky, V. N., and Dunker, A. K. (2010). Understanding protein non-folding. Biochim. Biophys. Acta 1804, 1231–1264. doi: 10.1016/j.bbapap.2010.01.017
Uversky, V. N., and Dunker, A. K. (2013). The case for intrinsically disordered proteins playing contributory roles in molecular recognition without a stable 3D structure. F1000 Biol. Rep. 5:1. doi: 10.3410/B5-1
Uversky, V. N., Gillespie, J. R., and Fink, A. L. (2000). Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41, 415–427.
Uversky, V. N., Oldfield, C. J., and Dunker, A. K. (2005). Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 18, 343–384. doi: 10.1002/jmr.747
Uversky, V. N., Oldfield, C. J., and Dunker, A. K. (2008). Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 37, 215–246. doi: 10.1146/annurev.biophys.37.032807.125924
Uversky, V. N., and Ptitsyn, O. B. (1994). “Partly folded” state, a new equilibrium state of protein molecules: four-state guanidinium chloride-induced unfolding of beta-lactamase at low temperature. Biochemistry 33, 2782–2791. doi: 10.1021/bi00176a006
Uversky, V. N., and Ptitsyn, O. B. (1996). Further evidence on the equilibrium “pre-molten globule state”: four-state guanidinium chloride-induced unfolding of carbonic anhydrase B at low temperature. J. Mol. Biol. 255, 215–228. doi: 10.1006/jmbi.1996.0018
Vacic, V., Oldfield, C. J., Mohan, A., Radivojac, P., Cortese, M. S., Uversky, V. N., et al. (2007). Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 6, 2351–2366. doi: 10.1021/pr0701411
van den Berg, B., Ellis, R. J., and Dobson, C. M. (1999). Effects of macromolecular crowding on protein folding and aggregation. EMBO J. 18, 6927–6933. doi: 10.1093/emboj/18.24.6927
Vucetic, S., Xie, H., Iakoucheva, L. M., Oldfield, C. J., Dunker, A. K., Obradovic, Z., et al. (2007). Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J. Proteome Res. 6, 1899–1916. doi: 10.1021/pr060393m
Walsh, C. T., Garneau-Tsodikova, S., and Gatto, G. J. Jr. (2005). Protein posttranslational modifications: the chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 44, 7342–7372. doi: 10.1002/anie.200501023
Wang, J. Z., Xia, Y. Y., Grundke-Iqbal, I., and Iqbal, K. (2013). Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimers Dis. 33(Suppl. 1), S123–S139.
Wang, Z., Gucek, M., and Hart, G. W. (2008). Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. U.S.A. 105, 13793–13798. doi: 10.1073/pnas.0806216105
Ward, J. J., Sodhi, J. S., Mcguffin, L. J., Buxton, B. F., and Jones, D. T. (2004). Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337, 635–645. doi: 10.1016/j.jmb.2004.02.002
Winogradoff, D., Echeverria, I., Potoyan, D. A., and Papoian, G. A. (2015). The acetylation landscape of the H4 histone tail: disentangling the interplay between the specific and cumulative effects. J. Am. Chem. Soc. 137, 6245–6253. doi: 10.1021/jacs.5b00235
Witze, E. S., Old, W. M., Resing, K. A., and Ahn, N. G. (2007). Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 4, 798–806. doi: 10.1038/nmeth1100
Wright, P. E., and Dyson, H. J. (1999). Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331. doi: 10.1006/jmbi.1999.3110
Xie, H., Vucetic, S., Iakoucheva, L. M., Oldfield, C. J., Dunker, A. K., Obradovic, Z., et al. (2007a). Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J. Proteome Res. 6, 1917–1932.
Xie, H., Vucetic, S., Iakoucheva, L. M., Oldfield, C. J., Dunker, A. K., Uversky, V. N., et al. (2007b). Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J. Proteome Res. 6, 1882–1898. doi: 10.1021/pr060392u
Xin, F., and Radivojac, P. (2012). Post-translational modifications induce significant yet not extreme changes to protein structure. Bioinformatics 28, 2905–2913. doi: 10.1093/bioinformatics/bts541
Xue, B., Dunker, A. K., and Uversky, V. N. (2012). Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 30, 137–149. doi: 10.1080/07391102.2012.675145
Yang, X. J. (2005). Multisite protein modification and intramolecular signaling. Oncogene 24, 1653–1662. doi: 10.1038/sj.onc.1208173
Yogesha, S. D., Mayfield, J. E., and Zhang, Y. (2014). Cross-talk of phosphorylation and prolyl isomerization of the C-terminal domain of RNA Polymerase II. Molecules 19, 1481–1511. doi: 10.3390/molecules19021481
Zhang, X., and Cheng, X. (2003). Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 11, 509–520. doi: 10.1016/S0969-2126(03)00071-6
Zimmerman, S. B., and Minton, A. P. (1993). Macromolecular crowding: biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 22, 27–65. doi: 10.1146/annurev.bb.22.060193.000331
Zimmerman, S. B., and Trach, S. O. (1991). Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 222, 599–620. doi: 10.1016/0022-2836(91)90499-V
Keywords: intrinsically disordered proteins, intrinsically disordered protein regions, posstranslational modifications, multifunctional proteins, protein–protein interaction (PPI)
Citation: Darling AL and Uversky VN (2018) Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front. Genet. 9:158. doi: 10.3389/fgene.2018.00158
Received: 16 February 2018; Accepted: 17 April 2018;
Published: 04 May 2018.
Edited by:
Amit Kumar Yadav, Translational Health Science and Technology Institute, IndiaReviewed by:
Andreas Zanzoni, INSERM UMR1090 Technologies Avancées pour le Génome et la Clinique, FranceGeorges Nemer, American University of Beirut, Lebanon
Copyright © 2018 Darling and Uversky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vladimir N. Uversky, vuversky@health.usf.edu