 Beverly S. Rubin
Beverly S. Rubin Cheryl M. Schaeberle
Cheryl M. Schaeberle Ana M. Soto
Ana M. Soto- Department of Immunology, Tufts University School of Medicine, Boston, MA, United States
Since the inception of the term endocrine disruptor, the idea that the environment is an important determinant of phenotype has motivated researchers to explore the effect of low dose exposure to BPA during organogenesis. The syndrome observed was complex, affecting various endpoints such as reproduction and reproductive tissues, behavior, mammary gland development and carcinogenesis, glucose homeostasis, and obesity. This constellation of impacted endpoints suggests the possibility of complex interactions among the multiple effects of early BPA exposure. One key finding of our rodent studies was alterations of energy and amino-acid metabolism that were detected soon after birth and continued to be present at all time points examined through 6 months of age. The classical manifestations of obesity and associated elements of metabolic disease took a longer time to become apparent. Here we examine the validity of the often-mentioned lack of reproducibility of obesogenic effects of BPA, starting from the known environmental causes of variation, which are diverse and range from the theoretical like the individuation process and the non-monotonicity of the dose-response curve, to the very pragmatic like housing, feed, and time and route of exposure. We then explore environmental conditions that may hinder reproducibility and discuss the effect of confounding factors such as BPA-induced hyperactivity. In spite of all the potential sources of variation, we find that some obesogenic or metabolic effects of BPA are reproducibly observed when study conditions are analogous. We recommend that study authors describe details of their study conditions including the environment, husbandry, and feed. Finally, we show that when experimental conditions are strictly maintained, reproducibility, and stability of the obese phenotype is consistently observed.
Introduction
The incidence of obesity and associated elements of metabolic disease has increased rapidly during the last 30–40 years (1–3) reaching epidemic proportions in the industrialized world. This phenomenon has stimulated scientists to ponder about novel environmental factors that could contribute to this sudden change (4). These include chemicals referred to as “obesogens” (5), that inappropriately foster lipid accumulation and adipogenesis and lead to obesity. Bisphenol A (BPA) is just one compound on a growing list of possible obesogens; however, due to the ubiquitous nature of BPA exposure, it may represent an important compound among the putative contributors to the obesity epidemic. In fact, BPA was detected in the urine of over 92% of a representative sample of the US population (6), and positive associations have been found between urinary BPA levels and several adverse outcomes, including obesity and associated elements of metabolic disease (7–9). In addition to a putative obesogen, based on the data available, BPA may also be considered a metabolic disruptor (10) and a diabesogenic compound (11).
There has been some level of controversy regarding the effects of BPA exposure on obesity and metabolic disease. The purpose of this review is to delineate potential factors underlying seemingly discrepant outcomes in animal studies. We posit that these factors encompass differences like the species and strain, developmental stage at the time of BPA exposure, the ages at which animals were examined, as well as the doses tested, the routes of exposure, and the environment surrounding the animals. Of particular importance is the shape of the dose-response curve, which for endocrinology is often non-monotonic regardless of whether the hormonally active agent is a natural hormone or an endocrine disruptor. In addition, there are theoretical reasons that can account for different responses.
A Brief Theoretical Primer
In contrast to inert objects, living objects are not generic (interchangeable); they are individuals. Hence, what we measure as variation is the composite of an intrinsic and irreducible variation, since each animal is a unique individual, and a measurement error. Biologists address this problem by trying to make biological objects as “generic” as possible by using clonal cell lines and inbred strains, removing runts, culling litters, providing food of standardized composition, controlling the number of animals/cage, etc (12, 13). These procedures force a type of “invariance” or conservation into the system, allowing researchers to apply the mathematical tool of statistics. However, there is no mathematical theory of individuals, and thus biologists use statistics knowing that it has no way to contend with intrinsic variation. In addition, differences that are not statistically significant could be of utmost biological relevance. For example, Dolly the sheep was one clone that materialized from an enormous number of attempts. This also applies to the goat studied by Slijper that was born with paralysis of its front legs and soon learned to move by hopping on its hind legs. This behavioral accommodation resulted in dramatic morphological changes in bones of the hind legs and pelvis and changes in the morphology of the pelvic muscles (14) that were reminiscent of those present in bipeds. The mere fact that these singular results happen is sufficient to establish their relevance.
In this manuscript we analyze the effect of perinatal BPA exposure on adiposity and associated elements of metabolic disease and will show that when experimental conditions are kept constant to reduce variation (Table 1), the effect of BPA is reproducible. We also demonstrate that consideration of intrinsic individual variation allowed separation of two distinct phenotypes among females exposed perinatally to BPA (15).
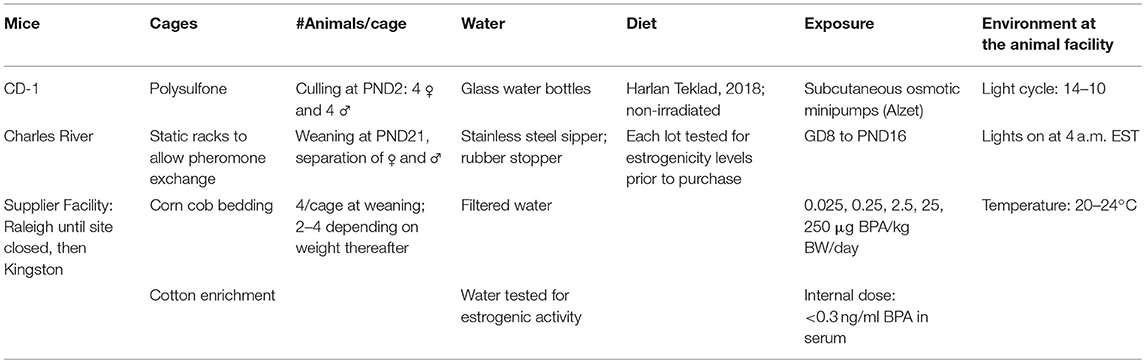
Table 1. Our experimental practices for consistent conditions.
BPA as an Obesogen: Our Laboratory
We initially chose BPA as the model endocrine disruptor for study in our lab because of its estrogenic activity and its structural resemblance to diethylstilbestrol (DES). Therefore, we expected to find alterations of the reproductive system and reproductive physiology in offspring exposed to BPA in utero and during lactation, as had been described in the DES-induced syndrome (16). We found reproductive alterations, but the earliest difference we noted between BPA-exposed and control offspring was on body weight (17), an effect that had been reported 2 years earlier in female mice by Howdeshell et al. (18) and was later reported in mice exposed neonatally to DES (19).
Body Weight and Adiposity
In the reproductive study mentioned above, we noticed differences in body weight in Sprague Dawley rats born to mothers that received BPA in their drinking water from gestational day 6 through weaning (17). Based on water consumption measurements, the exposure to the dams was estimated to be ~0.1 and 1.2 mg BPA/kg BW/day. The increase in body weight was modest, but significant. A similar study was performed by Somm et al. (20) using the same rat strain and only the lower BPA dose in drinking water. Both male and female BPA-exposed offspring were heavier at birth, and the females remained heavier through the termination of the study at 14 weeks. Increased perigonadal white adipose tissue weight and increased expression of adipogenic and lipogenic genes were observed in the females demonstrating that BPA exposure during gestation and lactation increased adipose storage and adipogenesis in a sex specific manner. Both BPA-exposed male and female offspring had increased body weights relative to controls when fed a high fat diet (HFD).
Subsequent studies in our lab using outbred CD-1 mice also examined the effects of perinatal BPA exposure on female reproduction and reproductive tissues (21–25). As in our previous rat study, we could not help but notice the increase in body weight in our BPA exposed mice. Also, when performing ovariectomies, we noted increased adiposity and ovarian fat pad size in the females born to mothers exposed to low levels of BPA. The BPA exposure for these studies was provided via osmotic minipumps that were implanted subcutaneously into pregnant females on GD-8 and released BPA through postnatal day 16; they provided continuous delivery of low levels of BPA (ranging from 0.025 μg/kg to 250 μg/kg BW/day) with great precision. Levels of unconjugated BPA in blood samples were below the detectability of the BPA assay (0.3 ng/ml) at all doses tested (15), and thus they are below or within human levels of exposure as measured in serum or plasma by analytical chemistry [adults: a range of 0–1 ng/ml [reviewed in Vandenberg et al. 26]; umbilical cord blood: median = 1.03 ng/ml (27).
In later studies undertaken to examine the effects of perinatal BPA exposure on body weight and adiposity, we continued to observe increased body weight and fat pad weights, adipocyte hypertrophy, and an increased number of white adipocytes in the intrascapular brown fat depot. Most recently we reported increased body weight and fat mass measured by echoMRI in male and female mice exposed perinatally to BPA. This outcome was exacerbated in females exposed to BPA both perinatally and peripubertally, particularly when exposures were 2.5 and 25 μg BPA/kg BW/day (15). The additional peripubertal exposure increased insulin resistance, fat mass, BW, and inflammation in females in a dose dependent manner. Although the males showed significant increases in body weight and fat mass with perinatal BPA exposure, they did not show increased detrimental effects with the additional peripubertal exposure. This suggests that the specific extended period of BPA treatment was more damaging in females, or that females were more sensitive to harmful metabolic effects of BPA during the peripubertal window of exposure. Evidence of inflammation of fat pads was observed in BPA exposed male and female mice. Also observed was lipid accumulation in liver (15, 28) and increased serum leptin levels (15).
Metabolome Data
Metabolomics is the measurement of the set of small molecules (metabolites) that result from metabolism (29). The integration of metabolomics and physiopathological studies provides valuable information for understanding endocrine disruption and is expected to produce useful data for risk assessment of endocrine disruptors such as BPA. Metabolic fingerprints based on nuclear magnetic resonance (NMR) spectroscopy, can detect slight changes in the metabolome of cells, tissues, or organisms exposed to endocrine disruptors (EDs) opening the way to examine whether exposure to an ED results in global alterations of metabolism, whether these changes persist after cessation of exposure, and whether further changes arise in aging. Additionally, and in contrast with other omics, the interpretation of metabolomics is straightforward as most metabolic pathways are known, and metabolic network analysis can be used to understand how a given set of metabolites in a metabolomic profile are linked (30).
Metabolomics was used in our studies to examine global metabolites in our BPA exposed mice and rats (31, 32). In all cases, at all ages studied, metabolomic profiles differed in BPA exposed and control animals. Our first metabolome study involved male mice exposed to 0.025, 0.25, or 25 μg BPA/kg body weight/day from GD8 to PND16 (32). Aqueous extracts of PND2 animals and of livers, brains, and serum samples from PND21 animals were examined by 1H nuclear magnetic resonance spectroscopy. Endogenous metabolic fingerprints revealed changes in the global metabolism in PND2 extracts. Consistent with the findings of Alonso-Magdalena et al. (33) demonstrating that BPA exposure during fetal development in mice leads to disruption of glucose homeostasis that manifested at 6-months of age, the metabolomic profile in our study revealed that glucose was already affected by perinatal exposure to BPA at PND2. Additional alterations at PND2 included variations in pyruvate, amino acids, and neurotransmitters. Regarding PND21, statistical analysis successfully discriminated among treatment groups in liver, serum, and brain samples. Variations in glucose, pyruvate, amino acids, and neurotransmitters (γ-aminobutyric acid and glutamate) were identified. The fact that these alterations occurred well before the manifestation of the classical pathophysiological signs of metabolic disease suggests that metabolic pathways are an early target of BPA, rather than a consequence of obesity. Studies in female rats, also revealed alterations of energy, and amino-acid metabolism in the BPA exposed animals at all ages examined (31).
Factors That Can Influence Effects of BPA Exposure on Body Weight and Adiposity
Although our studies in CD-1 mice have consistently suggested that BPA is an obesogen, there is considerable controversy in the literature that has caused many to question whether there is adequate evidence of the obesogenic properties of BPA. Several animal studies have noted increased BW and/or adiposity in their early BPA exposure models (15, 17, 18, 20, 34–43) while other studies have reported a decrease in BW and/or adiposity (40, 44–47) or no change (48, 49). Reviews of the data from human studies have also generated some controversy regarding the association between BPA exposure and obesity and metabolic disease (50–52). It is clear from the mouse studies in our lab that the effects of perinatal exposure to BPA on body weight, and fat mass, and associated parameters of metabolic disease are sex-dependent, dose dependent, and influenced by the precise window of exposure as well as the time of examination.
Route of exposure, diet, and housing conditions as well as other environmental factors undoubtedly play a role in the manifestation of increased adiposity and could potentially mask effects of BPA exposure. For example, accidental BPA exposure from damaged polycarbonate cages (53) led to meiotic disturbances including aneuploidy in oocytes in the mouse colony maintained by Pat Hunt. This finding demonstrates how external conditions could interfere with the ability to detect the effects of early BPA exposure on BW and adiposity or any other measurements. In order to facilitate comparisons of data from different laboratories, the factors mentioned above should be included in materials and methods (Table 2). Our practices for keeping the living conditions of our animals as constant as possible are summarized in Table 1.

Table 2. Conditions that should be made explicit in Materials and Methods sections of research papers.
Gender
There are several reports of increased adipogenesis in BPA exposed females relative to males particularly at young ages (20, 43). We initially noticed increased body weight and fat mass in our CD-1 mice with females showing more of an increase than males; however, that finding was influenced by changes in the environment and in housing conditions. For example, when males were singly housed to facilitate measurements of food intake and metabolism, the male data showed less variability allowing a clearer pattern to emerge. Whether the decrease in variability was influenced by the absence of a dominance hierarchy typically observed in group housed males, remains to be determined. In contrast to the males, when females were individually housed at 8 weeks of age, many of the BPA exposed females showed a tendency to lose weight. Overall compilation of recent data revealed that increased body weight and fat mass were more consistently apparent in perinatally exposed male than female CD-1 mice. This finding may be attributed, in part, to the hyperactivity observed in a subset of our BPA exposed females that represents a strong confounding factor when studying body weight and body composition and will be discussed below (15).
A recent study by Desai et al. (42), reported significant increases in BW and fat mass at 3 and 24 weeks of age in male but not female Sprague Dawley rats born to mothers exposed to BPA in their drinking water from 2 weeks prior to mating through pregnancy and lactation (5 mg BPA/liter which provided 500–900 μg/kg BW/day during pregnancy and ~1500 μg/kg BW/day during lactation). Further study of the BPA exposed males at 3 weeks of age revealed an increase in adipocyte size, lipogenic factors (SrebP1 and C/EBP alpha), and inflammation in the retroperitoneal fat pad. Sex also has a large impact on the effects of early BPA exposure on elements of metabolic disease. This is particularly apparent regarding the regulation of glucose/insulin homeostasis which has been extensively documented by Nadal and colleagues (54–56). They have clearly shown effects of gestational exposure to BPA on glucose regulation in male offspring that is not apparent in their female siblings at the times examined (33). Wistar rat offspring born to mothers exposed to 50 μg BPA/kg BW/day from GD0–PND 21 (by oral gavage) showed increased body weight on a chow diet. When fed a high fat diet (HFD), the body weight of males was increased before females (9 weeks vs. 15 weeks). This pattern was also seen in serum insulin levels that were increased at 15 weeks in males and 26 weeks in females (34).
Hyperactivity
As mentioned above, increased activity has been reported in rodents exposed perinatally to BPA (15, 40, 44). Hyperactivity represents a serious confound to the assessment of increased body weight and adiposity and metabolic parameters in BPA exposed offspring. Although others have reported hyperactivity in both BPA- exposed males and females (57–60), in our studies, hyperactivity was more extreme and far more prevalent in BPA exposed females. A subset of females showed flipping or running behavior; both are repetitive behaviors that served no obvious function but were persistent in the affected animals (15). They remained lean despite a significant increase in food intake. If their data were included with the other females, no differences were noted in mean BW and mean estimates of adiposity as there was too much variability to allow statistically significant findings. If the data from the flippers were eliminated as outliers, the remaining data showed a pattern of increased body weight and adiposity in the non-affected females (15). It will be important to identify the factors involved in causing hyperactivity in a subset of the BPA exposed female offspring and to determine how widespread this phenomenon may be in other laboratories. Of interest, in their studies of C57Bl6 mice, van Esterik et al. reported increased body weights in their BPA exposed males and not their females although they did mention increased activity in their females (40). Similarly, Anderson et al. (44), reported decreased body weight and adiposity in their BPA exposed Avy mice that was most pronounced in females, and when their movements were tracked, the females showed clear evidence of hyperactivity (44). In a study by Ryan et al. (49), when examined at 9–14 weeks of age, perinatally BPA exposed female CD-1 mice (but not their male siblings) consumed more calories and had less body fat than control females. These data would be consistent with increased activity levels or increased energy expenditure in the BPA exposed females. Hyperactivity was not measured in this study, and it can easily be overlooked as the hours of observation typically fall during the light cycle when the animals are sleeping.
Early BPA exposure has been found to alter cortical organization (61) and change dopaminergic projections in the neocortex which may play a role in activity changes. In our studies it is not clear why a subset of the females with identical BPA treatments show pronounced hyperactivity while others including their female siblings do not. This is an important observation to pursue and understand, particularly when one is attempting to study obesity and associated metabolic disease as it will have profound effects on the interpretation of the compiled data. Consistent with the results in rodents discussed above, it should be noted that there is a growing body of evidence suggesting an association between urinary BPA levels and increased activity in children (57, 62, 63).
Diet
We have routinely used a soy protein-based diet for our studies (Harlan-Teklad 2018). The estrogenicity of all food batches are tested in our laboratory prior to purchase using the ESCREEN assay (64). Only those found to have minimal estrogenic activity (<20 pmol of estrogen equivalents per gram) were purchased. Based on evaluation of available studies in the literature, casein based diets appear to be less likely to show the effects of BPA on several parameters (65) including body weight and adiposity (44, 48, 49, 66). Increased body weight and fat mass in BPA exposed rodents tended to be observed in animals eating a soy-based diet and not necessarily in a casein diet (44, 48, 49, 66). Ruhlen et al. have reported that a casein based diet elevated body weights of control animals and could therefore mask the effects of BPA on BW and adiposity in mice (65). In contrast, Cao et al. have reported that in their study in rats, it was soy and not BPA that caused increased body weight (48). However, in our studies with soy-based chow, we saw consistent effects of BPA. Unfortunately, many studies in the literature fail to provide information on diet; these omissions make it difficult to compare data between studies to formulate accurate conclusions.
Exposure Level: Low Doses, Monotonic and Non-monotonic Dose Response Curves (NMDRCs)
Exposure level is very important when studying the effects of early BPA exposure. BPA does not show a monotonic dose response curve for many endpoints including effects on BW and adiposity. The importance of the BPA dose on body weight and adiposity is clearly illustrated in a study by Wei et al. in rats (34). In that study, the lowest BPA dose administered by oral gavage through gestation and lactation (50 μg/kg BW/day, the current EPA reference dose) was obesogenic and predisposed the animals to elements of metabolic disease. The two higher doses (250 and 1250 μg/kg BW/day) did not produce these effects. Therefore, if only the higher doses had been examined, the report would conclude that BPA had no effect on BW, adiposity, and associated elements of metabolic syndrome. In another study in male CD-1 mice, animals were provided with 1 of 5 doses of BPA during the period of preadipocyte differentiation (GD9-GD 18) ranging from 5 μg to 5,000 μg/kg BW/day (35). The lower doses were more likely to increase BW and adiposity including adipocyte number and adipocyte size. In fact, no significant effects on these parameters were noted at the highest dose. Of interest, the studies of Tyl in mice (46) and in rats (47) revealed a very significant decrease in body weights in males and females exposed to the highest levels of BPA (500 mg or 600 mg/kg BW/day) during development. These levels were extremely high relative to the current EPA reference dose (67) and would not be considered environmentally relevant exposure levels. The toxicological model that presumes that the highest doses will provide the greatest effects is not typically applicable to endocrine disruptors or natural hormones.
A dose-response curve plots the intensity of a given effect over a range of doses examined. A monotonic response is characterized by a slope that does not change sign. In contrast, a non-monotonic dose response curve is characterized by a slope that changes sign within the dose-range tested. Some curves are U-shaped, others have an inverted U-shape and in others, the sign of the curve may change in multiple points along the range of doses examined. Non-monotonic dose-response curves are frequently seen in endocrinology. They range from proliferative effects observed in cell culture studies to whole organ, organismal effects and are even observed in epidemiological studies (68, 69).
Some processes leading to non-monotonicity have been identified in cell culture. Natural hormones like estrogens (70) and androgens (71–73) affect proliferation in a biphasic dose-response. At low doses, for example, sex steroids induce cell proliferation by neutralizing a plasma-borne inhibitor. At high doses, sex steroids block cell proliferation by inducing intracellular inhibitors (74) or by inducing cell death (75).
Natural hormones and endocrine disruptors also display non-monotonic dose-response curves when assessed in vivo. The vom Saal group observed non monotonic effects of estradiol and DES on prostate weight (76). Our group demonstrated that estradiol produces a monophasic dose response curve when the endpoint measured is gene induction or the expression of a protein. In contrast, at higher levels of organization the uterus displays a monotonic dose–response whereas the mammary gland exhibits a non-monotonic response to increasing levels of estradiol (77). From a theoretical point of view, monotonic phenomena can be easily modeled due to their simplicity, by assuming that each step in a linear pathway behaves according to the law of mass action (78); thus, high doses will give predictable high effects. On the contrary, modeling a non-monotonic effect is not possible without first obtaining results from experiments using a complex experimental design to separate the components of such NMDRCs.
Exposure Window
Exposure windows must be considered when reviewing data on early exposure to BPA. Liu et al. published a study in C57Bl6 mice (38) in which the BPA exposure windows were dissected to include the following time intervals: GD 1-6; GD6-PND0; PND0-PND21; and GD6-PND21. Gestational exposure from GD6 to birth resulted in decreased body weights in males and females, and the postnatal exposure from birth to weaning resulted in a significant increase in body weight in the males. In contrast, for glucose tolerance and for insulin tolerance, the most consistent and persistent problem was seen in animals exposed from GD6 to PND0. This study delineates distinct differences in the exposure windows that affect BW and glucose homeostasis. These windows are reminiscent of the work of Cederroth and Nef (79) who observed that the beneficial effects of dietary soy and phytoestrogens on adiposity were noted only in postnatally treated CD-1 mice whereas improvements in glucose tolerance were restricted to animals that had fetal exposure.
When considering obesity and metabolic disease, it is important to realize that the metabolic circuits for food intake and metabolism in the hypothalamus are established primarily after birth. Therefore, neonatal BPA exposure could influence the development of these important circuits. The exquisitely timed postnatal leptin surge is essential for the proper development of neural circuits for food intake and metabolism (80), and MacKay et al. have provided evidence that peak leptin secretion is delayed in mice exposed perinatally to BPA (81). Disruption to the timing of the leptin surge impaired development of the melanocortin system resulting in alterations in the hypothalamic feeding circuitry that could contribute to metabolic disturbances in adulthood.
Route of Exposure
The route of exposure to BPA may have a significant effect on the outcomes observed. Although diet is considered the main source of BPA exposure in humans and ingestion the main route of intake, it is not the only source of exposure (82, 83). BPA exposure can occur through food and water, dermal absorption as occurs with BPA-laden register receipts (84–87), and via inhalation of air and dust. Moreover, BPA levels remain measurable in fasting individuals (88) suggesting the potential for non-oral sources of exposure, longer half- life, or storage in the body that differs from the idea of a relatively rapid clearance rate for this compound following ingestion. Of interest, urinary BPA excretion was elevated 84% in volunteers who handled thermal receipts and the delayed return to normal pre-exposure levels suggested a potential difference in bioavailability of BPA following dermal absorption relative to oral exposure (89). Some suggest that the non-oral exposures, although they are considered to occur at significantly lower levels, may be more potent as they avoid the first pass metabolism of BPA by conjugation and therefore dermal sources may have far higher toxicological relevance than expected (83). In a controlled experiment, handling of cash register receipt paper plus still wet hand sanitizer elevated unconjugated BPA serum levels (84) to those found after oral exposure to 50 μg/kg BW (90). Dermal contact may be the main contributor when both unconjugated and conjugated BPA are considered (82).
Exposure routes for BPA differ widely in the numerous studies reported. Whether animals receive their BPA once a day as a bolus or throughout the day could conceivably alter the impact of the same daily dose. Studies that use a single daily dose have provided BPA in a single subcutaneous injection, or by oral gavage, or less intrusively, in a drop of oil on the tongue or dispensed on a cookie. Although most studies aim to provide BPA levels in the range of human exposures, humans are not exposed to a single daily dose of BPA. In contrast, other studies provide BPA throughout the day by incorporating it into the food or the drinking water or via silastic capsules or osmotic minipumps. Although they may afford the same daily dose as that provided by bolus delivery, these more continuous or intermittent exposure paradigms could have a different effect on the animal (91, 92). In most of our studies, animals were exposed to continuous low levels of BPA via osmotic minipumps. This method allowed us to provide very low BPA doses (beginning at 0.025 μg BPA/kg BW/day) with a precision that would not be technically possible in other non-bolus dosing regimens.
In one study that compared subcutaneous and oral exposure of BPA, neonatal male rats (3-day old) received 10 μg BPA/kg BW in oil. The same preparation was administered as a subcutaneous injection or as an oral bolus. As expected, the maximal serum concentration of free and total BPA was higher with subcutaneous administration (91). A second study compared an oral bolus of BPA with administration via diet in mice. The authors reported different patterns of BPA concentration. Bolus administration was found to peak faster (1 h) than administration through food (6 h); however, bioavailability in the form of unconjugated BPA was higher when administered in food (92). Therefore, the route of administration may have significant effects on study outcomes.
Age of Outcome Measurement
Many effects of early BPA exposure do not manifest until later in life while others are apparent early in life. For example in the study by Somm (20), both male and female pups exposed to BPA in utero were heavier at birth than controls; however, at PND 21, only the females remained significantly heavier. Both males and females were heavier than controls when fed a high fat diet. Ryan et al. (49) found that males and females were heavier at weaning, but the increase in BW did not persist through week 14 when the study was terminated Several studies end at weaning or soon after and report no differences in BW or adiposity (45), but it is possible that such findings might have been observed in adulthood if they were examined. Malaise et al. reported (93) that relative to controls, mice exposed perinatally to BPA (50 μg/kg BW/day) were significantly leaner at PND 25, and they had decreased gonadal fat at PND 45. However, additional measurements from PND 70 to the end of the study at PND 170 revealed a significant increase in body weight and adiposity in BPA exposed mice relative to controls. Similarly in the study by Wei et al. (34), rats exposed perinatally to BPA (50 μg/kg BW/day) showed significant increases in body weight on a standard chow diet beginning at 17–19 weeks of age.
In Summary
The purpose of this article was to examine the controversy surrounding BPA as an obesogen. To address this issue, we focused on obesity and adiposity and only peripherally touched on its metabolic effects on glucose homeostasis and lipid metabolism, as these subjects are addressed by other articles in this issue. To assess the controversy, we focused on perinatal exposures in rodents since our own data from this window of exposure could be used to illustrate consistent results over 10 years of attempting to maintain a constant environment. Moreover, we found that departing from these stringent conditions resulted in measurable changes.
If one examines the existing publications on exposure to a relatively “low dose” of BPA during a window that includes gestational and lactational exposure in rodents, most references show some indication of increased BW and/or increased adiposity and/or elements of obesity-associated metabolic disease. This is true even though the endpoints collected, and the windows of observation may be diverse. Some papers not showing such an outcome reported potential confounds that could preclude increased body weight and adiposity such as hyperactivity, a feature that may go undetected unless animals are observed close to the start of the dark cycle. We also found that the trajectory of increased body weight and adiposity was more clearly defined when males were singly housed rather than group housed. Diet appears to be another contributor to phenotype. A review of the literature suggests that animals exposed to a soy-based diet were more likely to reveal the obesogenic properties of BPA than those fed a casein-based diet. Moreover, in some studies, obesogenic properties of BPA were enhanced or brought out by feeding a HFD. From our overall analysis, we posit that these interactions may explain the lack of concordance among apparently similar experiments, pointing to the need for investigators to publish detailed descriptions of housing, diet, caging materials, culling practices, and other crucial methodological details that are often omitted from the materials and methods (Table 2).
One striking result from our metabolomic studies is that as early as 2 days after birth there are clear changes in amino-acid and energy metabolism, including glucose in the BPA exposed animals. These alterations precede the clinical manifestations of obesity and associated elements of metabolic disease including altered glucose homeostasis. Differences in the metabolic fingerprints between control and BPA exposed offspring persist through life. The precocity of altered metabolomic profiles suggest that they may play a causal role in the manifestation of elements of metabolic disease in adulthood.
The shape of the dose response curve may sometimes explain divergence between two studies, because non-monotonicity is a frequent feature of BPA effects. If the effect studied has a non-monotonic dose response curve, and only one or two BPA doses are tested, it is likely that one or more of the multiple effects of BPA could be missed. It is expected that because BPA targets distinct receptors and various organs the resulting syndrome would be complex (94). However, it is important to keep in mind that animals are individuals, and therefore, not all animals will be affected identically. If one considers all the sources of variation described above, the fact that obesity and/or alterations of metabolism are observed in the majority of studies with some equivalency of exposure times and levels argues for the robustness of the effect.
Although we reviewed studies of early (fetal and neonatal) BPA exposure here, studies of BPA exposure in adult rodents have revealed effects on adiposity and elements of metabolic disease (95–97). One particularly interesting finding was observed in females exposed to BPA (10 or 100 μg/kg BW/day) during days 9–16 of pregnancy (33, 95). When examined on days 16–18 of pregnancy, females exposed to BPA revealed glucose intolerance and elevated levels of plasma insulin, triglycerides, and leptin relative to controls (33). By 4 months postpartum, BPA exposed mothers had increased body weights and at 6.5 months they had increased body weights, and increased perigonadal fat pad weights (95). Alterations in glucose and insulin tolerance were observed by 4 months postpartum, worsened at 5 months, and became even more pronounced at 6 months. In contrast, non-pregnant females exposed to the same level and length of exposure to BPA showed no changes in glucose or insulin tolerance when examined at 4, 5, or 6 months after treatment. These data reveal harmful long- term implications for BPA exposure during pregnancy.
Epidemiological studies have found a positive association between urinary BPA levels and obesity and diabetes in adults, (7–9, 98–100) children, and adolescents (101–108); others have questioned the strength of the association (50, 51). Of particular interest, a recent and rare planned exposure study in humans provided oral administration of the reference BPA dose to volunteers and measured the insulin/C-peptide response to administered glucose. Although this study had a small sample size, BPA exposure was found to alter this response (90).
What are the important messages that one can glean from these studies? The most urgent one concerns public health. The studies addressed here bolster the notion that BPA is an obesogen and metabolic disruptor. The incidence of obesity and obesity-associated metabolic disease including diabetes is increasing to alarming proportions, and human data shows a positive association between BPA level and health effects (109). Given the fact that BPA exposure is ubiquitous, we need to conclude that BPA is a public health concern and take action to limit our exposure. European countries are reducing exposure by banning BPA in food packing materials, and a new law in France will ban the use of plastic in direct contact with food in school cafeterias (110).
A second issue in need of exploration is how BPA triggers these effects. The biological explanation of the syndrome produced by early BPA exposure is undoubtedly complex. It is worth remembering that the path from receptor binding to phenotype is not an obvious straight line. This complexity suggests it will take a long time to determine precisely how the BPA phenotype is determined. Organogenesis involves more than molecular interactions. As organs form, we observe supracellular phenomena such as cell to cell interactions, electrical gradients, mechanical forces, cell migration, and more (111). Additionally, the external environment co-constructs the phenotype (112) and the factors discussed above can modulate and affect the response to BPA. Therefore, we need to explore multiple pathways and processes that include all relevant levels of biological organization and we need to carefully control and report the conditions of our animals in the process. Unraveling the complexity of the BPA syndrome will provide us with a global understanding of how a very simple molecule derails development creating phenotypes prone to the development of multiple pathologies.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Support was provided by Grant R01ES08314, 5RC2ES18781 and R21ES026283 from NIEHS. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the U. S. National Institute of Environmental Health Sciences or NIH.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Thank you to Nafis Hasan for his critical reading of the manuscript.
References
1. Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA (2012) 307:491–7. doi: 10.1001/jama.2012.39
2. Hales CM, Caroll MD, Fryar CD, Ogden CL. Prevalence of obesity among adults and youth: United States 2015-2016. NCHS Data Brief (2017) 1–8. Available online at: https://www.cdc.gov/nchs/data/databriefs/db288.pdf
3. Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Alter Complement Med. (2002) 8:185–92. doi: 10.1089/107555302317371479
4. Heindel JJ, Blumberg B. Environmental obesogens: mechanisms and controversies. Annu Rev Pharmacol Toxicol. (2018) 59:89–106. doi: 10.1146/annurev-pharmtox-010818-021304
5. Grun F, Blumberg B. Endocrine disrupters as obesogens. Mol Cell Endocrinol. (2009) 304:19–29. doi: 10.1016/j.mce.2009.02.018
6. Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL. Exposure of the U.S. population to bisphenol A and 4-tertiary-Octylphenol: 2003-2004. Environ Health Perspect. (2008) 116:39–44. doi: 10.1289/ehp.10753
7. Carwile JL, Michels KB. Urinary bisphenol A and obesity: NHANES 2003-2006. Environ Res. (2011) 111:825–30. doi: 10.1016/j.envres.2011.05.014
8. Lang IA, Galloway TS, Scarlett A, Henley WE, Depledge M, Wallace RB, Melzer D. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. J Am Med Assoc. (2008) 300:1303–10. doi: 10.1001/jama.300.11.1303
9. Wang T, Li M, Chen B, Xu M, Xu Y, Huang Y, et al. Urinary bisphenol A (BPA) concentration associates with obesity and insulin resistance. J Clin Endocrinol Metab. (2012) 97:E223–7. doi: 10.1210/jc.2011-1989
10. Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, et al. Metabolism disrupting chemicals and metabolic disorders. Reprod Toxicol. (2017) 68:3–33. doi: 10.1016/j.reprotox.2016.10.001
11. Alonso-Magdalena P, Quesada I, Nadal A. Prenatal exposure to BPA and offspring outcomes: the diabesogenic behavior of BPA. Dose Response (2015) 13 15:59325815590395. doi: 10.1177/1559325815590395
12. Longo G, Soto AM. Why do we need theories? Prog Biophys Mol Biol. (2016) 122:4–10. doi: 10.1016/j.pbiomolbio.2016.06.005
13. Montévil M. Biological Time and Extended Critical Transitions:Towards an Objectivization of the Living State of Matter. École Doctorale 474 — Frontières du Vivant, Université de Paris V — René Descartes (2011). Paris.
14. West-Eberhard MJ. Phenotypic accommodation: adaptive innovation due to developmental plasticity. J Exp Zool B Mol Dev Evol. (2005) 304:610–8. doi: 10.1002/jez.b.21071
15. Rubin BS, Paranjpe M, DaFonte T, Schaeberle C, Soto AM, Obin M, et al. Perinatal BPA exposure alters body weight and composition in a dose specific and sex specific manner: the addition of peripubertal exposure exacerbates adverse effects in female mice. Reprod Toxicol. (2017) 68:130–44. doi: 10.1016/j.reprotox.2016.07.020
16. Newbold RR, Jefferson WN, Padilla-Banks E, Haseman J. Developmental exposure to diethylstilbestrol (DES) alters uterine response to estrogens in prepubescent mice: low versus high dose effects. Reprod Toxicol. (2004) 18:399–406. doi: 10.1016/j.reprotox.2004.01.007
17. Rubin BS, Murray MK, Damassa DA, King JC, Soto AM. Perinatal exposure to low doses of bisphenol-A affects body weight, patterns of estrous cyclicity and plasma LH levels. Environ Health Perspect. (2001) 109:675–80. doi: 10.1289/ehp.01109675
18. Howdeshell KL, Hotchkiss AK, Thayer KA, Vandenbergh JG, vom Saal FS. Exposure to bisphenol A advances puberty. Nature (1999) 401:763–4. doi: 10.1038/44517
19. Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN. Developmental exposure to estrogenic compounds and obesity. Birth Defects Res A Clin Mol Teratol. (2005) 73:478–80. doi: 10.1002/bdra.20147
20. Somm E, Schwitzgebel VM, Toulotte A, Cederroth CR, Combescure C, Nef S, et al. Perinatal exposure to bisphenol a alters early adipogenesis in the rat. Environ Health Perspect. (2009) 117:1549–55. doi: 10.1289/ehp.11342
21. Markey CM, Wadia PR, Rubin BS, Sonnenschein C, Soto AM. Long-term effects of fetal exposure to low doses of the xenoestrogen bisphenol-A in the female mouse genital tract. Biol Reprod. (2005) 72:1344–51. doi: 10.1095/biolreprod.104.036301
22. Cabaton NJ, Wadia PR, Rubin BS, Zalko D, Schaeberle CM, Askenase MH, et al. Perinatal exposure to environmentally relevant levels of Bisphenol-A decreases fertility and fecundity in CD-1 mice. Environ Health Perspect. (2011) 119:547–52. doi: 10.1289/ehp.1002559
23. Markey CM, Coombs MA, Sonnenschein C, Soto AM. Mammalian development in a changing environment: exposure to endocrine disruptors reveals the developmental plasticity of steroid-hormone target organs. Evolu Dev. (2003) 5: 67–75. doi: 10.1046/j.1525-142X.2003.03011.x
24. Acevedo N, Rubin BS, Schaeberle CM, Soto AM. Perinatal BPA exposure and reproductive axis function in CD-1 mice. Reprod Toxicol. (2018) 79:39–46. doi: 10.1016/j.reprotox.2018.05.002
25. Soto AM, Brisken C, Schaeberle CM, Sonnenschein C. Does cancer start in the womb? altered mammary gland development and predisposition to breast cancer due to in utero exposure to endocrine disruptors. J Mammary Gland Biol Neoplasia (2013) 18:199–208. doi: 10.1007/s10911-013-9293-5
26. Vandenberg LN, Chauhoud I, Heindel JJ, Padmanabhan V, Paumgartten FJ, Schoenfelder G. Urinary, circulating and tissue biomonitoring studies indicate widespread exposure to Bisphenol A. Environ Health Perspect. (2010) 118:1055–70. doi: 10.1289/ehp.0901716
27. Gerona RR, Woodruff TJ, Dickenson CA, Pan J, Schwartz JM, Sen S, et al. Bisphenol-A (BPA), BPA glucuronide, and BPA sulfate in midgestation umbilical cord serum in a northern and central California population. Environ Sci Technol. (2013) 47:12477–85. doi: 10.1021/es402764d
28. Shimpi PC, More VR, Paranjpe M, Donepudi AC, Goodrich JM, Dolinoy DC, et al. Hepatic lipid accumulation and Nrf2 expression following perinatal and peripubertal exposure to bisphenol A in a mouse model of nonalcoholic liver disease. Environ Health Perspect. (2017) 125:087005. doi: 10.1289/EHP664
30. Cottret L, Frainay C, Chazalviel M, Cabanettes F, Gloaguen Y, Camenen E, et al. MetExplore: collaborative edition and exploration of metabolic networks. Nucleic Acids Res. (2018) 46:W495–502. doi: 10.1007/s00204-015-1488-7
31. Tremblay-Franco M, Cabaton NJ, Canlet C, Gautier R, Schaeberle CM, Fabien J, et al. Dynamic metabolic disruption in rats perinatally exposed to low doses of bisphenol-A. PLoS ONE (2015) 10:e0141698. doi: 10.1371/journal.pone.0141698
32. Cabaton NJ, Canlet C, Wadia PR, Tremblay-Franco M, Gautier R, Molina J, et al. Effects of low doses of bisphenol A on the metabolome of perinatally exposed CD-1 mice. Environ Health Perspect. (2013) 121:586–93. doi: 10.1289/ehp.1205588
33. Alonso-Magdalena P, Vieira E, Soriano S, Menes L, Burks D, Quesada I, et al. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect. (2010) 118:1243–50. doi: 10.1289/ehp.1001993
34. Wei J, Lin Y, Li Y, Ying C, Chen J, Song L, et al. Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high-fat diet. Endocrinology (2011) 152:3049–61. doi: 10.1210/en.2011-0045
35. Angle BM, Do RP, Ponzi D, Stahlhut RW, Drury BE, Nagle SC, et al. Metabolic disruption in male mice due to fetal exposure to low but not high doses of bisphenol A (BPA): evidence for effects on body weight, food intake, adipocytes, leptin, adiponectin, insulin and glucose regulation. Reprod Toxicol. (2013) 42:256–68. doi: 10.1016/j.reprotox.2013.07.017
36. Nikaido Y, Yoshizawa K, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, et al. Effects of maternal xenoestrogen exposure on development of the reproductive tract and mammary gland in female CD-1 mouse offspring. Reprod Toxicol. (2004) 18:803–11. doi: 10.1016/j.reprotox.2004.05.002
37. Garc¡a-Arevalo M, Alonso-Magdalena P, Rebelo Dos Santos J, Quesada I, Carneiro EM, Nadal A. Exposure to bisphenol-A during pregnancy partially mimics the effects of a high-fat diet altering glucose homeostasis and gene expression in adult male mice. PLoS ONE (2014) 9:e100214. doi: 10.1371/journal.pone.0100214
38. Liu J, Yu P, Qian W, Li Y, Zhao J, Huan F, et al. Perinatal bisphenol A exposure and adult glucose homeostasis: identifying critical windows of exposure. PLoS ONE (2013) 10:e64143. doi: 10.1371/journal.pone.0064143
39. Patisaul HB, Bateman HL. Neonatal exposure to endocrine active compounds or an ERbeta agonist increases adult anxiety and aggression in gonadally intact male rats. Horm Behav. (2008) 53:580–8. doi: 10.1016/j.yhbeh.2008.01.008
40. van Esterik JC, Dollé ME, Lamoree MH, van Leeuwen SP, Hamers T, Legler J, et al. Programming of metabolic effects in C57BL/6JxFVB mice by exposure to bisphenol A during gestation and lactation. Toxicology (2014) 321:40–52. doi: 10.1016/j.tox.2014.04.001
41. Taylor JA, Shioda K, Mitsunaga S, Yawata S, Angle BM, Nagel SC, et al. Prenatal exposure to bisphenol A disrupts naturally occurring bimodal DNA methylation at proximal promoter of fggy, an obesity-relevant gene encoding a carbohydrate kinase, in gonadal white adipose tissues of CD-1 mice. Endocrinology (2018) 159:779–94. doi: 10.1210/en.2017-00711
42. Desai M, Ferrini MG, Jellyman JK, Han G, Ross MG. In vivo and in vitro bisphenol A exposure effects on adiposity. J Dev Orig Health Dis. (2018). doi: 10.1017/S2040174418000600. [Epub ahead of print].
43. Miyawaki J, Sakayama K, Kato H, Yamamoto H, Masuno H. Perinatal and postnatal exposure to bisphenol a increases adipose tissue mass and serum cholesterol level in mice. J Atheroscl Thromb. (2007) 14:245–52. doi: 10.5551/jat.E486
44. Anderson OS, Peterson KE, Sanchez BN, Zhang Z, Mancuso P, Dolinoy DC. Perinatal bisphenol A exposure promotes hyperactivity, lean body composition, and hormonal responses across the murine life course. FASEB J. (2013) 27:1784–92. doi: 10.1096/fj.12-223545
45. Ferguson SA, Law CD Jr, Abshire JS. Developmental treatment with bisphenol A or ethinyl estradiol causes few alterations on early preweaning measures. Toxicol Sci. (2011) 124:149–60. doi: 10.1093/toxsci/kfr201
46. Tyl RW, Myers CB, Marr MC, Sloan CS, Castillo NP, Veselica MM, et al. Two-generation reproductive toxicity study of dietary bisphenol A in CD-1 (Swiss) mice. Toxicol Sci. (2008) 104:362–84. doi: 10.1093/toxsci/kfn084
47. Tyl RW, Myers CB, Marr MC, Thomas BF, Keimowitz AR, Brine DR, et al. Three-generation reproductive toxicity study of dietary bisphenol A in CD Sprague-Dawley rats. Toxicol Sci. (2002) 68:121–46. doi: 10.1093/toxsci/68.1.121
48. Cao J, Echelberger R, Liu M, Sluzas E, McCaffery K, Buckley B, et al. Soy but not bisphenol A (BPA) or the phytoestrogen genistin alters developmental weight gain and food intake in pregnant rats and their offspring. Reprod Toxicol. (2015) 58:282–94. doi: 10.1016/j.reprotox.2015.07.077
49. Ryan KK, Haller AM, Sorrell JE, Woods SC, Jandacek RJ, Seeley RJ. Perinatal exposure to bisphenol-a and the development of metabolic syndrome in CD-1 mice. Endocrinology (2010) 151:2603–12. doi: 10.1210/en.2009-1218
50. Lakind JS, Goodman M, Mattison DR. Bisphenol A and indicators of obesity, glucose metabolism/type 2 diabetes and cardiovascular disease: a systematic review of epidemiologic research. Crit Rev Toxicol. (2014) 44:121–50. doi: 10.3109/10408444.2013.860075
51. Mirmira P, Evans-Molina C. Bisphenol A, obesity, and type 2 diabetes mellitus: genuine concern or unnecessary preoccupation? Transl Res. (2014) 164:13–21. doi: 10.1016/j.trsl.2014.03.003
52. Oppeneer SJ, Robien K. Bisphenol A exposure and associations with obesity among adults: a critical review. Public Health Nutr. (2015) 18:1847–63. doi: 10.1017/S1368980014002213
53. Hunt PA, Koehler KE, Susiarjo M, Hodges CA, Ilagan A, Voigt RC, et al. Bisphenol A exposure causes meiotic aneuploidy in the female mouse. Curr Biol. (2003) 13:546–53. doi: 10.1016/S0960-9822(03)00189-1
54. Ropero AB, Alonso-Magdalena P, Garcia-Garcia E, Ripoll C, Fuentes E, Nadal A. Bisphenol-A disruption of the endocrine pancreas and blood glucose homeostasis. Int J Androl. (2008) 31:194–200. doi: 10.1111/j.1365-2605.2007.00832.x
55. Nadal A Obesity: fat from plastics? Linking bisphenol A exposure and obesity. Nat Rev Endocrinol. (2013) 9:9–10. doi: 10.1038/nrendo.2012.205
56. Nadal A, Quesada I, Tudur E, Nogueiras R, Alonso-Magdalena P. Endocrine-disrupting chemicals and the regulation of energy balance. Nat Rev Endocrinol. (2017) 13:536–46. doi: 10.1038/nrendo.2017.51
57. Rochester JR, Bolden AL, Kwiatkowski CF. Prenatal exposure to bisphenol A and hyperactivity in children: a systematic review and meta-analysis. Environ Int. (2018) 114:343–56. doi: 10.1016/j.envint.2017.12.028
58. Nojima K, Takata T, Masuno H. Prolonged exposure to a low-dose of bisphenol A increases spontaneous motor activity in adult male rats. J Physiol Sci. (2013) 63:311–5. doi: 10.1007/s12576-013-0265-8
59. Ishido M, Masuo Y, Kunimoto M, Oka S, Morita M. Bisphenol A causes hyperactivity in the rat concomitantly with impairment of tyrosine hydroxylase immunoreactivity. J Neurosci Res. (2004) 76:423–33. doi: 10.1002/jnr.20050
60. Ishido M, Masuo Y, Terasaki M, Morita M. Rat hyperactivity by bisphenol A, but not by its derivatives, 3-hydroxybisphenol A or bisphenol A 3,4-quinone. Toxicol Lett. (2011) 206:300–5. doi: 10.1016/j.toxlet.2011.08.011
61. Komada M, Itoh S, Kawachi K, Kagawa N, Ikeda Y, Nagao T. Newborn mice exposed prenatally to bisphenol A show hyperactivity and defective neocortical development. Toxicology (2014) 323:51–60. doi: 10.1016/j.tox.2014.06.009
62. Harley KG, Gunier RB, Kogut K, Johnson C, Bradman A, Calafat AM, et al. Prenatal and early childhood bisphenol A concentrations and behavior in school-aged children. Environ Res. (2013) 126:43–50. doi: 10.1016/j.envres.2013.06.004
63. Braun JM, Kalkbrenner AE, Calafat AM, Yolton K, Ye X, Dietrich KN, et al. Impact of early-life bisphenol A exposure on behavior and executive function in children. Pediatrics (2011) 128:873–82. doi: 10.1542/peds.2011-1335
64. Soto AM, Sonnenschein C, Chung KL, Fernandez MF, Olea N, Olea-Serrano MF. The E-SCREEN assay as a tool to identify estrogens: an update on estrogenic environmental pollutants. Environ Health Perspect. (1995) 103:113–22.
65. Ruhlen RL, Howdeshell KL, Mao J, Taylor JA, Bronson FH, Newbold RR, et al. Low phytoestrogen levels in feed increase fetal serum estradiol resulting in the “fetal estrogenization syndrome” and obesity in CD-1 mice. Environ Health Perspect. (2008) 116:322–8. doi: 10.1289/ehp.10448
66. Mackay H, Patterson ZR, Khazall R, Patel S, Tsirlin D, Abidzaid A. Organizational effects of perinatal exposure to bisphenol-A and diethylstilbestrol on arcuate nucleus circuitry controlling food intake and energy expenditure in male and female CD-1 mice. Endocrinology (2013) 154:1465–75. doi: 10.1210/en.2012-2044
67. Krishnan K, Gagne M, Nong A, Aylward LL, Hays SM. Biomonitoring equivalents for bisphenol A (BPA). Regul Toxicol Pharmacol. (2010) 58:18–24. doi: 10.1016/j.yrtph.2010.06.005
68. Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR Jr, Lee DH, et al. Hormones and endocrine disrupting chemicals: low dose effects and non-monotonic dose responses. Endocr Rev. (2012) 33:378–455. doi: 10.1210/er.2011-1050
69. Vandenberg LN. Non-monotonic dose responses in studies of endocrine disrupting chemicals: bisphenol a as a case study. Dose Response (2014) 12:259–76. doi: 10.2203/dose-response.13-020.
70. Amara JF, Dannies PS. 17 á-Estradiol has a biphasic effect on GH cell growth. Endocrinology (1983) 112:1141–3. doi: 10.1210/endo-112-3-1141
71. Sonnenschein C, Olea N, Pasanen ME, Soto AM. Negative controls of cell proliferation: human prostate cancer cells and androgens. Cancer Res. (1989) 49:3474–81.
72. Geck P, Szelei J, Jimenez J, Lin TM, Sonnenschein C, Soto AM. Expression of novel genes linked to the androgen-induced, proliferative shutoff in prostate cancer cells. J Steroid Biochem Mol Biol. (1997) 63:211–8. doi: 10.1016/S0960-0760(97)00122-2
73. Soto AM, Lin TM, Sakabe K, Olea N, Damassa DA, Sonnenschein C. Variants of the human prostate LNCaP cell line as a tool to study discrete components of the androgen-mediated proliferative response. Oncol Res. (1995) 7:545–58.
74. Geck P, Maffini MV, Szelei J, Sonnenschein C, Soto AM. Androgen-induced proliferative quiescence in prostate cancer: the role of AS3 as its mediator. Proc Natl Acad Sci USA. (2000) 97:10185–90. doi: 10.1073/pnas.97.18.10185
75. Soto AM, Sonnenschein C. The two faces of Janus: sex steroids as mediators of both cell proliferation and cell death. J Ntl Cancer Institute (2001) 93:1673–5. doi: 10.1093/jnci/93.22.1673
76. vom Saal FS, Timms BG, Montano MM, Palanza P, Thayer KA, Nagel SC, et al. Prostate enlargement in mice due to fetal exposure to low doses of estradiol or diethylstilbestrol and opposite effects at high doses. Proc Natl Acad Sci USA. (1997) 94:2056–61. doi: 10.1073/pnas.94.5.2056
77. Vandenberg LN, Wadia PR, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. The mammary gland response to estradiol: monotonic at the cellular level, non-monotonic at the tissue-level of organization? J Steroid Biochem Mol Biol. (2006) 101:263–74. doi: 10.1016/j.jsbmb.2006.06.028
78. Kohn MC, Melnick RL. Biochemical origins of the non-monotonic receptor-mediated dose-response. J Mol Endocrinol. (2002) 29:113–23. doi: 10.1677/jme.0.0290113
79. Cederroth CR, Nef S. Fetal programming of adult glucose homeostasis in mice. PLoS ONE (2009) 4:e7281. doi: 10.1371/journal.pone.0007281
80. Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science (2004) 304:108–10. doi: 10.1126/science.1095004
81. Mackay H, Patterson ZR, Abizaid A. Perinatal exposure to low-dose bisphenol-A disrupts the structural and functional development of the hypothalamic feeding circuitry. Endocrinology (2017) 158:768–77. doi: 10.1210/en.2016-1718
82. Lu S, Yu Y, Ren L, Zhang X, Liu G, Yu Y. Estimation of intake and uptake of bisphenols and triclosan from personal care products by dermal contact. Sci Total Environ. (2018) 621:1398–6. doi: 10.1016/j.scitotenv.2017.10.088
83. von Goetz N, Pirow R, Hart A, Bradley E, Poças F, Arcella D, et al. Including non-dietary sources into an exposure assessment of the European Food Safety Authority: The challenge of multi-sector chemicals such as Bisphenol A. Regul Toxicol Pharmacol. (2017) 85:70–8. doi: 10.1016/j.yrtph.2017.02.004
84. Hormann AM, vom Saal FS, Nagel SC, Stahlhut RW, Moyer CL, Ellersieck MR, et al. Holding thermal receipt paper and eating food after using hand sanitizer results in high serum bioactive and urine total levels of bisphenol A (BPA). PLoS ONE (2014) 9:e110509. doi: 10.1371/journal.pone.0110509
85. Ehrlich S, Calafat AM, Humblet O, Smith T, Hauser R. Handling of thermal receipts as a source of exposure to bisphenol A. J Am Med Assoc. (2014) 311:859–60. doi: 10.1001/jama.2013.283735
86. Biedermann S, Tschudin P, Grob K. Transfer of Bisphenol A from thermal printer paper to the skin. Anal Bioanal Chem. (2010) 398:571–6. doi: 10.1007/s00216-010-3936-9
87. Zalko D, Jacques C, Duplan H, Bruel S, Perdu E. Viable skin efficiently absorbs and metabolizes bisphenol A. Chemosphere (2011) 82:424–30. doi: 10.1016/j.chemosphere.2010.09.058
88. Stahlhut RW, Welshons WV, Swan SH. Bisphenol A data in NHANES suggest longer than expected half-life, substantial nonfood exposure, or both. Environ Health Perspect. (2009) 117:784–9. doi: 10.1289/ehp.0800376
89. Lv Y, Lu S, Dai Y, Rui C, Wang Y, Zhou Y, et al. Higher dermal exposure of cashiers to BPA and its association with DNA oxidative damage. Environ Int. (2017) 98:69–74. doi: 10.1016/j.envint.2016.10.001
90. Stahlhut RW, Myers JP, Taylor JA, Nadal A, Dyer JA, vom Saal FS. Experimental BPA exposure and glucose-stimulated insulin response in adult men and women. J Endo Soc. (2018) 2: 1173–87. doi: 10.1210/js.2018-00151
91. Prins GS, Ye SH, Birch L, Ho SM, Kannan K. Serum bisphenol A pharmacokinetics and prostate neoplastic responses following oral and subcutaneous exposures in neonatal Sprague-Dawley rats. Reprod Toxicol. (2011) 31:1–9. doi: 10.1016/j.reprotox.2010.09.009
92. Sieli PT, Jasarevic E, Warzak DA, Mao J, Ellersieck MR, Liao C, et al. Comparison of serum bisphenol A concentrations in mice exposed to bisphenol A through the diet versus oral bolus exposure. Environ Health Perspect. (2011) 119:1260–5. doi: 10.1289/ehp.1003385
93. Malais Y, Menard S, Cartier C, Gaultier E, Lasserre F, Lencina C, et al. Gut dysbiosis and impairment of immune system homeostasis in perinatally-exposed mice to Bisphenol A precede obese phenotype development. Sci Rep. (2017) 7:14472. doi: 10.1038/s41598-017-15196-w
94. Sonnenschein C, Wadia PR, Rubin BS, Soto AM. Cancer as development gone awry: the case for bisphenol-A as a carcinogen. J Dev Orig Health Dis. (2011) 2:9–16. doi: 10.1017/S2040174411000043
95. Alonso-Magdalena P, Garc¡a-Arevalo M, Quesada I, Nadal A. Bisphenol-A treatment during pregnancy in mice: a new window of susceptibility for the development of diabetes in mothers later in life. Endocrinology (2015) 156:1659–70. doi: 10.1210/en.2014-1952
96. Yang M, Chen M, Wang J, Xu M, Sun J, Ding L, et al. Bisphenol A promotes adiposity and inflammation in a nonmonotonic dose-response way in five-week old male and female C57BL/6J mice fed a low-calorie diet. Endocrinology (2016) 157:2333–45. doi: 10.1210/en.2015-1926
97. Batista TM, Alonso-Magdalena P, Vieira E, Amaral ME, Cederroth CR, Nef S, et al. Short-term treatment with bisphenol-A leads to metabolic abnormalities in adult male mice. PLoS ONE. (2012) 7:e33814. doi: 10.1371/journal.pone.0033814
98. Klimentidis YC, Beasley TM, Lin HY, Murati G, Glass GE, Guyton M, et al. Canaries in the coal mine: a cross-species analysis of the plurality of obesity epidemics. Proc Biol Sci. (2011) 278:1626–32. doi: 10.1098/rspb.2010.1890
99. Shankar A, Teppala S, Sabanayagam C. Urinary bisphenol a levels and measures of obesity: results from the national health and nutrition examination survey 2003-2008. ISRN Endocrinol. (2012) 2012:965243. doi: 10.5402/2012/965243
100. Rancière F, Lyons JG, Loh HY, Botton J, Galloway T, Wang T, et al. Bisphenol A and the risk of cardiometabolic disorders: a systematic review with meta-analysis of the epidemiological evidence. Environ Health (2015) 14:46. doi: 10.1186/s12940-015-0036-5
101. Valvi D, Casas M, Mendez MA, Ballesteros-Gmez A, Luque N, Rubio S, et al. Prenatal bisphenol a urine concentrations and early rapid growth and overweight risk in the offspring. Epidemiology (2013) 24:791–9. doi: 10.1097/EDE.0b013e3182a67822
102. Vafeiadi M, Roumeliotaki T, Myridakis A, Chalkiadaki G, Fthenou E, Dermitzaki E, et al. Association of early life exposure to bisphenol A with obesity and cardiometabolic traits in childhood. Environ Res. (2016) 146:379–87. doi: 10.1016/j.envres.2016.01.017
103. Hoepner LA, Whyatt RM, Widen EM, Hassoun A, Oberfield SE, Mueller NT, et al. BPA and adiposity in inner city birth cohort. Environ Health Perspect. (2016) 124:1644–50. doi: 10.1289/EHP205
104. Trasande L, Attina TM, Blustein J. Association between urinary bisphenol A concentration and obesity prevalence in children and adolescents. J Am Med Assoc. (2012) 308:1113–21. doi: 10.1001/2012.jama.11461
105. Amin MM, Ebrahim K, Hashemi M, Shoshtari-Yeganeh B, Rafiei N, Mansourian M, et al. Association of exposure to Bisphenol A with obesity and cardiometabolic risk factors in children and adolescents. Int J Environ Health Res. (2018) 29:94–106. doi: 10.1080/09603123.2018.1515896
106. Li DK, Miao M, Zhou Z, Wu C, Shi H, Liu X, et al. Urine bisphenol-A level in relation to obesity and overweight in school-age children. PLoS ONE (2013) 8:e65399. doi: 10.1371/journal.pone.0065399
107. Bhandari R, Xiao J, Shankar A. Urinary bisphenol A and obesity in U.S. children. Am J Epidemiol. (2013) 177:1263–70. doi: 10.1093/aje/kws391
108. Eng DS, Lee JM, Gebrmariam A, Meeker JD, Peterson K, Padmanabhan V. Bisphenol A and chronic disease risk factors in US childrens. Pediatrics (2013) 132:e637–45. doi: 10.1542/peds.2013-0106
109. Legeay S, Faure S. Is bisphenol A an environmental obesogen? Fundam Clin Pharmacol. (2017) 31:594–609. doi: 10.1111/fcp.12300
110. French Law. French Law # 2018-938. 28. Available online at: https://www.legifrance.gouv.fr/eli/loi/2018/10/30/AGRX1736303L/jo/texte
111. Soto AM, Longo G, Miquel PA, Montévil M, Mossio M, Perret N, et al. Toward a theory of organisms: three founding principles in search of a useful integration. Prog Biophys Mol Biol. (2016) 122:77–82. doi: 10.1016/j.pbiomolbio.2016.07.006
Keywords: Bisphenol A, non-monotonic dose response, metabolome, adiposity, exposure windows, exposure route, perinatal
Citation: Rubin BS, Schaeberle CM and Soto AM (2019) The Case for BPA as an Obesogen: Contributors to the Controversy. Front. Endocrinol. 10:30. doi: 10.3389/fendo.2019.00030
Received: 01 October 2018; Accepted: 15 January 2019;
Published: 06 February 2019.
Edited by:
Angel Nadal, Universidad Miguel Hernández de Elche, SpainReviewed by:
Paloma Alonso-Magdalena, Universidad Miguel Hernández de Elche, SpainRichard Stahlhut, American College of Preventive Medicine, United States
Copyright © 2019 Rubin, Schaeberle and Soto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beverly S. Rubin, beverly.rubin@tufts.edu
Ana M. Soto, ana.soto@tufts.edu