 Sheerazed Boulkroun
Sheerazed Boulkroun Fabio Luiz Fernandes-Rosa1,2,3
Fabio Luiz Fernandes-Rosa1,2,3- 1UMRS_970, Paris Cardiovascular Research Center, Institut National de la Santé et de la Recherche Médicale (INSERM), Paris, France
- 2University Paris Descartes, Sorbonne Paris Cité, Paris, France
- 3Assistance Publique-Hôpitaux de Paris, Hôpital Européen Georges Pompidou, Service de Génétique, Paris, France
Primary aldosteronism (PA) is the most common form of secondary hypertension with an estimated prevalence of ~10% in referred patients. PA occurs as a result of a dysregulation of the normal mechanisms controlling adrenal aldosterone production. It is characterized by hypertension with low plasma renin and elevated aldosterone and often associated with hypokalemia. The two major causes of PA are unilateral aldosterone producing adenoma (APA) and bilateral adrenal hyperplasia, accounting together for ~95% of cases. In addition to the well-characterized effect of excess mineralocorticoids on blood pressure, high levels of aldosterone also have cardiovascular, renal, and metabolic consequences. Hence, long-term consequences of PA include increased risk of coronary artery disease, myocardial infarction, heart failure, and atrial fibrillation. Despite recent progress in the management of patients with PA, critical issues related to diagnosis, subtype differentiation, and treatment of non-surgically correctable forms still persist. A better understanding of the pathogenic mechanisms of the disease should lead to the identification of more reliable diagnostic and prognostic biomarkers for a more sensitive and specific screening and new therapeutic options. In this review, we will summarize our current knowledge on the molecular and cellular mechanisms of APA development. On one hand, we will discuss how various animal models have improved our understanding of the pathophysiology of excess aldosterone production. On the other hand, we will summarize the major advances made during the last few years in the genetics of APA due to transcriptomic studies and whole exome sequencing. The identification of recurrent and somatic mutations in genes coding for ion channels (KCNJ5 and CACNA1D) and ATPases (ATP1A1 and ATP2B3) allowed highlighting the central role of calcium signaling in autonomous aldosterone production by the adrenal.
Background
Aldosterone is synthesized from cholesterol by a series of specific enzymatic reactions in the zona glomerulosa of the adrenal cortex; the final steps are catalyzed by the aldosterone synthase (encoded by CYP11B2). Aldosterone production from the adrenal cortex is tightly controlled to maintain electrolyte and fluid homeostasis; the two principal secretagogues are the renin/angiotensin system and the extracellular concentration of potassium (K+). The stimulation by angiotensin II or K+ results in depolarization of the zona glomerulosa cell membrane and opening of voltage-gated calcium (Ca2+) channels, leading to an increase of intracellular Ca2+ concentration. Angiotensin II, by its binding to the angiotensin II type I receptor (AT1R), also acts by increasing inositol triphosphate formation leading to the release of Ca2+ from the endoplasmic reticulum. Activation of the calcium signaling pathway triggers a phosphorylation cascade, involving calmodulin and calmodulin-dependent kinase I/IV, leading to the activation of specific transcription factors (NURR1, NGF1B, CREB) that bind to the promoter region and positively regulate the transcription of CYP11B2 leading to an increase in aldosterone biosynthesis (Figure 1) (1). Hence, the activation of hormone synthesis is Ca2+ dependent, and the regulatory mechanism involves Ca2+ mediated processes.
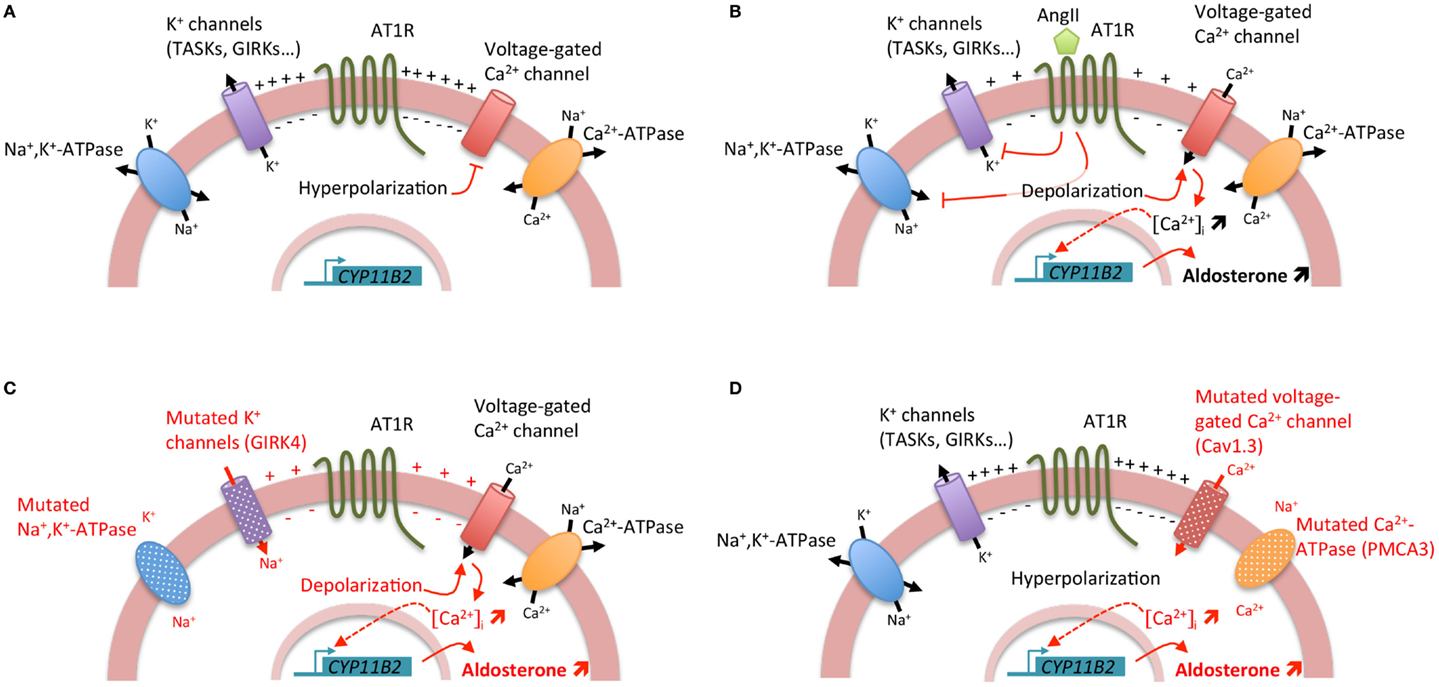
Figure 1. Regulation of aldosterone biosynthesis in normal and pathological conditions. (A) Under resting conditions, zona glomerulosa cells exhibit a strongly negative membrane potential (−80 mV) due to the expression of a large number of potassium channels. (B) Stimulation of aldosterone biosynthesis by AngII. The binding of AngII to the AngII type I receptor (AT1R) induces a cascade of events leading to the zona glomerulosa cell depolarization and the increase of intracellular Ca2+ concentration. The inhibition of potassium channels and Na+, K+-ATPase by AngII results in zona glomerulosa cell depolarization, opening of voltage-gated Ca2+ channels, and increase of intracellular Ca2+ concentration. Furthermore, activation of AT1R leads also to the increase of inositol triphosphate formation and consequently to the release of Ca2+ from the endoplasmic reticulum. Activation of the calcium signaling pathway triggers a phosphorylation cascade, involving calmodulin and calmodulin-dependent kinase I/IV, leading to the activation of specific transcription factors that bind to the promoter region and positively regulate the transcription of CYP11B2 leading to an increase in aldosterone biosynthesis. (C) Genetic alterations in KCNJ5 (coding for the potassium channel GIRK4) and ATP1A1 (encoding the α1 subunit of the Na+, K+-ATPase) genes lead to cell membrane depolarization triggering opening of voltage-gated Ca2+ channels and consequently positive regulation of CYP11B2. (D) Genetic alterations in ATP2B3 (coding for the plasma membrane Ca2+ ATPase, PMCA3) and CACNA1D (encoding the Cav1.3 subunit of the L-type voltage-gated Ca2+ channel) genes lead directly to the increase of intracellular Ca2+ concentration by affecting calcium recycling and influx, resulting in positive regulation of CYP11B2.
Deregulation of the mechanisms regulating aldosterone biosynthesis results in primary aldosteronism (PA), the most common form of secondary hypertension with an estimated prevalence of about 10% in referred patients and 4% in primary care (2) and as high as 20% in patients with resistant hypertension (3). PA is characterized by hypertension with elevated plasma aldosterone and low plasma renin levels, and often associated with hypokalemia. The two major causes of PA are unilateral aldosterone producing adenoma (APA) and bilateral adrenal hyperplasia (BAH), accounting together for ~95% of cases. The early detection of PA has an important impact on clinical outcome and survival given the major cardiovascular adverse effect of aldosterone excess, which is independent of blood pressure (BP). Patients with PA have been reported to exhibit more severe left ventricular hypertrophy and diastolic dysfunction than patients with essential hypertension and a high prevalence of myocardial infarction, stroke, and atrial fibrillation (4, 5). Despite the publications in 2008 of guidelines for the management of PA, there remain a few critical issues related to diagnosis, subtype differentiation, and treatment of non-surgically correctable forms (6). A better understanding of the pathogenic mechanisms of the disease should lead to the identification of more reliable diagnostic and prognostic biomarkers for a more sensitive and specific screening and new therapeutic options.
During the last few years, major advances have been made in understanding the genetic basis of APA, with the identification of mutations in genes coding for ion channels [KCNJ5, coding for the G protein-activated inward rectifier potassium channel 4 (GIRK4) (7) and CACNA1D, encoding the Cav1.3 channel (calcium channel, voltage-dependent, L type, alpha 1d subunit) (8, 9)] and ATPases [ATP1A1, coding for the α1 subunit of the Na+/K+-ATPase (9, 10) and ATP2B3 encoding the plasma membrane Ca2+-ATPase, type 3 (10)] in more than 50% of APA. Interestingly, all these mutations lead to the activation of calcium signaling, the major trigger for aldosterone production (Figure 1). However, if the role of these mutations in regulating aldosterone production has been clearly established, their implication in proliferation and APA formation are still matter of debate (11).
In this review, we will summarize our current knowledge on the molecular and cellular mechanism of APA development. We will discuss how various animal models have improved our understanding of the pathophysiology of excess aldosterone production. We will also summarize the major advances made during the last few years in the comprehension of the genetic basis of APA formation using omics approaches, highlighting the major role of the ionic equilibrium and regulation of cell membrane potential in autonomous aldosterone overproduction.
Ionic Equilibrium and Membrane Potential Regulation
The regulation of cell membrane potential of the zona glomerulosa is crucial to maintain the cell in a hyperpolarized state in the absence of a secretagogue stimulus. The zona glomerulosa cell membrane is selectively permeable to K+, giving it the characteristics of a K+ electrode over a wide range of extracellular K+ concentrations, due to the expression of a large number of potassium channels. However, their major role in the development of APA was highlighted only recently by the identification of somatic and germline mutations in genes coding for proteins involved in ionic equilibrium and membrane potential regulation but also by the establishment and analysis of mouse models in which the expression of specific potassium channels was invalidated.
Alteration of Ionic Equilibrium in APA
In 2011, by a whole exome sequencing approach, few recurrent somatic KCNJ5 mutations were identified (7). These mutations (p.Gly151Arg and p.Leu168Arg) are located near or within the selectivity filter of the channel GIRK4. Additional mutations in or surrounding the selectivity filter have been identified, including p.Gly151Glu, p.Thr158Ala, p.Glu141Gln, p.Ile157Ser, delIle157, InsThr149 (12–16). All these mutations result in a significant decrease in K+ selectivity and greater influx of Na+ into the cell, resulting in chronic cell depolarization followed by opening of voltage-dependent calcium channels and activation of calcium signaling and aldosterone production (11, 17). Germline KCNJ5 mutations were also identified as the causative event of Familial hyperaldosteronism type III (FH-III). FH-III was first described in 2008 in a father and two daughters with early-onset severe arterial hypertension resistant to medical treatment and hypokalemia (18). To control BP, a bilateral adrenalectomy was required for all three individuals; histology revealed massive hyperplasia of the adrenal cortex (18). Further exome sequencing performed on APA allowed the identification of other somatic mutations in genes coding for ATPases, namely ATP1A1 (9, 10) and ATP2B3 (10) and the Cav1.3 calcium channel, CACNA1D (8, 9). Whereas mutations in KCNJ5 and ATP1A1 affect adrenal zona glomerulosa cell membrane potential and intracellular ionic homeostasis, with chronic depolarization leading to opening of voltage-dependent calcium channels and activation of calcium signaling and aldosterone production (7, 9–11), mutations in ATP2B3 and CACNA1D modify directly intracellular calcium equilibrium, also leading to an activation of calcium signaling and aldosterone production (Figure 1) (8–10).
Prevalence of Somatic Mutations and Genotype/Phenotype Correlations
The prevalence of somatic mutations in APA has been extensively investigated in many studies (9, 10, 14, 19–22). KCNJ5 mutations are the most frequent genetic abnormalities reported in APA with a prevalence of ~40% in Caucasian population, and as high as 70% in series from Japan (21, 23). The mutations affecting ATP1A1 and ATP2B3 genes are less frequent with a reported prevalence of 5.3 and 1.7, respectively (9, 10, 20). Mutations in the CACNA1D gene are the second most frequent genetic alterations observed in APA with a prevalence comprised between 5 and 9.3% (8, 9, 20). Interestingly, whereas mutations in KCNJ5, ATP1A1, and ATP2B3 are located in specific “hot spots,” a large number of mutations were reported in different exons of the CACNA1D gene, affecting more frequently segment M4 and M6 of the protein, implying the necessity of a large genotyping of CACNA1D in APA. Different studies established correlations between clinical and biological parameters and the mutational status of the tumor (10, 19, 20). Hence, patients with KCNJ5 mutations were more frequently female and diagnosed younger than patients harboring CACNA1D mutations and non-carriers (20); and CACNA1D mutations associated with smaller adenoma size (9, 20). Some studies reported also association between the mutational status and cellular composition of the adenoma. APA harboring KCNJ5 mutations would be composed essentially of zona fasciculata-like cells whereas those carrying CACNA1D mutations of a majority of zona glomerulosa-like cells (9), although this association was not replicated in all series (20). The exploration of the relationship between adrenal cortex remodeling and KCNJ5 mutations revealed the absence of association between the KCNJ5 mutational status and the nodulation score in the peritumoral tissue, the vascularization and the presence of zona glomerulosa hyperplasia in the peritumoral cortex, suggesting that KCNJ5 mutations are not likely to be responsible for a specific microenvironment propitious to promote adrenal cortex remodeling and APA formation (24).
Lessons from Potassium Channel Knock-Out Mouse Models
Though the role of all these mutations in abnormal aldosterone secretion has been clearly established, their impact in adenoma formation still remains unclear. Indeed, whereas in HAC15 cells, the overexpression of GIRK4 carrying the p.Thr158Ala mutation was responsible for a significant increase in aldosterone production, it induced, in parallel, a decrease in cell proliferation, independently of intracellular Ca2+ concentration (11). Likewise, the overexpression of p.Glu151Arg or p.Glu151Gln in HEK293T cells resulted in rapid Na+-dependent lethality (15). More extensively, a still open question is to know whether a modification in the ionic equilibrium and the regulation of the cell membrane potential are also able to promote adenoma formation. Response elements came some years ago with the investigation of mouse models in which TASK1 and/or TASK3 potassium channels were invalidated to determine the contributions of TASK channels to background K+ currents in adrenal zona glomerulosa cells and test their role in the control of aldosterone production (25, 26). TASK1 and TASK3 are two-pore domain K+ channels (K2P) that contribute largely to the very high background conductance of zona glomerulosa cells, making of zona glomerulosa cells highly sensitive sensor for plasma K+ concentration. They clamp the cell membrane to hyperpolarized voltages, restraining the production of aldosterone in absence of stimulus. In mouse adrenal cortex, whereas TASK1 expression is found throughout the zona glomerulosa and fasciculata, TASK3 expression is restricted to zona glomerulosa (25). Deletions of task1 and task3, respectively, lead to the development of hyperaldosteronism or low-renin hypertension (25–28) In task1−/− mice, hyperaldosteronism was due to aberrant functional zonation of the adrenal cortex, with intense cyp11b2 expression being localized in zona fasciculata instead of the zona glomerulosa. Interestingly, young task1−/− mice exhibited PA both in males and females; after puberty, this phenotype was only observed in females. Hyperaldosteronism was modulated by sexual hormones, being corrected by testosterone administration in task1−/− females and triggered by castration in males (26), suggesting that after puberty other factors, including task3 potassium channels, could substitute for the absence of task1 and promote compensatory mechanisms in male task1−/− mice (26). Deletion of task3 in mice leads to low-renin salt-sensitive hypertension, with suppressed plasma renin and aldosterone secretion that is not suppressible by increasing salt intake (27). Primary cultures of adrenocortical cells of these mice were strongly depolarized when compared with wild-type mice, and in fresh adrenal slices, calcium signaling was abnormal in zona glomerulosa cells (28). Finally, deletion of both task1 and task3 results in a marked depolarization of the zona glomerulosa cell membrane potential and a mild hyperaldosteronism with plasma aldosterone levels stimulated by a low-sodium diet but not suppressed by a high-sodium diet and partially responsive to AngII blockade (25). Interestingly, invalidation of these different potassium channels leads to hyperaldosteronism due to abnormal depolarization of the zona glomerulosa cell membrane resulting in increased intracellular Ca2+ concentration and stimulation of aldosterone biosynthesis; however formation of adrenal tumors has never been observed in these models indicating that other mechanisms are required to promote increased cell proliferation in APA. Although the invalidation of task1 and task3 in mice resulted in hyperaldosteronism or low-renin hypertension, to date no mutation in KCNK3 or KCNK9 genes has been reported in APA. However a reduced expression of TASK2, encoded by KCNK5, has been recently described in APA compared with normal adrenal (29), and the expression in H295R cells of a TASK2 dominant-negative mutant resulted in increased aldosterone production and CYP11B2 and StAR expression. Comparison of gene expression profiles of adrenal glands of task1−/− female and male mice allowed the identification of a cluster of genes closely associated with hyperaldosteronism (30), among them dickkopf3 (Dkk3), a member of the dickkopf family of Wnt signaling modulators. Inactivation of dkk3 in task1−/− mice resulted in the extension of the phenotype of hyperaldosteronism to male animals, without inducing abnormal zonation of the adrenal cortex (30). Interestingly, the expression of Dkk3 was found to be frequently downregulated in almost any cancer entity and emerged as a potential key player in tumor suppression (31). These results suggest that the Wnt/β-catenin pathway could play a role in the development of APA.
Activation of Sonic HedgeHog and Wnt/β-Catenin Pathway: Common Features of APA
The role of specific mutations of channels and ATPases in affecting aldosterone biosynthesis is now clearly established, whereas the question of the mechanism responsible for abnormal proliferation leading to adenoma formation is still open. In 2011, Lifton suggested that KCNJ5 mutations could be responsible for both autonomous aldosterone production and abnormal cell proliferation (7); however it has been rapidly shown that cells expressing mutated KCNJ5 channels were less proliferative (11), raising the questions as to the events leading to abnormal cell proliferation and adenoma formation? Two specific pathways are known to play a crucial role in adrenal development: the Sonic HedgeHog and the Wnt/β-catenin pathways.
Wnt Signaling Pathway in Proliferation and/or Aldosterone Biosynthesis
The Canonical and Non-Canonical Wnt/β-Catenin Pathway
Wnt signaling has been shown to be a key signaling pathway in both normal adrenal development and tumorigenesis. The “canonical Wnt signaling pathway” acts through the regulation of the amount of the transcriptional regulator β-catenin, which controls the expression of specific genes involved in development. In the absence of Wnt, β-catenin is a part of the axin complex consisting of adenomatous polyposis coli (APC), axin, glycogen synthase kinase-3β (GSK-3β), and casein kinase-1β (CK-1β). CK-1β and GSK-3β sequentially phosphorylate β-catenin in its N-terminal part resulting in its ubiquitination and degradation by the proteasome, thus preventing β-catenin from translocation to the nucleus and activation of specific Wnt target genes. The Wnt/β-catenin activation occurs through the binding of Wnt ligand to its cell surface receptor consisting of a frizzled receptor and its co-receptor, the low-density lipoprotein receptor related protein (LRP) 6 or LRP5. Activation of the receptor leads, through an unknown mechanism, to the phosphorylation of the disheveled (Dvl) protein, which prevents GSK-3β from phosphorylating specific substrates such as axin, APC, and β-catenin. Hence, the binding of Wnt ligands to their receptor results in the inhibition of β-catenin phosphorylation, dissociation from the axin complex, accumulation in the cytoplasm and translocation to the nucleus where it serves as a transcriptional coactivator of transcription factors of the T-cell factor (TCF)/lymphocyte enhancer factor (LEF) family. TCF/LEF target genes are involved in regulating cell proliferation, stem cells maintenance, and differentiation. To increase the complexity of the system, Wnt signaling independent of β-catenin has been described as “non-canonical Wnt signaling pathway.” It implicates small GTPases/jun N-terminal kinase (JNK) and intracellular calcium signaling (32). Finally, the activation of the Wnt pathway can be antagonized by specific natural molecules including secreted Frizzled-related proteins (sFrps) and Dickkopk (Dkk) family members. SFrps and Dkk are secreted proteins acting on different components of the Wnt signaling pathway. sFrps display a high sequence homology with the Wnt binding site of Frizzled allowing sFRP proteins to directly bind to Wnts, thus functioning as Wnt antagonists for both “canonical” and “non-canonical” pathways (33), whereas Dkk members are not only able to inhibit the Wnt coreceptors LRP5 and 6 but also to bind with high affinity to the transmembrane proteins Kremen 1 and 2, which also modulate Wnt signaling (33, 34).
Role of Wnt/β-Catenin Pathway in Adrenal Function
The Wnt/β-catenin pathway plays an important role in embryonic development, stem cell maintenance, and differentiation in many tissues. During the two last decades, the role of Wnt/β-catenin in adrenal development has been highlighted by the exploration of different mouse models in which expression of different components of the pathway were disrupted (35–37) or constitutively activated (38). The first element indicating a role of this pathway in adrenal is the localization of some of its components (i.e., β-catenin, wnt4, dkk3, sfrp1…) specifically in the subcapsular zone and in zona glomerulosa (39, 40). The loss of Wnt4 was associated with abnormal differentiation of the definitive zone of the adrenal cortex and aberrant migration of adrenocortical cells into the developing gonad (35) and with a decrease of the number of zona glomerulosa cells which results in a decrease of aldosterone production (36). Interestingly, the expression of Wnt4 mRNA has been reported to be higher in APA than in normal adult adrenocortical cells (41). Overexpression of WNT4 in human adrenocortical cells resulted in an increase of aldosterone biosynthesis, whereas DKK3 had an inhibitory effect, suggesting that Wnt/β-catenin pathway could be also involved in glomerulosa specific functions (42).
Modulation of β-Catenin Expression or Activation in the Adrenal Gland
The disruption of β-catenin specifically in adrenocortical cells, through the use of a sf-1 (steroidogenic factor-1)-Cre mouse, resulted in complete adrenal aplasia or defects in maintenance of the adult cortex resulting in depletion of adrenocortical cells (37). Inversely, the constitutive activation of β-catenin in the adrenal cortex resulted in profound adrenocortical zonation defects characterized by an ectopic activation of the zona glomerulosa differentiation program and inhibition of orthotopic zona fasciculata differentiation. Interestingly, at the age of 10 months, these mice develop hyperaldosteronism (38) similarly to mice expressing a defective APC allele (43), suggesting that constitutive activation of the Wnt/β-catenin pathway could play a role in the development of APA. In human adrenal, while β-catenin expression was found in the entire cortex, its activated form was restricted to zona glomerulosa cells (44), suggesting that restriction of β-catenin activation to sub-capsular regions and in zona glomerulosa is necessary for the development of functional zonation in the human adrenal cortex. Studies in human adrenocortical cells have indicated that Wnt signaling molecules may also have multiple actions on steroidogenesis, particularly in regulating aldosterone biosynthesis (36, 45). All these results suggest that aberrant Wnt signaling may be driving the development of APA. Recently, the activation of Wnt/β-catenin has been reported in two-thirds of APA (44, 46). Whereas activating mutations of the β-catenin are found in a wide variety of human cancers including adrenocortical tumors and adrenocortical adenoma, only few mutations were reported in APA (8, 47) strongly suggesting that the activation of β-catenin was not associated with the presence of mutation (47–49). Moreover, in adrenocortical carcinoma, the activation of β-catenin was associated with a poor prognosis (49), whereas in APA it was not associated with specific tumor characteristics. Thus Wnt/β-catenin activation may play distinct roles in APA compared to adrenal cortex carcinoma, contributing to aldosterone hypersecretion rather than to autonomous cell proliferation (46). The activation of β-catenin was not only associated with an increased expression of specific target genes, i.e., AXIN2 and LEF1, but also with down regulation of SFRP2, a member of the SFRP family of Wnt signaling inhibitors (46). Interestingly, sfrp2 knockout mice exhibit an increase in plasma aldosterone concentration, associated with ectopic expression of cyp11b2 in adrenal cortex, similarly to what observed in mice expressing the constitutive active form of β-catenin in adrenal cortex (46).
Shh Signaling Pathway in APA
Similarly to the Wnt/β-catenin signaling pathway, Sonic HedgeHog signaling (shh) is essential for adrenal gland development and maintenance. Shh encodes a secreted signal that belongs to the Hedgehog family. The activation of shh signaling occurs through its binding to a receptor complex formed by the twelve transmembrane domain protein patched-1 (PTCH1) and the G-protein coupled receptor Smoothened (SMO). In the presence of Shh, SMO is released from PTCH1 inhibition and activates the transcription factors GLI1, GLI2, and GLI3 (50). In rodent adult adrenals, Shh is expressed exclusively in the subcapsular region of the cortex in cells also expressing sf1, indicating their commitment to steroidogenic cells (51–53). Similarly, in human adult adrenals, the expression of SHH was found to be restricted to a few numbers of cells of the subcapsular region, where stem/progenitor cells are supposed to be localized (44, 54). Mice invalidated for Shh, specifically in Sf1 positive cells, exhibit reduced proliferation of capsular cells and a significant reduction of adrenocortex thickness and adrenal size but no modification of adrenal zonation. Moreover, the remaining adrenal cortex was able to synthetize steroids, indicating that shh is essential for expansion of the adrenal cortex but not for zonation and differentiation (55). Interestingly, the expression of Shh was found in APA as well as in the entire hyperplasic zona glomerulosa, with a similar pattern of expression than CYP11B2 and Dab2 (44). The activation of the SHH signaling pathway in APA was confirmed by transcriptomic analysis (44). These results suggest that APA have acquired some characteristics of stem/precursor cells or, alternatively, that reexpression of fetal markers from the definitive zone in the adrenal cortex could underlie excessive proliferation and APA formation. Remarkably, the antagonism of hedgehog signaling has been shown to inhibit the proliferation of H295R cells (56) and to decrease cell viability (57). Moreover, the inhibition of shh signaling pathway results in the inhibition of wnt/β-catenin signaling (57). Interestingly, the activation of shh signaling pathway was found to be increased in adult adrenocortical carcinoma (57) as well as in non-producing adenoma (56), suggesting a role in tumor formation or development.
Clock Genes in the Control of Aldosterone Production
Many physiological functions such as metabolism, BP, and renal function are regulated by the circadian clock (58–60). Up to 10% of the transcriptome has been estimated to be under the control of the circadian clock and a number of diseases are associated with clock gene disorders (61). The circadian timing system is organized in central and peripheral clocks. The central circadian clock is composed of specialized neurons in suprachiasmatic nuclei in the hypothalamus and is synchronized to the daily light/dark cycle through the retino-hypothalamic tract (62). The peripheral circadian clocks, found in most peripheral tissues, are synchronized to geophysical time through a wide range of master clock-dependent stimuli (62, 63). Four canonical proteins are the components of the circadian time clocks: period (Per)1–3, cryptochrome (Cry)1–2, Bmal1, and Clock. Clock and Bmal1 form a heterodimer that interacts with E-boxes to transcriptionally upregulate clock-controlled genes, which include Per and Cry (62, 64). Cry proteins act as potent transcriptional repressors that downregulate the transcription of E-box (CACGTG) enhancer-containing clock genes (including Per- and Cry-encoding genes) as well as a wide variety of clock-controlled genes (65, 66).
Different circadian mutant mice models show abnormalities in BP regulation and/or plasma aldosterone concentration. BP is decreased in clock knockout mice, accompanied by changes in circadian rhythms of urinary sodium and potassium excretion, and loss of the circadian rhythmicity of plasma aldosterone (67). A mouse model carrying a conditional allele of the circadian clock gene Bmal1 and expressing Cre recombinase under the endogenous Renin promoter (Bmal1lox/lox/Ren1dCre) loose the BMAL1 protein expression in the renin-secreting granular cells of the juxtaglomerular apparatus. These mice exhibit decreased BP, increased urine volume, changes in the circadian rhythm of urinary sodium excretion, and significantly reduced plasma aldosterone (63). Mice lacking the core clock components Cry1 and Cry2 (referred as Cry-null mice) show disrupted rhythmic behavior, physiology, and metabolism (68, 69). Interestingly, Cry-null mice exhibit salt-sensitive hypertension due to increased aldosterone production by the adrenal gland (70). Investigation of steroidogenic alterations in Cry-null mice showed chronic overexpression of Hsd3b6 mRNA and chronically enhanced 3β-hydroxysteroid dehydrogenase activity in adrenal cortex. Hsd3b6 encodes a dehydrogenase-isomerase specifically expressed in zona glomerulosa, which catalyzes the conversion of pregnenolone into progesterone, an enzymatic reaction required for aldosterone biosynthesis. The inactivation of Cry genes leads to chronically enhanced mineralocorticoid production, which, in turn, renders BP salt sensitive (70). On the other hand, it has been previously shown that Per1 and Cry2 modulate opposing actions on Per1 target gene expression in some tissues (71). Remarkably, Per1 knockout mice exhibit lower BP when compared to wild-type mice (60). To verify the hypothesis that Per1 plays a role in the regulation of aldosterone levels, Richards et al. have performed RNA silencing and pharmacological blockade of Per1 nuclear entry in the NCI-H295R human adrenal cell line, demonstrating that Hsd3b6 expression is decreased after Per1 knockdown in vitro (72). In addition, they have demonstrated that Per1 heterozygous mice exhibited lower plasma aldosterone levels and reduced Hsd3b6 mRNA expression in vivo, with a significant blunted circadian expression of this gene (72). In the human adrenal, two 3β-hydroxysteroid dehydrogenase isoform are expressed, namely HSD3B1 and HSD3B2 (70). In the adrenal cortex, expression of HSD3B1 is specific to the zona glomerulosa (70), suggesting its potential involvement in adrenal zona glomerulosa pathophysiology. Both HSD3B1 and HSD3B2 are found to be express in APA; and whereas HSD3B2 expression was higher than that of HSD3B1 in APA, only the level of HSD3B1 expression was correlated with plasmatic aldosterone concentration and CYP11B2 expression in APA, suggesting that HSD3B1 may contribute to autonomous aldosterone production in APA (73).
Conclusion
Despite major advances performed these last years in our understanding of the pathophysiology of APA development, the natural history of APA formation is still a matter of debate. Our current knowledge is not enough advanced to explain the mechanisms involved in APA formation. However, the identification, in about 50% of APA, of recurrent somatic mutations in genes coding for ionic channels and ATPases has elucidated the mechanism responsible for the autonomous aldosterone production. On the other hand, activation of the Wnt/β-catenin pathway or reexpression of stem/precursor cell markers, i.e., shh, could explain the abnormal proliferation leading to the formation of an adenoma. It is possible that APA formation is the result of the combination of two events: (1) the activation of signaling pathways such as wnt/β-catenin or shh pathways driving abnormal cell proliferation and creating a favorable environment for (2) the occurrence of recurrent somatic mutations responsible for autonomous aldosterone production.
Although it was suggested that genetic alterations leading to abnormal calcium signaling are sufficient for both abnormal proliferation and inappropriate aldosterone production in APA, there are some evidences suggesting that mutations in potassium and calcium channel and ATPases may not be sufficient for promoting cell proliferation and tumor formation. It could be speculated that some groups of cells start to abnormally proliferate creating a propitious environment for the emergence of specific mutations affecting ionic channels and ATPases leading to increased aldosterone production. Further mechanistic insight may come from specific mouse models developing a phenotype of hyperaldosteronism in the context of an APA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Sources of funding: This work was funded through institutional support from INSERM and by the Agence Nationale pour la Recherche (ANR Blanc 2011, No.: 11-BSV1 005 03, ANR-13-ISV1-0006-01), the Fondation pour la Recherche Médicale (ING20101221177, DEQ20140329556), the Programme Hospitalier de Recherche Clinique (PHRC grant AOM 06179), and by grants from INSERM and Ministère Délégué à la Recherche et des Nouvelles Technologies.
References
1. Spat A, Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev (2004) 84:489–539. doi:10.1152/physrev.00030.2003
2. Hannemann A, Wallaschofski H. Prevalence of primary aldosteronism in patient’s cohorts and in population-based studies – a review of the current literature. Horm Metab Res (2012) 44:157–62. doi:10.1055/s-0031-1295438
3. Bakris G, Calhoun D, Egan B, Hellmann C, Dolker M, Kingma I. Orlistat improves blood pressure control in obese subjects with treated but inadequately controlled hypertension. J Hypertens (2002) 20:2257–67. doi:10.1097/00004872-200211000-00026
4. Mulatero P, Monticone S, Bertello C, Viola A, Tizzani D, Iannaccone A, et al. Long-term cardio- and cerebrovascular events in patients with primary aldosteronism. J Clin Endocrinol Metab (2013) 98:4826–33. doi:10.1210/jc.2013-2805
5. Savard S, Amar L, Plouin PF, Steichen O. Cardiovascular complications associated with primary aldosteronism: a controlled cross-sectional study. Hypertension (2013) 62:331–6. doi:10.1161/HYPERTENSIONAHA.113.01060
6. Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2008) 93:3266–81. doi:10.1210/jc.2008-0104
7. Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science (2011) 331:768–72. doi:10.1126/science.1198785
8. Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, et al. Lifton, somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet (2013) 45:1050–4. doi:10.1038/ng.2695
9. Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet (2013) 45:1055–60. doi:10.1038/ng.2716
10. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet (2013) 45:e1–2. doi:10.1038/ng.2550
11. Oki K, Plonczynski MW, Luis Lam M, Gomez-Sanchez EP, Gomez-Sanchez CE. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology (2012) 153:1774–82. doi:10.1210/en.2011-1733
12. Charmandari E, Sertedaki A, Kino T, Merakou C, Hoffman DA, Hatch MM, et al. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. J Clin Endocrinol Metab (2012) 97(8):E1532–9. doi:10.1210/jc.2012-1334
13. Murthy M, Azizan EA, Brown MJ, O’Shaughnessy MK. Characterization of a novel somatic KCNJ5 mutation delI157 in an aldosterone-producing adenoma. J Hypertens (2012) 30(9):1827–33. doi:10.1097/HJH.0b013e328356139f
14. Akerstrom T, Crona J, Delgado Verdugo A, Starker LF, Cupisti K, Willenberg HS, et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS One (2012) 7:e41926. doi:10.1371/journal.pone.0041926
15. Scholl UI, Nelson-Williams C, Yue P, Grekin R, Wyatt RJ, Dillon MJ, et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U S A (2012) 109:2533–8. doi:10.1073/pnas.1121407109
16. Mulatero P, Tauber P, Zennaro MC, Monticone S, Lang K, Beuschlein F, et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension (2012) 59:235–40. doi:10.1161/HYPERTENSIONAHA.111.183996
17. Monticone S, Hattangady NG, Nishimoto K, Mantero F, Rubin B, Cicala MV, et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells. J Clin Endocrinol Metab (2012) 97:E1567–72. doi:10.1210/jc.2011-3132
18. Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab (2008) 93:3117–23. doi:10.1210/jc.2008-0594
19. Boulkroun S, Beuschlein F, Rossi GP, Golib-Dzib JF, Fischer E, Amar L, et al. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension (2012) 59:592–8. doi:10.1161/HYPERTENSIONAHA.111.186478
20. Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension (2014) 64:354–61. doi:10.1161/HYPERTENSIONAHA.114.03419
21. Kitamoto T, Suematsu S, Matsuzawa Y, Saito J, Omura M, Nishikawa T. Comparison of cardiovascular complications in patients with and without KCNJ5 gene mutations harboring aldosterone-producing adenomas. J Atheroscler Thromb (2014) 22(2):191–200. doi:10.5551/jat.24455
22. Rossi GP, Cesari M, Letizia C, Seccia TM, Cicala MV, Zinnamosca L, et al. KCNJ5 gene somatic mutations affect cardiac remodelling but do not preclude cure of high blood pressure and regression of left ventricular hypertrophy in primary aldosteronism. J Hypertens (2014) 32:1514–21. doi:10.1097/HJH.0000000000000186
23. Taguchi R, Yamada M, Nakajima Y, Satoh T, Hashimoto K, Shibusawa N, et al. Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J Clin Endocrinol Metab (2012) 97:1311–9. doi:10.1210/jc.2011-2885
24. Boulkroun S, Golib Dzib JF, Samson-Couterie B, Rosa FL, Rickard AJ, Meatchi T, et al. KCNJ5 mutations in aldosterone producing adenoma and relationship with adrenal cortex remodeling. Mol Cell Endocrinol (2013) 371:221–7. doi:10.1016/j.mce.2013.01.018
25. Davies LA, Hu C, Guagliardo NA, Sen N, Chen X, Talley EM, et al. TASK channel deletion in mice causes primary hyperaldosteronism. Proc Natl Acad Sci U S A (2008) 105:2203–8. doi:10.1073/pnas.0712000105
26. Heitzmann D, Derand R, Jungbauer S, Bandulik S, Sterner C, Schweda F, et al. Invalidation of TASK1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. EMBO J (2008) 27:179–87. doi:10.1038/sj.emboj.7601934
27. Guagliardo NA, Yao J, Hu C, Schertz EM, Tyson DA, Carey RM, et al. TASK-3 channel deletion in mice recapitulates low-renin essential hypertension. Hypertension (2012) 59:999–1005. doi:10.1161/HYPERTENSIONAHA.111.189662
28. Penton D, Bandulik S, Schweda F, Haubs S, Tauber P, Reichold M, et al. Task3 potassium channel gene invalidation causes low renin and salt-sensitive arterial hypertension. Endocrinology (2012) 153:4740–8. doi:10.1210/en.2012-1527
29. Lenzini L, Caroccia B, Campos AG, Fassina A, Belloni AS, Seccia TM, et al. Lower expression of the TWIK-related acid-sensitive K+ channel 2 (TASK-2) gene is a hallmark of aldosterone-producing adenoma causing human primary aldosteronism. J Clin Endocrinol Metab (2014) 99:E674–82. doi:10.1210/jc.2013-2900
30. El Wakil A, Bandulik S, Guy N, Bendahhou S, Zennaro MC, Niehrs C, et al. Dkk3 is a component of the genetic circuitry regulating aldosterone biosynthesis in the adrenal cortex. Hum Mol Genet (2012) 21:4922–9. doi:10.1093/hmg/dds333
31. Veeck J, Dahl E. Targeting the Wnt pathway in cancer: the emerging role of Dickkopf-3. Biochim Biophys Acta (2012) 1825:18–28. doi:10.1016/j.bbcan.2011.09.003
32. Rao TP, Kuhl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res (2010) 106:1798–806. doi:10.1161/CIRCRESAHA.110.219840
33. Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci (2003) 116:2627–34. doi:10.1242/jcs.00623
34. Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene (2006) 25:7469–81. doi:10.1038/sj.onc.1210054
35. Jeays-Ward K, Hoyle C, Brennan J, Dandonneau M, Alldus G, Capel B, et al. Endothelial and steroidogenic cell migration are regulated by WNT4 in the developing mammalian gonad. Development (2003) 130:3663–70. doi:10.1242/dev.00591
36. Heikkila M, Peltoketo H, Leppaluoto J, Ilves M, Vuolteenaho O, Vainio S. Wnt-4 deficiency alters mouse adrenal cortex function, reducing aldosterone production. Endocrinology (2002) 143:4358–65. doi:10.1210/en.2002-220275
37. Kim AC, Reuter AL, Zubair M, Else T, Serecky K, Bingham NC, et al. Targeted disruption of beta-catenin in Sf1-expressing cells impairs development and maintenance of the adrenal cortex. Development (2008) 135:2593–602. doi:10.1242/dev.021493
38. Berthon A, Sahut-Barnola I, Lambert-Langlais S, de Joussineau C, Damon-Soubeyrand C, Louiset E, et al. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum Mol Genet (2010) 19:1561–76. doi:10.1093/hmg/ddq029
39. El Wakil A, Lalli E. The Wnt/beta-catenin pathway in adrenocortical development and cancer. Mol Cell Endocrinol (2011) 332:32–7. doi:10.1016/j.mce.2010.11.014
40. Drelon C, Berthon A, Mathieu M, Martinez A, Val P. Adrenal cortex tissue homeostasis and zonation: a WNT perspective. Mol Cell Endocrinol (2014) 408:156–64. doi:10.1016/j.mce.2014.12.014
41. Kuulasmaa T, Jaaskelainen J, Suppola S, Pietilainen T, Heikkila P, Aaltomaa S, et al. WNT-4 mRNA expression in human adrenocortical tumors and cultured adrenal cells. Horm Metab Res (2008) 40:668–73. doi:10.1055/s-2008-1078716
42. Chen M, Hornsby PJ. Adenovirus-delivered DKK3/WNT4 and steroidogenesis in primary cultures of adrenocortical cells. Horm Metab Res (2006) 38:549–55. doi:10.1055/s-2006-950500
43. Bhandaru M, Kempe DS, Rotte A, Rexhepaj R, Kuhl D, Lang F. Hyperaldosteronism, hypervolemia, and increased blood pressure in mice expressing defective APC. Am J Physiol Regul Integr Comp Physiol (2009) 297:R571–5. doi:10.1152/ajpregu.00070.2009
44. Boulkroun S, Samson-Couterie B, Golib-Dzib JF, Amar L, Plouin PF, Sibony M, et al. Aldosterone-producing adenoma formation in the adrenal cortex involves expression of stem/progenitor cell markers. Endocrinology (2011) 152:4753–63. doi:10.1210/en.2011-1205
45. Schinner S, Willenberg HS, Krause D, Schott M, Lamounier-Zepter V, Krug AW, et al. Adipocyte-derived products induce the transcription of the StAR promoter and stimulate aldosterone and cortisol secretion from adrenocortical cells through the Wnt-signaling pathway. Int J Obes (Lond) (2007) 31:864–70. doi:10.1038/sj.ijo.0803508
46. Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, et al. WNT/beta-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum Mol Genet (2014) 23:889–905. doi:10.1093/hmg/ddt484
47. Tadjine M, Lampron A, Ouadi L, Bourdeau I. Frequent mutations of beta-catenin gene in sporadic secreting adrenocortical adenomas. Clin Endocrinol (Oxf) (2008) 68:264–70. doi:10.1111/j.1365-2265.2007.03033.x
48. Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagnere AM, et al. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res (2005) 65:7622–7. doi:10.1158/0008-5472.CAN-05-0593
49. Gaujoux S, Grabar S, Fassnacht M, Ragazzon B, Launay P, Libe R, et al. Beta-catenin activation is associated with specific clinical and pathologic characteristics and a poor outcome in adrenocortical carcinoma. Clin Cancer Res (2011) 17:328–36. doi:10.1158/1078-0432.CCR-10-2006
50. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev (2001) 15:3059–87. doi:10.1101/gad.938601
51. Ching S, Vilain E. Targeted disruption of sonic hedgehog in the mouse adrenal leads to adrenocortical hypoplasia. Genesis (2009) 47(9):628–37. doi:10.1002/dvg.20532
52. King P, Paul A, Laufer E. Shh signaling regulates adrenocortical development and identifies progenitors of steroidogenic lineages. Proc Natl Acad Sci U S A (2009) 106:21185–90. doi:10.1073/pnas.0909471106
53. Guasti L, Paul A, Laufer E, King P. Localization of sonic hedgehog secreting and receiving cells in the developing and adult rat adrenal cortex. Mol Cell Endocrinol (2011) 336:117–22. doi:10.1016/j.mce.2010.11.010
54. Kim AC, Barlaskar FM, Heaton JH, Else T, Kelly VR, Krill KT, et al. In search of adrenocortical stem and progenitor cells. Endocr Rev (2009) 30:241–63. doi:10.1210/er.2008-0039
55. Huang CC, Miyagawa S, Matsumaru D, Parker KL, Yao HH. Progenitor cell expansion and organ size of mouse adrenal is regulated by sonic hedgehog. Endocrinology (2010) 151:1119–28. doi:10.1210/en.2009-0814
56. Werminghaus P, Haase M, Hornsby PJ, Schinner S, Schott M, Malendowicz LK, et al. Hedgehog-signaling is upregulated in non-producing human adrenal adenomas and antagonism of hedgehog-signaling inhibits proliferation of NCI-H295R cells and an immortalized primary human adrenal cell line. J Steroid Biochem Mol Biol (2014) 139:7–15. doi:10.1016/j.jsbmb.2013.09.007
57. Gomes DC, Leal LF, Mermejo LM, Scrideli CA, Martinelli CE Jr, Fragoso MC, et al. Sonic hedgehog signaling is active in human adrenal cortex development and deregulated in adrenocortical tumors. J Clin Endocrinol Metab (2014) 99:E1209–16. doi:10.1210/jc.2013-4098
58. Agarwal R. Regulation of circadian blood pressure: from mice to astronauts. Curr Opin Nephrol Hypertens (2010) 19:51–8. doi:10.1097/MNH.0b013e3283336ddb
59. Richards J, Greenlee MM, Jeffers LA, Cheng KY, Guo L, Eaton DC, et al. Inhibition of alphaENaC expression and ENaC activity following blockade of the circadian clock-regulatory kinases CK1delta/epsilon. Am J Physiol Renal Physiol (2012) 303:F918–27. doi:10.1152/ajprenal.00678.2011
60. Stow LR, Richards J, Cheng KY, Lynch IJ, Jeffers LA, Greenlee MM, et al. The circadian protein period 1 contributes to blood pressure control and coordinately regulates renal sodium transport genes. Hypertension (2012) 59:1151–6. doi:10.1161/HYPERTENSIONAHA.112.190892
61. Hastings MH, Reddy AB, Maywood ES. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci (2003) 4:649–61. doi:10.1038/nrn1177
62. Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol (2010) 72:517–49. doi:10.1146/annurev-physiol-021909-135821
63. Tokonami N, Mordasini D, Pradervand S, Centeno G, Jouffe C, Maillard M, et al. Local renal circadian clocks control fluid-electrolyte homeostasis and BP. J Am Soc Nephrol (2014) 25:1430–9. doi:10.1681/ASN.2013060641
64. Albrecht U. The mammalian circadian clock: a network of gene expression. Front Biosci (2004) 9:48–55. doi:10.2741/1196
65. Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science (2003) 302:255–9. doi:10.1126/science.1086271
66. Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, et al. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell (1999) 98:193–205. doi:10.1016/S0092-8674(00)81014-4
67. Nikolaeva S, Pradervand S, Centeno G, Zavadova V, Tokonami N, Maillard M, et al. The circadian clock modulates renal sodium handling. J Am Soc Nephrol (2012) 23:1019–26. doi:10.1681/ASN.2011080842
68. van der Horst GT, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, et al. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature (1999) 398:627–30. doi:10.1038/19323
69. Vitaterna MH, Selby CP, Todo T, Niwa H, Thompson C, Fruechte EM, et al. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc Natl Acad Sci U S A (1999) 96:12114–9. doi:10.1073/pnas.96.21.12114
70. Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, et al. Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med (2010) 16:67–74. doi:10.1038/nm.2061
71. Richards J, All S, Skopis G, Cheng KY, Compton B, Srialluri N, et al. Opposing actions of Per1 and Cry2 in the regulation of Per1 target gene expression in the liver and kidney. Am J Physiol Regul Integr Comp Physiol (2013) 305:R735–47. doi:10.1152/ajpregu.00195.2013
72. Richards J, Cheng KY, All S, Skopis G, Jeffers L, Lynch IJ, et al. A role for the circadian clock protein Per1 in the regulation of aldosterone levels and renal Na+ retention. Am J Physiol Renal Physiol (2013) 305:F1697–704. doi:10.1152/ajprenal.00472.2013
Keywords: primary aldosteronism, aldosterone producing adenoma, somatic mutations, potassium channels, calcium channels, ATPase, wnt/β-catenin pathway, shh signaling pathway
Citation: Boulkroun S, Fernandes-Rosa FL and Zennaro M-C (2015) Molecular and cellular mechanisms of aldosterone producing adenoma development. Front. Endocrinol. 6:95. doi: 10.3389/fendo.2015.00095
Received: 27 March 2015; Accepted: 26 May 2015;
Published: 11 June 2015
Edited by:
Pierre Val, Centre national de la recherche scientifique, FranceReviewed by:
Yewei Xing, University of Michigan, USAJohn Watson Funder, Prince Henry’s Institute, Australia
Copyright: © 2015 Boulkroun, Fernandes-Rosa and Zennaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheerazed Boulkroun, UMRS_970, Paris Cardiovascular Research Center (PARCC), Institut National de la Santé et de la Recherche Médicale (INSERM), 56 rue Leblanc, Paris 75015, France, sheerazed.boulkroun@inserm.fr